

Abstract
More than 120 million people worldwide are chronically infected with hepatitis C virus (HCV), making HCV infection the leading cause of liver transplantation in developed countries. Treatment is limited, and efficacy depends upon the infecting strain and the initial viral load. The HCV envelope glycoproteins (E1 and E2) are involved in receptor binding, virus-cell fusion, and entry into the host cell. HCV infection proceeds by endosomal acidification, suggesting that fusion of the viral envelope with cellular membranes is a pH-triggered event. E2 consists of an amino-terminal ectodomain, an amphipathic helix that forms a stem region, and a carboxy-terminal membrane-associating segment. We have devised a novel expression system for the production of a secreted form of E2 ectodomain (eE2) from mammalian cells and performed a comprehensive biochemical and biophysical characterization. eE2 is properly folded, as determined by binding to human CD81, blocking of infection of cell culture-derived HCV, and recognition by antibodies from patients chronically infected with different genotypes of HCV. The glycosylation pattern, number of disulfide bonds, oligomerization state, and secondary structure of eE2 have been characterized using mass spectrometry, size exclusion chromatography, circular dichroism, and analytical ultracentrifugation. These results advance the understanding of E2 and may assist in the design of an HCV vaccine and entry inhibitor.
Since its initial discovery in the late 1980s, hepatitis C virus (HCV) has been identified in all parts of the world, with at least 6 major genotypes and about 50 subtypes isolated. Currently, 2 to 3% of the human population is chronically infected, making HCV a global health problem (55; J. F. Perz, L. A. Farrington, C. Pecoraro, Y. J. F. Hutin, and G. L. Armstrong, presented at the 42nd Annual Meeting of the Infectious Disease Society of America, Boston, MA, 2004). HCV infection is the leading cause of liver transplantation in the developed world and results in 10,000 to 20,000 deaths annually in the United States (7). Infection leads to chronic liver disease, cirrhosis, and in many cases hepatocellular carcinoma. The only approved treatment is combination therapy with pegylated interferon and ribavirin, which has various efficacies depending upon the genotype and the initial viral load (17).
HCV is the only member of the Hepacivirus genus within the Flaviviridae family (39). Its genome consists of a 9.6-kb positive-sense, single-stranded RNA with a single open reading frame. The viral genome is translated in a cap-independent manner via an internal ribosome entry site located within the 5′ nontranslated region (1). Translation generates a viral polyprotein that is proteolytically processed into 10 separate proteins by cellular and virus-encoded proteases. The N-terminal region of the polyprotein is cleaved by cellular signal peptidase and signal peptide peptidase to yield the structural components of the virus particle (core and envelope proteins E1 and E2) and a putative ion channel (p7). The mature nonstructural proteins (NS2, NS3, NS4A, NS4B, NS5A, and NS5B) are liberated by two essential virus-encoded enzymes: the NS2-3 cysteine protease and the NS3-4A serine protease (1). NS3-NS5B constitutes the minimal RNA replication machinery. Replication occurs in association with perinuclear and endoplasmic reticulum (ER) membranes and requires the synthesis of a negative-strand RNA intermediate; this provides the template for positive-strand RNA synthesis for new virion packaging (42). It is thought that genomic RNA is encapsulated by the core and buds into the ER, deriving the lipid envelope and embedded glycoproteins. The newly created HCV particles progress through the secretory pathway and are released at the cell membrane.
The HCV envelope protein E2 is found on the outer shell of the virus particle, mediates virus attachment by interacting with several cellular receptors, and contains hypervariable regions that are likely to facilitate immune evasion (21). Upon binding to the target cell, infection proceeds by endosomal acidification, suggesting that fusion of the viral envelope with cellular membranes is a pH-triggered event (38, 46, 57, 61). Numerous candidate cellular receptors have been identified, including CD81 (50), scavenger receptor class B type I (SR-BI) (54), claudin-1 (22), and occludin (41, 51). CD81 and SR-BI have been shown to directly interact with E2 (29, 31). CD81 is an integral membrane protein of the tetraspanin family, and the E2 binding site has been mapped to the larger of the two extracellular loops (large extracellular loop [LEL]) (19). The addition of exogenous human CD81-LEL or antibodies against CD81 has been shown to inhibit infection (32). SR-BI is highly expressed on hepatocytes, and antibodies against SR-BI and small interfering RNA-mediated downregulation of SR-BI expression result in a significant inhibition of HCV infectivity (9, 36).
E2 is a type I transmembrane protein with an amino-terminal ectodomain and a carboxy-terminal membrane-associating segment. The ectodomain and transmembrane helix are linked by a stem region that is proposed to be an amphipathic helix with a heptad repeat sequence (18). During genome translation, E2 is targeted to the ER lumen by a signal sequence located at the carboxy terminus of E1 (37). The transmembrane and stem regions of E2 are involved in ER retention (12) and in the formation of noncovalent E1/E2 heterodimers (18, 47). Since HCV is thought to bud into the ER, retention of the glycoproteins at the ER membrane ensures their placement on the virion particles. The E2 ectodomain (eE2) is defined as the minimal carboxy-terminal deletion that results in secretion of properly folded protein and has been mapped to the first 334 amino acids (45). E2 is highly modified posttranslationally with numerous N-linkage and O-linkage (23) glycosylation consensus sites and may have as many as 9 disulfide bonds from 18 absolutely conserved cysteines.
Folding of the envelope proteins is slow and requires the ER chaperone machinery, including calnexin (11). Studies of recombinant E2 expression have yielded two different forms of the molecule: (i) a glycosylated protein with intramolecular disulfide bonds that is believed to be the active form and (ii) high-molecular-weight aggregates caused by intermolecular disulfide bonds. In fact, aggregation is so common that a nonproductive folding pathway has been proposed as a physiologically relevant part of the HCV life cycle (20). The formation of disulfide-bonded aggregates and misfolded protein has limited functional, structural, and biophysical studies of the HCV glycoproteins. Described here is a method to produce significant quantities of eE2 using human cells. The resulting protein is not aggregated, is recognized by antibodies from chronically infected HCV patient sera, and blocks HCV infection in vitro. Furthermore, we have extensively characterized the glycosylation pattern, oxidation state, and oligomeric nature of eE2 using a variety of biophysical techniques. Our purified eE2 protein constitutes an exceptional tool for probing E2 function and may facilitate the design of an entry inhibitor or HCV vaccine.
MATERIALS AND METHODS
Production of stable cell lines.
HEK293T cells were cultured in Dulbecco's modified Eagle medium supplemented with 10% FBS. A six-well plate was seeded with 0.5 × 106 cells per well, and the pPro-eE2-Fc vector (from the J6 strain) was transfected the following day using FuGene-HD (Roche Diagnostics, Basel, Switzerland). After 3 days, the cells were placed under hygromycin (Calbiochem, San Diego, CA) selection at 75 μg/ml. Individual colonies were selected, expanded, and tested for eE2-Fc expression using an anti-Fc enzyme-linked immunosorbent assay (ELISA).
ELISA for eE2-Fc.
MaxiSorp plates (Nunc, Thermo Fisher Scientific, Rochester, NY) were coated with 100 μl of supernatant for 2 h at room temperature. The wells were washed 3× with 200 μl of phosphate-buffered saline (PBS) plus 0.05% Tween 20 (PBS-T) and then blocked with 200 μl of 2% bovine serum albumin in PBS-T for 1 h at room temperature. After three more washes with PBS-T, 100 μl of goat anti-Fc antibody (Pierce, Thermo Fisher Scientific, Rochester, NY) at a 1:15,000 dilution (in PBS-T) was incubated for 1 h at room temperature. The ELISA was developed with the tetramethylbenzedene substrate (Pierce) and quantified using the SpectraMAX 250 plate reader and SOFTMax 2.6 software.
Western blotting for eE2-Fc.
Supernatants from mock-transfected or eE2-expressing cell lines were boiled in 2× sample buffer containing β-mercaptoethanol and run on a 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel. The gel was transferred to a nitrocellulose membrane and blocked for 1 h in 5% nonfat dry milk in PBS-T. The blot was then incubated in peroxidase-conjugated goat anti-human immunoglobulin G (Fcγ) (Thermo Scientific) at a 1:1,000 dilution in 5% nonfat dry milk in PBS-T for 1 h at room temperature. After three 10-min washes in PBS-T, the blot was developed using the SuperSignal West Pico chemiluminescent substrate (Pierce).
Expression and purification of eE2 and eE2-C656S.
Supernatants from stable cell lines expressing either eE2 or eE2-C656S were harvested, centrifuged to remove cellular debris, and filtered through a 0.22-μm membrane. The eE2-Fc (or eE2-C656S-Fc) protein was applied to protein A-conjugated resin (GE Healthcare, Piscataway, NJ) overnight with gentle rocking. The resin was washed with buffer (50 mM HEPES [pH 7.5], 150 mM KCl, 5% glycerol) and incubated with thrombin protease to release the protein from the Fc tag. After cleavage, the protein eluate was consolidated and the concentration was determined by Bradford assay, yielding 1 to 2 mg of eE2 per liter of supernatant.
Deglycosylation of eE2.
eE2 was deglycosylated using either endoglycosidase H (Endo H) or peptide-N-glycosidase F (PNGase F) according to the manufacturer's protocol (New England Biolabs, Ipswich, MA). Briefly, 20 μg of eE2 was denatured, and 10 U of Endo H or PNGase F was added. The reaction mixture was incubated at 37°C for 1 h and analyzed by SDS-PAGE followed by Coomassie staining.
Mapping the N-linked glycosylation sites.
The eE2 protein sample was denatured in 6 M urea and then reduced with 10 mM dithiothreitol (DTT) for 30 min at 60°C. After denaturation, 20 mM iodoacetamide was added to alkylate sulfhydryl groups and incubated in the dark for 1 h at room temperature. Following this treatment, the sample was buffer exchanged into 50 mM NH4HCO3. The sample was digested using either sequencing-grade trypsin (Promega, Madison, WI) or chymotrypsin (Roche Diagnostics) according to the manufacturer's protocol. Digested samples were dried via vacuum centrifugation and reconstituted in 50 mM NH4HCO3. Samples were deglycosylated with 50 U of PNGase F (New England Biolabs, Massachusetts) by incubation for 1 h at 37°C, and the reaction was stopped with 0.1% trifluoroacetic acid.
All liquid chromatography-tandem mass spectrometry (LC-MS/MS) experiments were performed using the U3000 high-performance LC system (Dionex, Sunnyvale, CA) in nano-LC mode on line with the liquid trap quadrupole mass spectrometer (Thermo Fisher Scientific). Samples were first solubilized in 0.1% trifluoroacetic acid and loaded onto a 75-μm by 12-cm emitter column self-packed with Magic C18AQ resin (3 μm, 200 Å; Michrom Bioresources Inc., Aubum, CA). The sample was eluted using a linear gradient from 98% of 0.1% formic acid in water to 45% of 0.1% formic acid in acetonitrile over 30 min. MS data were acquired using a data-dependent acquisition procedure with a full-scan cyclic series. This was followed by zoom scans and MS/MS scans of the five most intense ions with a repeat count of 2 and a dynamic exclusion duration of 60 s.
The LC-MS/MS data were searched against a human database using a local version of the Global Proteome Machine (local implementation of the Global Protome Machine [13]). Carbamidomethylation of cysteine was used as the fixed modification, while oxidation of methionine and deamination of asparagine were used as potential modifications. Manual interpretation and peak integration were performed on all peptide peaks covering potential glycosylation sites (NXT/S).
Gel filtration analysis of eE2 and eE2-C656S.
Purified eE2 protein was loaded onto a Superdex200 gel filtration column (GE Healthcare, Piscataway, NJ) equilibrated with HEPES buffer (50 mM HEPES [pH 7.5], 150 mM KCl, 5% glycerol).
Free cysteine analysis.
To label free cysteines, the protein sample was incubated with a 20-fold molar excess of N-ethylmaleimide (NEM) and 6 M guanidine-HCl at room temperature for 1 h in the dark. The sample was then buffer exchanged to 6 M guanidine-HCl using a spin filter and washed three times with 400 μl of 6 M guanidine-HCl to remove the NEM. Disulfide bonds were reduced by adding 10 mM DTT at 60°C for 30 min. The newly generated free sulfhydryl groups were alkylated with 20 mM iodoacetamide (IAM) at room temperature for 1 h in the dark. After buffer exchange into 50 mM NH4HCO3, the sample was digested with trypsin protease at 37°C overnight. The sample was then deglycosylated with PNGase F (100 U) at 37°C for 3 h and acidified prior to LC-MS/MS analysis.
The LC-MS/MS data were searched using the Sequest software program against the sequence of the target protein. +57 Da (alkylation by IAM) and +125 Da (alkylation by NEM) on cysteine, oxidation of methionine (+16 Da), and deamination of asparagine (+1 Da) were used as potential modifications. The identification was confirmed manually.
Analytical ultracentrifugation.
All sedimentation experiments were performed using a Beckman Optima XL-I ultracentrifuge at the Center for Analytical Ultracentrifugation of Macromolecular Assemblies at the University of Texas Health Science Center at San Antonio. Sedimentation velocity data were analyzed with the UltraScan software program (15), version 9.9 (14). All measurements were made at 230 nm in intensity mode, at 20°C, and at 37,000 rpm, using standard epon two-channel centerpieces. All samples were measured in 25 mM sodium phosphate buffer containing 50 mM KCl, adjusted to pH 7.0. Concentration dependency of the sedimentation data was assessed by sedimenting the sample at both high (∼ 0.8 optical density at 230 nm [OD230]) and low (∼0.25 OD230) concentrations. Hydrodynamic corrections for buffer density and viscosity were made according to methods outlined by Laue et al. (35) and as implemented in UltraScan. The data were analyzed by two-dimensional spectrum analysis (6) using the ASTFEM-RA solution (8) with simultaneous removal of time-invariant noise. Molecular weight and shape distributions obtained in the two-dimensional spectrum analysis were further refined by Monte Carlo analysis (16). The calculations were performed on the Lonestar cluster at the Texas Advanced Computing Center at the University of Texas at Austin and at the Bioinformatics Core Facility at the University of Texas Health Science Center at San Antonio.
CD.
The protein sample was buffer exchanged into 25 mM sodium phosphate (pH 7.0 or pH 5.0) and 50 mM KCl. The circular dichroism (CD) spectra in the wavelength range of 195 to 260 nm were measured at 0.5-nm intervals on an Aviv spectropolarimeter, model 400 (Lakewood, NJ), at 25°C. A quartz cell with a path length of 0.1 cm was used. The data are presented in millidegrees.
eE2 ELISA using human sera.
Ninety-six-well enzyme immunoassay/radioimmunoassay (Corning, Lowell, MA) were coated with 100 μl of a 1 μg/ml solution of eE2 in NaHCO3 overnight at 4°C. The plates were washed twice with 200 μl/well PBS-T and then blocked with a 10% solution of normal goat serum in PBS-T (Jackson ImmunoResearch, West Grove, PA) for 1 h at 37°C. Human serum was isolated from whole-blood samples (IRB no. 1358-2004; Emory University School of Medicine, principal investigator Arash Grakoui) collected in SST tubes (Becton Dickenson, Franklin Lakes, NJ) via centrifugation and frozen in aliquots at −80°C. Ten-fold serial dilutions were made for each serum sample using binding buffer composed of 0.1% normal goat serum in PBS-T. One hundred microliters of the dilutions was added to each well of the plates and incubated for 90 min at room temperature. The plates were washed eight times with PBS. One hundred microliters of goat anti-human immunoglobulin G-biotin conjugate (Biosource, Camarillo, CA) diluted 1:20,000 in binding buffer was added and allowed to incubate for 90 min at room temperature. Finally, 100 μl streptavidin-horseradish peroxidase (HRP) conjugate (Biosource) was added to each well at a 1:2,000 dilution and incubated for 45 min at room temperature. Using tetramethylbenzidine substrate solution (Ebioscience, San Diego, CA), absorbance was measured using a VersaMax microplate reader and SoftMax Pro software (Molecular Devices, Sunnyvale, CA).
Cell culture-derived HCV (HCVcc) infection in presence of purified proteins.
Approximately 100 50% tissue culture infective doses of Cp7 (44) viruses were incubated with twofold dilutions of the purified eE2, eE2-C656S, glutathione S-transferase (GST), GST-CD81-LEL, or GST-mCD81 starting at 200 μg/ml. Huh-7.5 cells (6.0 × 103) were seeded into a collagen-coated 96-well plate. The virus-protein mixture was incubated with the cells for 3 days at 37°C. Cells were stained by immmunohistochemistry as previously described (44).
Cytotoxicity.
Huh-7.5 cells were incubated with various dilutions of the purified proteins as described above. Three days later, cells were washed twice with PBS, harvested by trypsinization, and resuspended in 100 μl of PBS. Cells were stained with BD Via-Probe (BD Biosciences, San Jose, CA) according to the manufacturer's instructions and counted using FACSCalibur (BD Biosciences) equipment and FlowJo (v8) analysis software.
Expression and purification of GST and GST-CD81-LEL.
CD81-LEL was expressed with an amino-terminal GST tag and a carboxy-terminal histidine tag. The protein was expressed and purified as described previously (25). The GST tag alone was expressed and purified using the same method.
RESULTS
Expression of eE2 in Escherichia coli (40, 60), yeast (43), insect cells (52), CHO cells (5), and various other eukaryotic and viral recombinant expression systems has consistently resulted in the formation of insoluble, misfolded and disulfide-linked, aggregrated protein. We sought to develop a system for the expression of HCV eE2 that would yield large amounts of highly purified, active protein for functional studies. Our approach was to utilize cell lines that have been shown to produce functional E2 while adding an affinity tag to promote eE2 folding and facilitate purification. HEK293T cells were chosen owing to their ability to produce functional E2 in the form of HCV pseudoparticles (HCVpp) (4), ease of handling, robust growth rate, and excellent transfectability. We expressed the J6 (genotype 2a) E2 ectodomain (amino acids 384 to 664) because this fragment of E2 has been shown to be the minimal functional unit for binding to CD81 (53) and SR-BI (54) (Fig. 1A). eE2 is preceded by a signal sequence and signal peptidase cleavage site to promote targeting and trafficking through the secretory pathway and followed by a thrombin cleavage site and Fc tag (eE2-Fc) (2). The Fc tag was chosen since it is glycosylated and disulfide bonded, which may assist similar posttranslational modifications on eE2. An HEK293T cell line was created that stably secretes eE2-Fc into the media. The presence of the fusion protein is detectable by Western blotting of the cell supernatants (Fig. 1B) and cell lysate (data not shown). eE2-Fc was purified using protein A resin, and eE2 was subsequently separated from the immobilized Fc tag via thrombin protease digestion, leaving the Fc tag and contaminants bound to the resin (Fig. 1C). The protein that eluted from the resin by thrombin cleavage was confirmed to be eE2 by protease digestion followed by high-resolution MS (see below) and amino-terminal sequencing (data not shown). The calculated molecular mass of the J6 eE2 protein is 33 kDa, although it migrates around 60 kDa in reducing SDS-PAGE. This molecular mass discrepancy and the diffuse nature of the band are observations consistent with glycosylated proteins.
FIG. 1.

(A) Sequence of eE2 (residues 384 to 664, genotype J6), highlighting the conserved cysteine residues (underlined) and the potential N-linked (bold) and O-linked (italics) glycosylation sites. (B) Supernatants from HEK293T cells transfected with green fluorescent protein (mock) or eE2-Fc were separated by reducing SDS-PAGE, followed by Western blot analysis with an anti-Fc antibody. eE2 migrates at 100 kDa, which is the approximate molecular mass of glyscosylated E2 with a carboxy-terminal Fc tag. (C) Purification of eE2-Fc on protein A-Sepharose. Supernatants from cell lines expressing eE2-Fc were clarified by centrifugation (Sup loaded) and applied to the resin. After incubation, the column was extensively washed to remove unbound material (Flowthrough). eE2 was eluted off the column in five fractions (elutions) following incubation with thrombin protease. Also shown is the resin before (Bound resin) and after (Post-cleavage resin) elution. Samples were analyzed by SDS-PAGE and stained with Coomassie blue.
Analysis of glycosylation.
Glycosylation of viral envelope proteins is critical for folding, structural integrity, and immune evasion. The number and conservation of glycosylation sites vary across different HCV genotypes (28). J6-E2 contains 11 potential N-linked glycosylation sites (N-X-T/S, where X is any amino acid except proline) along with three potential O-linkage consensus sites (Fig. 1A). We investigated the extent of eE2 N-linked glycosylation and the type of oligosaccharide at each site using endoglycosidases. High mannose and complex N-linked oligosaccharides can be differentiated by Endo H sensitivity, since Endo H will cleave only high-mannose and some hybrid glycans. PNGase F will remove all types of N-linked glycosylation indiscriminately. Deglycosylation of eE2 with PNGase F under denatured, reducing conditions resulted in a faster-migrating band greater than the 31-kDa standard, consistent with its calculated molecular mass of 33 kDa (Fig. 2A). In contrast, eE2 was largely resistant to digestion with Endo H (Fig. 2A). This result suggests that the majority of the N-linked glycans on eE2 are of the complex form, in accordance with its mode of expression by export through the secretory pathway.
FIG. 2.

(A) Deglycosylation of eE2 with PNGase F and Endo H. Purified eE2 was deglycosylated with PNGase F or Endo H under denaturing and reducing conditions, followed by SDS-PAGE analysis. The positions of the glyscosylated eE2 (eE2/gly), deglycosylated eE2 (eE2/degly), and both enzymes (Endo H and PNGase F) are also shown. (B) Mapping the N-linked glycosylation sites. Each panel contains the extracted ion chromatograms of proteolytic peptides of eE2 with (bottom) or without (top) PNGase F digestion. The data are plotted with the elution time on the x axis, and relative abundance is plotted on the y axis. The peptide sequence and calculated molecular mass used for the extraction are provided at the top of each panel. The measured mass/charge ratio (m/z) and charge state for each spectrum are provided. Note that one panel has a peptide that contains two N-linked glycosylation sites. The identification of each peak was confirmed by MS/MS.
To investigate the glycosylation site usage in further detail, we employed high-resolution MS. eE2 was digested with either trypsin or chymotrypsin, and samples of the protein fragments were deglycosylated with either PNGase F or Endo H. PNGase F deaminates the asparagine residue to which the N-linked glycan is attached and converts it to aspartic acid. If the glycan is of the high-mannose form, it will be sensitive to Endo H, which leaves one N-acetylglucosamine (GlcNAc) bound to the Asn. Thus, the gain of 1 Da (Asn to Asp; nitrogen to oxygen) by PNGase F or 203 Da by the GlcNAc residue left by Endo H digestion can be resolved by MS of the peptides. From the resulting three spectra (untreated, Endo H, and PNGase F treated), we are able to map all the glycosylation sites, estimate the approximate usage of each site, and determine whether the glycan at a particular site was complex or high mannose. We achieved almost 100% coverage of the eE2 sequence using either trypsin or chymotrypsin proteases. Peptides from the 11 predicted N-linked glycosylation sites were shown to be fully glycosylated, since we were unable to detect unglycosylated peptides with Asn residues in them (Fig. 2B, upper spectra of each panel). Only one of the 11 glycosylation sites was found to be Endo H sensitive (VGGVEHRLTAACNF; data not shown for Endo H), suggesting that the majority of the glycans are complex in nature (Fig. 2B, lower spectra of each panel). Peptides containing the potential O-linked glycosylation sites were resolved and shown to be unmodified (data not shown).
Oligomeric state and secondary structure of eE2.
Since previous reports have shown that E2 tends to aggregate, we set out to define the oligomeric state of eE2, using nonreducing SDS-PAGE, size exclusion chromatography (SEC), and analytical ultracentrifugation. SDS-PAGE analysis of eE2 under nonreducing conditions demonstrated that eE2 consisted of a mixture of two components with approximate molecular masses of ∼120 kDa (dimer) and ∼60 kDa (monomer), with a small amount of larger-molecular-mass protein (Fig. 3A). To determine the oligomeric state of eE2 under native conditions, the protein was evaluated by SEC. eE2 yielded two major peaks and a slight peak found in the void volume of the column (Fig. 3B). The major and minor peaks were measured at ∼123 kDa and ∼75 kDa, respectively, by an inline static light-scattering detector (data not shown). Both nonreducing SDS-PAGE and native SEC clearly demonstrated that eE2 is not aggregated. The ratio of dimer to monomer in nonreduced SDS-PAGE (Fig. 3A) and that in gel filtration (Fig. 3B) are similar, suggesting that a portion of the dimer may result from an intermolecular disulfide bond.
FIG. 3.
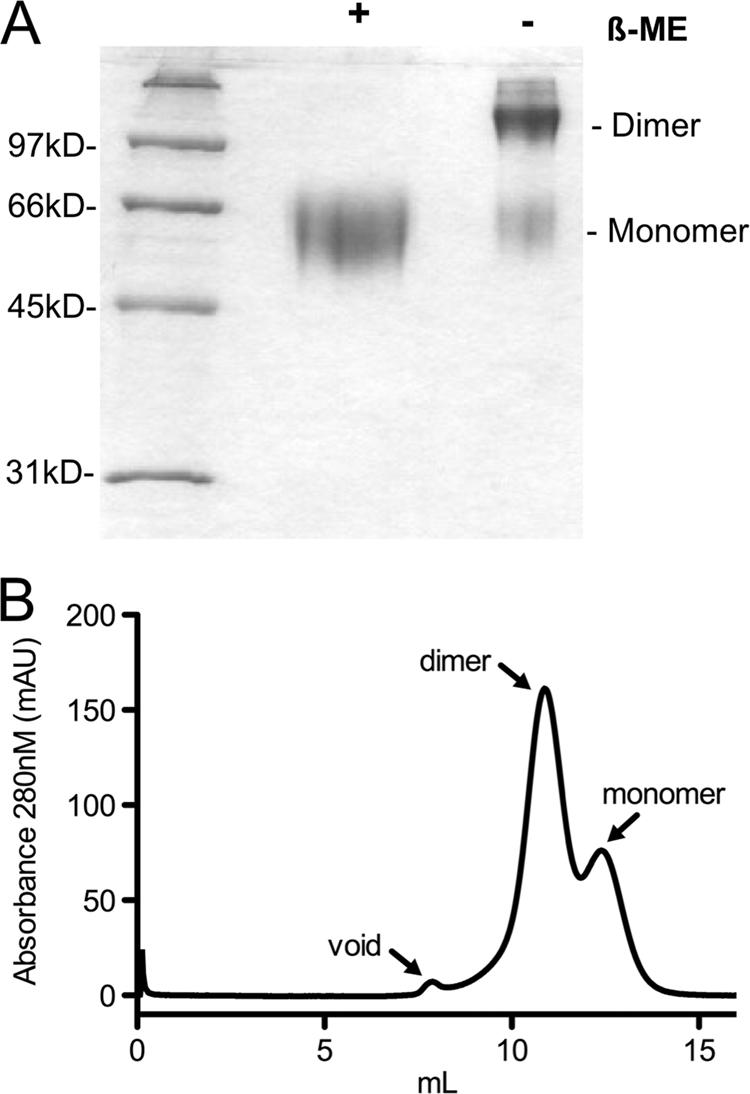
(A) SDS-PAGE analysis of purified eE2 in the presence and absence of β-mercaptoethanol (β-ME) and stained with Coomassie blue. (B) Purified eE2 was applied to a Superdex 200 size exclusion column equilibrated with 50 mM HEPES (pH 7.5), 150 mM KCl, 5% glycerol. The arrows denote the positions of the void volume, dimer, and monomer.
There are 18 conserved cysteines in full-length E2, which could result in the formation of 9 disulfide bonds. The eE2 fragment contains only 17 cysteines, leaving at least 1 unpaired. Therefore, we sought to determine the oxidation state of the conserved cysteine residues in eE2. A differential cysteine labeling method followed by high-resolution MS was employed to distinguish free cysteine residues from those involved in disulfide bonding. Briefly, eE2 was incubated with a molar excess of NEM under denaturing, nonreducing conditions to label all free cysteine residues. After disulfide bond reduction with DTT, the newly generated free cysteines were alkylated with IAM. The modified protein was digested with trypsin and deglycosylated with PNGase F, and the resulting peptides were resolved by MS. All cysteine-containing peptides were identified, and only one peptide (C656NLEDRDR) was modified by NEM (Fig. 4A). The expected unmodified molecular mass of this peptide is 1,020.45 Da, 1,077.45 Da if modified by IAM, or 1,145.45 Da if modified by NEM. In Fig. 4A, the spectrum shows a 573.75 m/z peak, corresponding to this peptide modified by NEM and carrying a +2 charge, while no peptide appears at a position corresponding to a modification by IAM (expected ∼538 m/z with a 2+ charge). All other cysteine-containing peptides were shown to have an addition of 57 Da, indicating that they were modified only after reduction with DTT. For example, the expected molecular mass of the unmodified SAC459RSIEAF peptide is 983.46 Da, 1,040.46 Da if modified by IAM, or 1,108.46 Da if modified by NEM. This peptide resolves at 521.32 m/z, which corresponds to the molecular mass when modified by IAM and carrying a +2 charge. It does not resolve as modified by NEM (expected ∼554 m/z with a 2+ charge) (Fig. 4B). From the results of nonreducing SDS-PAGE, cysteine-labeling experiment and SEC, we presumed that C656 may potentially be involved in the formation of an intermolecular disulfide bond.
FIG. 4.

Differential labeling of free and disulfide-linked cysteines. Free and disulfide-bonded cysteines were labeled with NEM and IAM, respectively. LC-MS/MS-extracted ion chromatograms for peptides containing C656 (A) or C459 (B) are shown. The extraction was done using the molecular masses of the peptides modified with IAM (upper panel) or NEM (bottom panel). C656 is free, while C459 is found in a disulfide bond. All of the other cysteine residues were labeled with IAM (data not shown), suggesting the formation of eight disulfide bonds. The data are plotted with the elution time on the x axis and relative abundance on the y axis. The measured mass/charge ratio (m/z) and charge state for each spectrum is provided. The identification of each peak was confirmed by MS/MS. (C) SDS-PAGE analysis of purified eE2-C656S in the presence and absence of β-mercaptoethanol (β-ME), followed by staining with Coomassie blue. (D) Purified eE2-C656S was applied to a Superdex 200 size exclusion column equilibrated with 50 mM HEPES (pH 7.5), 150 mM KCl, and 5% glycerol. The arrows denote the positions of the void, dimer, and monomer.
To test this hypothesis, C656 was mutated to a serine (eE2-C656S) to conserve some of the biochemical properties at this position. An HEK293T cell line that stably expresses eE2-C656S-Fc was generated, and the protein was purified as before. Results from nonreducing SDS-PAGE indicated that the proportion of monomer in eE2-C656S is substantially increased relative to wild-type eE2 (compare Fig. 3A and 4C). In order to determine the native behavior, eE2-C656S was analyzed by SEC under conditions identical to those for wild-type eE2. The results demonstrated that the mutant is predominantly monomeric at pH 7.5, with a lesser amount of dimer (Fig. 4D).
To substantiate our SEC results, we used sedimentation velocity experiments by analytical ultracentrifugation, which can determine mass distributions and globularity from mixtures of proteins without the use of standards or interactions with a sieving matrix. Sedimentation of eE2-C656S at pH 7 indicates the presence of two dominant species, a monomeric form (60 to 70 kDa) and a dimeric form (80 to 130 kDa) (Fig. 5A and B). There is a minor third species that has the approximate molecular mass of a trimer (150 to 200 kDa). The relative proportions of the three species are 65%:29%:6% (monomer:dimer:trimer). Since analytical ultracentrifugation failed to detect large aggregates, we assume that the trimer corresponds to the small peak that eluted in the void volume of SEC. The monomer has a wide distribution of frictional ratio in the range of 1.0 to 2.0, where a perfect sphere would have a frictional ratio of 1.0. It is interesting to note that the frictional ratio shows a decreasing trend with increasing molecular mass, suggesting a more globular shape for the oligomeric forms. To determine if the ratio of oligomers or protein globularity was dependent on the amount of protein measured, the analysis was performed at two different concentrations (Fig. 5). There is no appreciable difference in the molecular mass, ratio of the different species, or shape distributions at the two concentrations used in this analysis (OD230s of 0.25 and 0.8), indicating little or no effect due to mass action at these concentrations.
FIG. 5.

Analytical ultracentrifugation data for eE2-C656S at pH 7. Two-dimensional spectrum/Monte Carlo analysis of HCN sedimentation velocity data. Measurements of eE2 were made at a low concentration (0.25 OD230) (A) or at a higher concentration (0.8 OD230) (B). Both samples show the presence of a trimer species. Larger species appear more globular than smaller species according to the frictional coefficient ratios. The units of the color gradient are in OD230s.
The HCV glycoproteins are predicted to be class II fusion proteins due to their relatedness to proteins of alphaviruses and flaviviruses. Class II fusion proteins are composed of mostly β-sheet structure and do not undergo major rearrangements in secondary structure upon exposure to low pH (34). CD was employed to determine the secondary structure of eE2 and whether there are any changes upon lowering of the pH. CD spectra of both eE2 and eE2-C656S measured at pH 7.0 exhibited a minimum at about 203 nm (Fig. 6A and B). Multilinear regression analysis (data not shown) suggested that the eE2 spectrum is consistent with a protein composed of predominantly β-sheet and random coil secondary structure with little to no alpha-helical content. The CD spectra measured at pH 5 are superimposable on those measured at pH 7 for both eE2 and eE2-C656S. From these data, we conclude that eE2 has mostly β-sheet and random coil structure and does not undergo major changes in secondary structure, similar to the flavivirus envelope protein.
FIG. 6.

CD spectroscopy of eE2 (A) or eE2-C656S (B) at pH 7 and pH 5. CD spectra are shown as millidegrees versus wavelength (nm). Error bars for each data point are given.
J6 eE2 is recognized by antibodies from patients chronically infected with different genotypes of HCV.
The presence of high levels of anti-E2 antibodies in HCV-infected human serum has been reported (3). In order to further examine if purified eE2 is conformationally similar to the E2 present on infectious HCV particles, we tested whether eE2 was recognized by antibodies from infected patient sera. An ELISA plate was coated with eE2 and probed with serum from patients chronically infected with either genotype 1, 2, or 3. Serum from a healthy donor was tested in parallel as a negative control. Anti-human HRP was used to quantify the result. Serum from the three infected patients bound to J6 eE2 (genotype 2a) at similar titers regardless of infecting genotype, while the serum of the uninfected donor responded at background levels (Fig. 7A). This illustrates the capacity of eE2 to be recognized by antibodies in patient sera, while also pointing out the maintenance of cross-reactive epitopes.
FIG. 7.

Functional analysis of eE2 and eE2-C656S. (A) ELISA plates were coated with eE2 and probed with a series of 10-fold dilutions of serum from patients infected with HCV (genotype 1, 2, or 3) or a healthy donor. Antibodies in HCV-infected patient sera could detect eE2 at up to a 1:100,000 dilution. (B) Cells were incubated with HCVcc plus GST, GST-mCD81-LEL, GST-hCD81-LEL, eE2, or eE2-C656S. Three days postinfection, the cells were fixed, focus forming units were determined, and the percentage of inhibition was calculated. eE2, eE2-C656S, and hCD81 inhibit HCVcc infection. Error bars represent standard errors of the means for two independent experiments. Each experiment was performed in duplicate. (C) Cells were incubated with eE2, GST, or GST-hCD81-LEL at two concentrations (200 or 100 μg/ml). Three days later, cells were analyzed for viability using flow cytometry. The results demonstrate that eE2 is not toxic when applied to cells at the concentration that inhibits HCVcc infection. (D) Enzyme-linked immunoassay for CD81-LEL binding. Tissue culture supernatants of eE2-Fc fusion (no dilution or 1:10 or 1:100 dilution) were incubated in plates coated with either GST, GST-mCD81-LEL, or GST-hCD81-LEL. After washing, bound eE2-Fc was detected with anti-human Fc-HRP. PBS, medium from wild-type HEK293T cells, and wells without any coating were used as controls. Both eE2 and eE2-C656S bind only to hCD81.
eE2 blocks HCVcc entry.
It was shown previously that properly folded, purified E2 ectodomain from pestiviruses, bovine viral diarrhea virus, and classical swine fever virus was able to block viral infection (33, 49). In order to confirm correct folding and function of HCV eE2, we performed a similar assay using HCVcc. Recombinant human CD81 LEL, which has been shown to inhibit HCVcc infection, was used as a positive control for blocking infection (38). About 100 50% tissue culture infective doses of HCVcc were incubated with serial twofold dilutions of purified eE2, eE2-C656S, GST-human-CD81-LEL (hCD81), GST-mouse-CD81-LEL (mCD81), or GST. eE2, eE2-C656S, and hCD81 reduced the number of focus-forming units in a concentration-dependent manner, while the mCD81 and GST proteins had no effect. This experiment yielded a 50% blocking efficiency in the range of 25 to 150 μg/ml for eE2, eE2-C656S, and hCD8-LEL (Fig. 7B). Thus, HCVcc infection can be effectively blocked by eE2 or eE2-C656S at concentrations similar to those of hCD81-LEL. In order to rule out the possibility that inhibition of viral infection was due to toxicity, we quantified cell death after incubation with purified protein using fluorescence-activated cell sorting analysis. Cells were incubated for 3 days with twofold dilutions of eE2, GST, and hCD81-LEL, followed by staining with Via-Probe to estimate viability. As shown in Fig. 7C, purified eE2 and hCD81 are not toxic at 200 μg/ml (the concentration with the highest level of inhibition).
Inhibition of HCVcc entry by eE2 is thought to occur by sequestering of cellular receptors. To confirm this, we analyzed the ability of eE2 to bind hCD81 in vitro by adapting an ELISA first described by Flint et al. for the detection of properly folded E2 based on hCD81 binding (25). Plates were coated with GST, mCD81, and hCD81, probed with supernatants from cell lines expressing eE2-Fc or eE2-C656S-Fc, and then developed with HRP-conjugated anti-Fc. The assay was executed in triplicate using undiluted cell culture supernatant and two 10-fold dilutions with medium. Both eE2-Fc and eE2-C656S-Fc specifically bind hCD81 but not mCD81 or GST alone (Fig. 7D). Identical results were obtained using purified eE2 and eE2-C656S and an anti-E2 antibody (data not shown). Taken together, these data confirm that the eE2 protein made in the system described here maintains many of the functional characteristics of E2 found on virus.
DISCUSSION
HCV E1 and E2 are primary determinants of entry and pathogenicity (21). Deletion mutagenesis has defined the ectodomain of HCV E2 to comprise amino acids 384 to 661 of E2 from genotype H77 (45). Functional and biophysical studies of HCV E2 have been hindered by the formation of mostly aggregated, misfolded material. Here we describe a method for production of milligram quantities of eE2, which do not contain large disulfide-linked aggregates. The resulting protein can compete with HCVcc to inhibit infection, is recognized by antibodies from chronically infected patients, and can specifically bind the large extracellular loop of human CD81. These results demonstrate that the eE2 protein maintains many of the functionalities associated with E2 found on virions. Using purified eE2, we have conducted a series of biochemical and biophysical studies aimed at characterizing the glycosylation, oxidation state, and oligomeric nature of eE2.
HCV is thought to bud into the ER and egress through the cellular secretory pathway. As the virion passes through the ER and the Golgi apparatus, the exposed glycans associated with E1 and E2 can be modified by processing enzymes, resulting in hybrid or complex glycans. Indeed, the E1 and E2 glycans of HCVpp are mostly insensitive to Endo H digestion, suggesting that the glycans are hybrid or complex (48). One of the advantages of the expression system described here is the use of the same mammalian cell line (HEK293T) employed for HCVpp production, presumably yielding similar glycan modifications. In fact, an Endo H digest comparison of eE2 (Fig. 2A) and HCVpp E2 (Fig. 2A in the work of Op De Beeck et al. [48]) reveals remarkable similarity in the digestion pattern. All 11 of the predicted N-linked glycosylation sites in eE2 are utilized, which is consistent with previous data for HCVpp (27). Ten of the 11 sites have complex glycans attached, while only the most carboxy-terminal is of the high mannose form. This observation suggests that the glycan at this position is concealed from the modification enzymes in the secretory pathway, possibly due to steric effects within E2 or blocking of the site by the Fc tag attached to the carboxy terminus. As reported by Goffard et al., mutation of the carboxy-terminal glycosylation site does not affect folding, secretion, or E1/E2 heterodimer formation but does result in less than 50% infectivity when incorporated into HCVpp (27).
Recently it has been reported that E2 contains O-linked glycans (23). Peptides containing the putative O-linked glycosylation sites (Fig. 1A) were not modified, as determined by LC-MS/MS. However, at this time we cannot rule out that O-linked glycosylation occurs at a low level on eE2 or on full-length E2 when expressed as part of the HCV polyprotein.
Full-length HCV E2 has 18 highly conserved cysteines, although the eE2 construct as defined by Michalak et al. and used in this study has only the first 17 (45). Nonreducing SDS-PAGE and SEC under native conditions demonstrated that about 60 to 70% of wild-type eE2 exists as a dimer through an intermolecular disulfide bond. Differential labeling of free and disulfide-bonded cysteines demonstrated the presence of eight disulfide bonds and one free cysteine (C656) in eE2. We reasoned that since C656 is the only free cysteine, it might be involved in the intermolecular disulfide. Mutating C656 did not affect inhibition of HCVcc entry (Fig. 7B), CD81 binding (Fig. 7D), folding as determined by CD (Fig. 6), or binding to HCV patient antibodies (data not shown). Nonreducing SDS-PAGE and SEC of eE2-C656S showed a dramatic shift toward a mostly monomeric form.
Our results are consistent with recently published data, showing that all 18 cysteine residues of E2 are in disulfide bonds and reduction of up to half of these is compatible with HCV entry as well as antibody and CD81 binding (24). The 18th cysteine is located in the stem region found between the ectodomain and the transmembrane helix (18). The stem consists of a heptad repeat and has been proposed to be an amphipathic alpha helix with a hydrophilic and hydrophobic surface. In this model, the 18th cysteine would be facing away from the membrane. Taken together, the data suggest that the ectodomain of E2 might be tethered to the stem region by an intramolecular disulfide bond between the 17th (C656) and 18th cysteine residues.
The HCV glycoproteins, like those from related alphaviruses and flaviviruses, are predicted to be class II fusion proteins. This class of proteins is characterized by mostly β-sheet structures that do not undergo changes in secondary structure upon exposure to low pH (34). The flavivirus E protein (comprised of three domains) is responsible for receptor binding and membrane fusion. Flaviviruses have icosahedral symmetry, with E arranged as 90 dimers. Cryo-electron microscopy has demonstrated that the E protein lies flat on the surface of the viral lipid bilayer (30). Upon exposure to low pH, there is a slight rotation of domain III, resulting in dissociation of the E dimers and rearrangement into trimers via a monomeric intermediate stage. The oligomeric nature of E2 on the surface of HCV remains unknown. It is assumed that the native form of the molecule contains intramolecular disulfide bonds and heterodimerizes with E1 through their respective transmembrane regions. The ectodomain of HCV E2 with the C656S mutation appears mostly as a monomer and dimer with a much smaller amount of trimer. Sedimentation velocity measurements showed that the ratio of the three oligomers did not change at the two concentrations tested. Isolation of the monomer by SEC and reanalysis did not suggest that the various states were in equilibrium (data not shown). However, the oligomeric state of E2 may be influenced by the deleted carboxy-terminal portion, the high concentration found on virus particles, and/or heterodimerization with E1.
CD spectroscopic analysis of HCV eE2 had a pronounced minimum at about 203 nm, consistent with a protein containing mostly β-sheet and random coil. Exposure to low pH did not cause any significant changes in the CD spectrum, which suggests the lack of major rearrangement of secondary structure. However, CD cannot rule out the possibility of structural rearrangements that preserve the overall proportion of β-sheet and random coil. In fact, changes in certain conformational epitopes on HCVpp E2 upon lowering of the pH have been reported (48).
Recombinant CD81-LEL, eE2, and eE2-C656S are capable of blocking HCVcc infection (Fig. 7B). The concentration of GST-CD81-LEL needed to block HCVcc infection in our assay is comparable with previously published data from Lindenbach et al. (38). Interestingly, the amount of hCD81-LEL, eE2, or eE2-C656S required to prevent 50% inhibition of HCVcc infection was approximately the same, with a slight increase needed for eE2-C656S. Since the molecular mass of GST-CD81-LEL is about 37 kDa, the total amount of eE2 and CD81-LEL added was within twofold. It is thought that CD81-LEL would bind to virus, while eE2 is presumed to bind to the cellular receptors. Therefore, the result that CD81 and eE2 can block infection at similar concentrations is surprising since the mechanism of blocking of infection is thought to be different. There are many factors that contribute to the similar inhibition results seen with the E2 and CD81 proteins, including the total number of CD81 and eE2 binding sites, the relative affinities of the interactions, and the kinetics of binding. At the moment, we are not certain which of these or other factors are responsible for the similar inhibition results.
Based on the results shown in this study and in accordance with previous studies of the HCV envelope proteins, eE2 is potentially a vaccine candidate. Chimpanzees immunized with E1/E2 heterodimeric proteins are protected from infection with low doses of homologous HCV (10). Weiner et al. identified hypervariable regions in E2, while it was later shown that deletion of hypervariable region 1 attenuated infection in chimpanzees (26, 58). Youn et al. also showed that an E2 antibody response correlates with lower viral titers in chimpanzees (59). Most recently, rodents injected with HCV envelope glycoproteins have been shown to produce antibodies that are broadly cross-reactive in their neutralization properties (56). The production of large quantities of functional and properly folded E2 ectodomain has allowed us to perform comprehensive biochemical and biophysical analysis of eE2, which was previously difficult due to production of mostly misfolded proteins in other expression systems. The next challenge is to use our newfound understanding of eE2 protein properties and our ability to generate milligram quantities of the eE2 protein to dissect E2 function in vivo and to examine its interaction with E1 and other cellular factors, with the eventual goal of facilitating the design of an HCV vaccine and entry inhibitor.
Acknowledgments
We thank Stephen K. Burley, Brian Chait, Matthew Evans, Alla Kostyukova, Peter Lobel, Jeremy Mann, Dimitar Nikolov, Aaron Shatkin, Virgil Schirf, Rong Xiao, Caifeng Zhao, and Haiyan Zheng for helpful advice, support, and technical assistance. In addition, we thank Charles Rice for providing DNA for the J6 structural proteins and for many helpful discussions.
The work was supported by the Busch Biomedical Foundation (grant to J.M.), the Life Sciences Research Foundation (grant to J.M.), a grant from the Schering-Plough Corporation (to J.M.), EVC/CFAR Flow Cytometry Core, P30 AI050409 (to A.G.), a Cancer Research Institute Investigator Award (to A.G.), Yerkes Research Center Base Grant RR-00165 (to A.G.), and Public Health Service grant AI070101 (to A.G.). The development of the UltraScan software is supported by the National Institutes of Health through grant RR022200 (to principal investigator B.D.). Supercomputer time allocations were provided through National Science Foundation grant TG-MCB070038 (to B.D.).
Footnotes
Published ahead of print on 26 August 2009.
REFERENCES
- 1.Bartenschlager, R., M. Frese, and T. Pietschmann. 2004. Novel insights into hepatitis C virus replication and persistence. Adv. Virus Res. 63:71-180. [DOI] [PubMed] [Google Scholar]
- 2.Barton, W., D. Tzvetkova, and D. Nikolov. 2005. Structure of the angiopoietin-2 receptor binding domain and identification of surfaces involved in Tie2 recognition. Structure 13:825-832. [DOI] [PubMed] [Google Scholar]
- 3.Bartosch, B., J. Bukh, J. C. Meunier, C. Granier, R. E. Engle, W. C. Blackwelder, S. U. Emerson, F. L. Cosset, and R. H. Purcell. 2003. In vitro assay for neutralizing antibody to hepatitis C virus: evidence for broadly conserved neutralization epitopes. Proc. Natl. Acad. Sci. USA 100:14199-14204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartosch, B., J. Dubuisson, and F. Cosset. 2003. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J. Exp. Med. 197:633-642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brazzoli, M., A. Helenius, S. K. Foung, M. Houghton, S. Abrignani, and M. Merola. 2005. Folding and dimerization of hepatitis C virus E1 and E2 glycoproteins in stably transfected CHO cells. Virology 332:438-453. [DOI] [PubMed] [Google Scholar]
- 6.Brookes, E., W. Cao, and B. Demeler. A two-dimensional spectrum analysis for sedimentation velocity experiments of mixtures with heterogeneity in molecular weight and shape. Eur. Biophys. J., in press. [DOI] [PubMed]
- 7.Brown, R. S. 2005. Hepatitis C and liver transplantation. Nature 436:973-978. [DOI] [PubMed] [Google Scholar]
- 8.Cao, W., and B. Demeler. 2008. Modeling analytical ultracentrifugation experiments with an adaptive space-time finite element solution for multicomponent reacting systems. Biophys. J. 95:54-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Catanese, M. T., R. Graziani, T. von Hahn, M. Moreau, T. Huby, G. Paonessa, C. Santini, A. Luzzago, C. M. Rice, R. Cortese, A. Vitelli, and A. Nicosia. 2007. High-avidity monoclonal antibodies against the human scavenger class B type I receptor efficiently block hepatitis C virus infection in the presence of high-density lipoprotein. J. Virol. 81:8063-8071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choo, Q. L., G. Kuo, R. Ralston, A. Weiner, D. Chien, G. Van Nest, J. Han, K. Berger, K. Thudium, C. Kuo, et al. 1994. Vaccination of chimpanzees against infection by the hepatitis C virus. Proc. Natl. Acad. Sci. USA 91:1294-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choukhi, A., S. Ung, C. Wychowski, and J. Dubuisson. 1998. Involvement of endoplasmic reticulum chaperones in the folding of hepatitis C virus glycoproteins. J. Virol. 72:3851-3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cocquerel, L., C. Wychowski, F. Minner, F. Penin, and J. Dubuisson. 2000. Charged residues in the transmembrane domains of hepatitis C virus glycoproteins play a major role in the processing, subcellular localization, and assembly of these envelope proteins. J. Virol. 74:3623-3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Craig, R., and R. C. Beavis. 2004. TANDEM: matching proteins with tandem mass spectra. Bioinformatics 20:1466-1467. [DOI] [PubMed] [Google Scholar]
- 14.Demeler, B. 2009, posting date. UltraScan: a comprehensive data analysis software package for analytical ultracentrifugation experiments. http://www.ultrascan.uthscsa.edu.
- 15.Demeler, B. 2005. UltraScan: a comprehensive data analysis software package for analytical ultracentrifugation experiments, p. 210-229. In D. J. Scott, S. E. Harding, and A. J. Rowe (ed.), Analytical ultracentrifugation: techniques and methods. Royal Society of Chemistry, London, United Kingdom.
- 16.Demeler, B., and E. Brookes. 2008. Monte Carlo analysis of sedimentation experiments. Colloid Polymer Sci. 286:129-137. [Google Scholar]
- 17.Dhillon, R., S. Rossi, and S. K. Herrine. 2008. Pegylated interferon 2a and 2b in combination with ribavirin for the treatment of chronic hepatitis C in HIV infected patients. Ther. Clin. Risk Manag. 4:789-796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Drummer, H. E., and P. Poumbourios. 2004. Hepatitis C virus glycoprotein E2 contains a membrane-proximal heptad repeat sequence that is essential for E1E2 glycoprotein heterodimerization and viral entry. J. Biol. Chem. 279:30066-30072. [DOI] [PubMed] [Google Scholar]
- 19.Drummer, H. E., K. A. Wilson, and P. Poumbourios. 2002. Identification of the hepatitis C virus E2 glycoprotein binding site on the large extracellular loop of CD81. J. Virol. 76:11143-11147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dubuisson, J. 2000. Folding, assembly and subcellular localization of hepatitis C virus glycoproteins. Curr. Top. Microbiol. Immunol. 242:135-148. [DOI] [PubMed] [Google Scholar]
- 21.Dubuisson, J. 2007. Hepatitis C virus proteins. World J. Gastroenterol. 13:2406-2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Evans, M. J., T. von Hahn, D. M. Tscherne, A. J. Syder, M. Panis, B. Wölk, T. Hatziioannou, J. A. McKeating, P. D. Bieniasz, and C. M. Rice. 2007. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 446:801-805. [DOI] [PubMed] [Google Scholar]
- 23.Falkowska, E., F. Kajumo, E. Garcia, J. Reinus, and T. Dragic. 2007. Hepatitis C virus envelope glycoprotein E2 glycans modulate entry, CD81 binding, and neutralization. J. Virol. 81:8072-8079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fenouillet, E., D. Lavillette, S. Loureiro, G. Krashias, G. Maurin, F. L. Cosset, I. M. Jones, and R. Barbouche. 2008. Contribution of redox status to hepatitis C virus E2 envelope protein function and antigenicity. J. Biol. Chem. 283:26340-26348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Flint, M., T. von Hahn, J. Zhang, M. Farquhar, C. T. Jones, P. Balfe, C. M. Rice, and J. A. McKeating. 2006. Diverse CD81 proteins support hepatitis C virus infection. J. Virol. 80:11331-11342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Forns, X., R. Thimme, S. Govindarajan, S. U. Emerson, R. H. Purcell, F. V. Chisari, and J. Bukh. 2000. Hepatitis C virus lacking the hypervariable region 1 of the second envelope protein is infectious and causes acute resolving or persistent infection in chimpanzees. Proc. Natl. Acad. Sci. USA 97:13318-13323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goffard, A., N. Callens, B. Bartosch, C. Wychowski, F. L. Cosset, C. Montpellier, and J. Dubuisson. 2005. Role of N-linked glycans in the functions of hepatitis C virus envelope glycoproteins. J. Virol. 79:8400-8409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goffard, A., and J. Dubuisson. 2003. Glycosylation of hepatitis C virus envelope proteins. Biochimie 85:295-301. [DOI] [PubMed] [Google Scholar]
- 29.Grove, J., T. Huby, Z. Stamataki, T. Vanwolleghem, P. Meuleman, M. Farquhar, A. Schwarz, M. Moreau, J. S. Owen, G. Leroux-Roels, P. Balfe, and J. A. McKeating. 2007. Scavenger receptor BI and BII expression levels modulate hepatitis C virus infectivity. J. Virol. 81:3162-3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harrison, S. C. 2008. Viral membrane fusion. Nat. Struct. Mol. Biol. 15:690-698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Helle, F., and J. Dubuisson. 2008. Hepatitis C virus entry into host cells. Cell Mol. Life Sci. 65:100-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hsu, M., J. Zhang, M. Flint, C. Logvinoff, C. Cheng-Mayer, C. M. Rice, and J. A. McKeating. 2003. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc. Natl. Acad. Sci. USA 100:7271-7276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hulst, M. M., and R. J. Moormann. 1997. Inhibition of pestivirus infection in cell culture by envelope proteins E(rns) and E2 of classical swine fever virus: E(rns) and E2 interact with different receptors. J. Gen. Virol. 78:2779-2787. [DOI] [PubMed] [Google Scholar]
- 34.Jardetzky, T. S., and R. A. Lamb. 2004. Virology: a class act. Nature 427:307-308. [DOI] [PubMed] [Google Scholar]
- 35.Laue, T. M., B. D. Shah, T. M. Ridgeway, and S. L. Pelleteir. 1992. Computer-aided interpretation of analytical sedimentation data for proteins, p. 90-125. In S. E. Harding, A. J. Rowe, and J. C. Horton. (ed.), Analytical ultracentrifugation in biochemistry and polymer science. Royal Society of Chemistry, Cambridge, United Kingdom.
- 36.Lavillette, D., A. W. Tarr, C. Voisset, P. Donot, B. Bartosch, C. Bain, A. H. Patel, J. Dubuisson, J. K. Ball, and F. L. Cosset. 2005. Characterization of host-range and cell entry properties of the major genotypes and subtypes of hepatitis C virus. Hepatology 41:265-274. [DOI] [PubMed] [Google Scholar]
- 37.Lin, C., B. D. Lindenbach, B. M. Pragai, D. W. McCourt, and C. M. Rice. 1994. Processing in the hepatitis C virus E2-NS2 region: identification of p7 and two distinct E2-specific products with different C termini. J. Virol. 68:5063-5073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lindenbach, B., M. Evans, A. Syder, B. Wölk, T. Tellinghuisen, C. Liu, T. Maruyama, R. Hynes, D. Burton, J. McKeating, and C. Rice. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623-626. [DOI] [PubMed] [Google Scholar]
- 39.Lindenbach, B. D., H.-J. Thiel, and C. M. Rice. 2007. Flaviviridae: the viruses and their replication, p. 1101-1152. In D. M. Knipe et al. (ed.), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA.
- 40.Liu, J., L. Zhu, X. Zhang, M. Lu, Y. Kong, Y. Wang, and G. Li. 2001. Expression, purification, immunological characterization and application of Escherichia coli-derived hepatitis C virus E2 proteins. Biotechnol. Appl. Biochem. 34:109-119. [DOI] [PubMed] [Google Scholar]
- 41.Liu, S., W. Yang, L. Shen, J. R. Turner, C. B. Coyne, and T. Wang. 2009. Tight junction proteins claudin-1 and occludin control hepatitis C virus entry and are downregulated during infection to prevent superinfection. J. Virol. 83:2011-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lohmann, V., F. Korner, J. Koch, U. Herian, L. Theilmann, and R. Bartenschlager. 1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285:110-113. [DOI] [PubMed] [Google Scholar]
- 43.Martinez-Donato, G., Y. Capdesuner, N. Acosta-Rivero, A. Rodriguez, J. Morales-Grillo, E. Martinez, M. Gonzalez, J. C. Alvarez-Obregon, and S. Duenas-Carrera. 2007. Multimeric HCV E2 protein obtained from Pichia pastoris cells induces a strong immune response in mice. Mol. Biotechnol. 35:225-235. [DOI] [PubMed] [Google Scholar]
- 44.Mateu, G., R. O. Donis, T. Wakita, J. Bukh, and A. Grakoui. 2008. Intragenotypic JFH1 based recombinant hepatitis C virus produces high levels of infectious particles but causes increased cell death. Virology 376:397-407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Michalak, J. P., C. Wychowski, A. Choukhi, J. C. Meunier, S. Ung, C. M. Rice, and J. Dubuisson. 1997. Characterization of truncated forms of hepatitis C virus glycoproteins. J. Gen. Virol. 78:2299-2306. [DOI] [PubMed] [Google Scholar]
- 46.Op De Beeck, A., L. Cocquerel, and J. Dubuisson. 2001. Biogenesis of hepatitis C virus envelope glycoproteins. J. Gen. Virol. 82:2589-2595. [DOI] [PubMed] [Google Scholar]
- 47.Op De Beeck, A., R. Montserret, S. Duvet, L. Cocquerel, R. Cacan, B. Barberot, M. Le Maire, F. Penin, and J. Dubuisson. 2000. The transmembrane domains of hepatitis C virus envelope glycoproteins E1 and E2 play a major role in heterodimerization. J. Biol. Chem. 275:31428-34137. [DOI] [PubMed] [Google Scholar]
- 48.Op De Beeck, A., C. Voisset, B. Bartosch, Y. Ciczora, L. Cocquerel, Z. Keck, S. Foung, F. L. Cosset, and J. Dubuisson. 2004. Characterization of functional hepatitis C virus envelope glycoproteins. J. Virol. 78:2994-3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pande, A., B. V. Carr, S. Y. Wong, K. Dalton, I. M. Jones, J. W. McCauley, and B. Charleston. 2005. The glycosylation pattern of baculovirus expressed envelope protein E2 affects its ability to prevent infection with bovine viral diarrhoea virus. Virus Res. 114:54-62. [DOI] [PubMed] [Google Scholar]
- 50.Pileri, P., Y. Uematsu, S. Campagnoli, G. Galli, F. Falugi, R. Petracca, A. J. Weiner, M. Houghton, D. Rosa, G. Grandi, and S. Abrignani. 1998. Binding of hepatitis C virus to CD81. Science 282:938-941. [DOI] [PubMed] [Google Scholar]
- 51.Ploss, A., M. J. Evans, V. A. Gaysinskaya, M. Panis, H. You, Y. P. de Jong, and C. M. Rice. 2009. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 457:882-886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rodriguez-Rodriguez, M., D. Tello, B. Yelamos, J. Gomez-Gutierrez, B. Pacheco, S. Ortega, A. G. Serrano, D. L. Peterson, and F. Gavilanes. 2009. Structural properties of the ectodomain of hepatitis C virus E2 envelope protein. Virus Res. 139:91-99. [DOI] [PubMed] [Google Scholar]
- 53.Rosa, D., S. Campagnoli, C. Moretto, E. Guenzi, L. Cousens, M. Chin, C. Dong, A. J. Weiner, J. Y. Lau, Q. L. Choo, D. Chien, P. Pileri, M. Houghton, and S. Abrignani. 1996. A quantitative test to estimate neutralizing antibodies to the hepatitis C virus: cytofluorimetric assessment of envelope glycoprotein 2 binding to target cells. Proc. Natl. Acad. Sci. USA 93:1759-1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Scarselli, E., H. Ansuini, R. Cerino, R. M. Roccasecca, S. Acali, G. Filocamo, C. Traboni, A. Nicosia, R. Cortese, and A. Vitelli. 2002. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 21:5017-5025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shepard, C. W., L. Finelli, and M. J. Alter. 2005. Global epidemiology of hepatitis C virus infection. Lancet Infect. Dis. 5:558-567. [DOI] [PubMed] [Google Scholar]
- 56.Stamataki, Z., S. Coates, M. J. Evans, M. Wininger, K. Crawford, C. Dong, Y. L. Fong, D. Chien, S. Abrignani, P. Balfe, C. M. Rice, J. A. McKeating, and M. Houghton. 2007. Hepatitis C virus envelope glycoprotein immunization of rodents elicits cross-reactive neutralizing antibodies. Vaccine 25:7773-7784. [DOI] [PubMed] [Google Scholar]
- 57.Tscherne, D. M., C. T. Jones, M. J. Evans, B. D. Lindenbach, J. A. McKeating, and C. M. Rice. 2006. Time- and temperature-dependent activation of hepatitis C virus for low-pH-triggered entry. J. Virol. 80:1734-1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Weiner, A. J., G. Kuo, D. W. Bradley, F. Bonino, G. Saracco, C. Lee, J. Rosenblatt, Q. L. Choo, and M. Houghton. 1990. Detection of hepatitis C viral sequences in non-A, non-B hepatitis. Lancet 335:1-3. [DOI] [PubMed] [Google Scholar]
- 59.Youn, J. W., S. H. Park, D. Lavillette, F. L. Cosset, S. H. Yang, C. G. Lee, H. T. Jin, C. M. Kim, M. T. Shata, D. H. Lee, W. Pfahler, A. M. Prince, and Y. C. Sung. 2005. Sustained E2 antibody response correlates with reduced peak viremia after hepatitis C virus infection in the chimpanzee. Hepatology 42:1429-1436. [DOI] [PubMed] [Google Scholar]
- 60.Yurkova, M. S., A. H. Patel, and A. N. Fedorov. 2004. Characterisation of bacterially expressed structural protein E2 of hepatitis C virus. Protein Expr. Purif. 37:119-125. [DOI] [PubMed] [Google Scholar]
- 61.Zhong, J., P. Gastaminza, G. Cheng, S. Kapadia, T. Kato, D. R. Burton, S. F. Wieland, S. L. Uprichard, T. Wakita, and F. V. Chisari. 2005. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. USA 102:9294-9299. [DOI] [PMC free article] [PubMed] [Google Scholar]