

Abstract
Agents inducing O6-methylguanine (O6MeG) in DNA such as N-methyl-N’-nitro-N-nitrosoguanidine (MNNG) are cytotoxic and a deficiency in mismatch repair (MMR) results in lack of sensitivity to this genotoxin (termed alkylation tolerance). Here, we show that ING2, a member of the inhibitor of growth family, is required for cell death induced by MNNG. We further observe that following MNNG treatment increases cellular protein levels of ING2 that is dependent on intact MMR function and that MNNG-induced ING2 localizes and associates with p73α in the nucleus. Suppression of ING2 by short hairpin RNA (shRNA) in MMR-proficient colorectal cancer cells decreased its sensitivity to MNNG, and in addition, abrogated MNNG-induced stabilization and acetylation of p73α. Interestingly, suppression of p73α had a greater impact on MNNG-induced cell death than ING2 leading us to conclude that ING2 regulates the cell death response, in part, through p73α. Inhibition of c-Abl by STI571 or suppression of c-Abl expression by shRNA blocked ING2 induction and p73α acetylation induced by this alkylator. Similarly, suppression of MMR (MLH1) by shRNA abrogated ING2 induction/ p73α acetylation. Taken together, these results demonstrate that MLH1/ c-Abl-dependent activation of ING2>p73α signaling regulates cell death triggered by MNNG and further suggests that dysregulation of this event may, in part, be responsible for alkylation-tolerance observed in MMR compromised cells.
Keywords: Apoptosis, DNA damage, Inhibitor of growth family member, mismatch repair, cell-cycle, p73
Introduction
The monofunctional SN1 alkylating (methylating) agent, N-methyl-N’-nitro-N-nitrosoguanidine (MNNG) is an extremely mutagenic, carcinogenic agent that evokes multiple cellular responses including DNA repair, cell-cycle arrest, apoptosis and necrosis [1-4]. Although MNNG reacts with DNA to form several adducts, O-6-alkyl guanine, generated by alkylation of the DNA base, is the predominant cytotoxic and mutagenic lesion in both prokaryotes and eukaryotes [5]. The enzyme, methylguanine-DNA methyl-transferase (MGMT) normally repairs O6MeG lesion by direct reversal [6,7]. As a result, lost or diminished MGMT activity increases MNNG-induced lesion and raises cellular sensitivity to MNNG [8-10]. O6MeG lesions are also recognized and repaired by the mismatch repair (MMR) system [11,12], an evolutionarily conserved DNA repair mechanism principally responsible for resolving post-replicative mismatches in DNA during S phase of cell cycle [13, 14]. It has been proposed that processing of O6MeG lesions by MMR generates DNA strand breaks that eventually activate signaling events which trigger abortive replication and eventual cell death [15].
Although numerous observations showed that MMR-mediated signaling events govern cell death induced by MNNG the precise mechanism by which this alkylator induces cell death is not known [16]. MNNG induces p53 in a MMR-dependent manner; however, its requirement in cell death is cell-type specific [17, 18]. For example, lymphoblastoid cells expressing either a mutant form or ablated complement of p53 displayed heightened resistance to this alkylator [19]. In p53 mutated gliomas, the same DNA lesion triggers mitochondrial apoptotic pathway [20]. A recent study showed that MNNG-induced intrinsic apoptosis in colorectal cancer cells (RKO) is stimulated by MLH1/c-Abl/p73α-mediated retrograde signaling [18]. O6MeG-triggered apoptosis in TK6 lymphoblastoid cells is shown to require Fas/CD95/Apo-1 receptor activation [21]. MNNG exposure activates MAPK signaling that requires MLH1, c-Abl and ATM [22]. Other studies showed that MNNG activates the PI-3 kinase, ATM [23] and the cell cycle checkpoint kinases, Chk1 and Chk2 [24] and dysregulated activation of these kinases have been attributed to defects in G2/M checkpoint activation observed in MMR-deficient cells following MNNG treatment [24, 25]. Biochemical studies revealed that components of the MMR system interact with the damage-responsive kinases ATM/ATR potentially facilitating phosphorylation and activation of Chk1/Chk2 kinases [24, 26]. Collectively, these studies demonstrated that the absence of MMR confers chemoresistance to SN1 methylators because of failure to recognize and consequently respond to DNA lesions that normally activate a robust cellular response [27-29].
Inhibitor of growth (ING) family of proteins is a group of tumor suppressors that control cell growth/proliferation, and senescence [29]. In S. cerevisiae, three ING proteins, namely Pho23, Yng1, and Yng2, co-purified as stable components of the Sin3/Rpd3 HDAC, NuA3, and NuA4 HAT complexes, respectively [30-32]. In higher organisms, ING1 is observed to reside within the mSin3A-HDAC complex [33]. ING1 can also associate with the p300 HAT [34]. Similarly, ING3 is documented to form a stable complex withTip60/NuA4 HAT [35]. ING4 and ING5 have been found to associate with HBO1 HAT complex [36]. Biochemical analysis showed that ING4 is essential for bulk histone H4 acetylation. All members of the ING family harbor a highly conserved plant homeodomain (PHD) that is commonly found in various chromatin remodeling proteins [37]. The PHD of ING2 regulates p53-dependent apoptosis through phosphoinositide signaling [38]. Genetic and crystal structure analyses revealed that ING proteins bind to tri-methylated lysine of histone H3 in yeast and mammalian cells via the PHD finger [39-41]. Deletion of ING2-leucine zipper (LZL) domain abrogated its association with p53, but not with p300 [43]. ING2 is also shown to enhance NER via inducement of histone H4 acetylation and chromatin relaxation [42]. Conceivably, ING2 modulates p53-dependent chromatin remodeling, apoptosis and DNA repair by functioning as a scaffold protein mediating interaction between p53 and p300.
In this study, we evaluated the role of ING2 in mediating cellular responses induced by the alkylating agent, MNNG. Our results show that treatment with MNNG increased the cellular levels of ING2 protein. Further, we observed that MNNG-induced ING2 translocates into the nucleus where it associates with and facilitates acetylation of p73α. We further demonstrate that ING2 regulates apoptotic cell death, induced by MNNG, in part, through activation of p73α function. Induction/ acetylation of p53 induced by MNNG, however, did not require ING2. Additionally, suppression of p53 did not affect cell death induced by MNNG. Finally, the requirement of MMR- and c-Abl for MNNG-activated ING2>p73α signaling lead us to conclude that MMR/c-Abl-dependent induction of ING2 regulates the cell death response to MNNG.
Materials and Methods
Materials
MNNG and Caffeine were obtained from Sigma-Aldrich (St. Louis, MO). STI 571 [Imatinib (Gleevec™)] was a gift from Novartis (Basel, Switzerland). Antibodies specific for p53 (DO-1), caspase-3, PARP and β-tubulin were obtained from Cell Signaling (Danvers, MA). Monoclonal p73α (SPM431) antibody was from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies specific for caspase-9, acetyl-p53 (K373/K382) and acetyl-lysine were from Upstate Biotechnology (Lake Placid, NY). HRP-conjugated secondary antibodies were purchased from Novus Biologicals (Littleton, CO).
Cell lines
HeLa, HEK-293, U2OS and HCT116 were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS). HCT116+ch2 (H2) is an MLH1-deficient derivative of colorectal cancer cell line, HCT116 that has a portion of human chromosome 2 introduced by microcell fusion. HCT116+ch3 (H3) was created by the stable transfer of a portion of human chromosome 3 bearing a wild-type copy of the hMLH1 gene into HCT116 [43]. H2 and H3 cells were maintained in DMEM containing 10% FBS supplemented with 400 μg/ml geneticin (G418) as described [26]. All cells were grown at 37° C in a humidified 5% CO2 incubator.
Short hairpin RNA (shRNA)
Overlapping synthetic oligonucleotides corresponding to sequences specific for the human ING2 (5′-AGAGAGCACTAATTAATAG-3′), MLH1 (5′-GGTTCACTACTAGTAAACTG-3′) and c-Abl (5′-GGATCAACACTGCTTCTGAT-3′) transcripts were hybridized and cloned into pSIREN-RETRO-Q (Clontech, La Jolla, CA). The recombinant pSiren plasmid was co-transfected with pCL-ampho plasmid encoding the packaging viral DNA into the packaging cell line, 293T (Clontech) using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). At 36 h post-transfection, supernatant containing the viral DNA was collected, filtered, and used to infect H3 (HCT116+ch3) cells in polybrene-supplemented medium. Cells were incubated with puromycin (1 μg/ml) for 4 days and downregulation of target gene expression was confirmed by immunoblotting.
Transfections and SiRNA
Shp53/ING2 and shING2/p73α double knocked down cells were obtained by transient transfection of siRNA specific for p53 (5-GACUCCAGUGGUAAUCUACTT-3) or p73α (5′-CCAUCCUGUACAACUUCAUGU G-3′) into MMR-proficient H3 (HCT116+ch3) cells stably expressing shRNA against p53, p73α or ING2 using Lipofectamine 2000™ (Invitrogen). Transfection complexes were prepared in 100 μl serum-free, antibiotic-free F12-K media containing 8 μl of HiPerFect transfection reagent and 40 pmol of siRNA. The mixture was incubated at room temperature for 20-30 min to allow for efficient complex formation. Transfection complexes were then mixed with 700 μl of complete medium and added to 30-40 × 103 cells. Transfected cells were cultured for 48 h prior to MNNG treatment.
Treatments
A stock concentration of 10 mM MNNG was prepared in 0.1M Na-acetate (pH 5.0) and stored at -80° C. MNNG was added to cell culture at the indicated concentration for 1 h at 37° C in a CO2 incubator. Cells were then rinsed extensively with PBS, re-fed on complete growth media and returned to the incubator. MNNG treatments were performed in media lacking antibiotics or selection agents.
Immunoblotting
Immunoblotting was performed as previously outlined [23]. Cells were harvested by scraping, washed with ice-cold PBS, and lysed in ice-cold 1X lysis buffer [10 mM Tris HCl pH 8.0, 240 mM NaCl, 5 mM EDTA, 1 mM DTT, 0.1 mM PMSF, 1% Triton X-100, 1 mM sodium vanadate, and 1 μg/ml of leupeptin, pepstatin, and aprotinin] by incubating on ice for 20 min. Lysates were clarified by centrifugation (1500 × g/2 min) and protein concentration was determined using Bradford dye (BioRad). After adjusting the lysates for equal protein content, the proteins were resolved on a 4-12% SDS-PAGE and then electro-transferred onto Immobilon-P (Millipore, Billerica, MA). The membrane was probed with primary antibody, subsequently with HRP-conjugated secondary antibody, and developed using Luminol reagent (PerkinElmer, Inc). For reprobing, the membrane was stripped by incubating in stripping buffer containing 65 mM Tris-HCl pH 6.7, 100 mM β-mercaptoethanol (BME) and 2% SDS at 40°C for 30 min. The membrane was then probed with indicated antibody.
Immunoprecipitation
Approximately, 1×106 cells were lysed in 500 μl of 1X lysis buffer (20 mM Tris-HCl, pH7.5 / 150 mM NaCl / 5 mM EDTA / 0.5% NP40 / 1 mM NaF / 1 mM DTT / 1 mM Na-Vanadate) and clarified by centrifugation. Lysates were adjusted for equal protein content and then incubated with 5 μl of anti- p73α antibody at 4°C for 4 h. 20 μl of protein A-Sepharose beads (Amersham-Pharmacia Biotech) in PBS were added and incubation continued for another 2 hours. Immune-complexes were then collected by quick spin, washed three times in 1X lysis buffer containing 300 mM NaCl, once with 1X lysis buffer, and subsequently analyzed by SDS-PAGE followed by immunoblotting.
Fractionation
Cells were fractionated as previously described [22]. Briefly, HCT116+Ch3 (H3) cells were either mock-treated or exposed to 10 μM MNNG for 1 h and harvested 6 h later. Cells were washed with PBS, resuspended in hypotonic buffer containing 10 mM Hepes-KOH, pH 7.5/10 mM NaCl/1 mM KH2PO4/5 mM NaHCO3/1 mM CaCal2/0.5 mM MgCl 2 and incubated on ice for 5 min. Cells were subsequently homogenized by 12 strokes in a Dounce homogenizer and cell lysis was verified by trypan blue staining. The homogenate was centrifuged at 1,000 × g for 5 min and the nuclear pellet was removed. The supernatant was again centrifuged at 100,000 × g for 90 min at 4° C to generate the cytosolic fraction. The nuclear pellet was washed twice in PBS and resuspended in nuclear isolation buffer (10 mM Tris, pH 7.5/300 mM sucrose/0.1% Nonidet P-40). The pellet was subsequently homogenized 20 times using a Dounce homogenizer and harvested by centrifugation. Both cytoplasmic and nuclear fractions were subjected to immunoblot analysis or immunoprecipitation using rat polyclonal ING2 antibody (a gift from Dr. Or Gozani at Stanford University).
Flow cytometry
Mock- and MNNG-treated cells were washed twice with 1X PBS, fixed by incubating in ice-cold 70% ethanol for 30 min and stored at 4° C. Prior to analysis, cells were incubated in PBS containing RNase A (1 mg/ml) and propidium iodide (40 μg/ml) or PI+ Annexin V-FITC or Annexin V for 30 min in dark at 37° C and then analyzed by flow cytometry. For each sample, more than 3 × 104 cells were counted and cells with lower DNA content (sub-G1) than those of G0/G1 phase were referred to as apoptotic cells. DNA histograms were analyzed using ModFit (Verity Software House, Topsham, ME) software. All flow cytometry experiments were performed in triplicate. The paired Student’s t test was used to determine the statistical significance. StatView software (Abacus Concepts, Berkeley, CA) was used. A P value less than 0.05 represents a statistically significant difference between the values of two group means.
Microscopy
Cells treated with DMSO or MNNG (10 μM; 1h) were washed with Hank’s Balanced Salt Solution (HBSS) containing 10 mM Hepes, 2 mM CaCl2, and 4 mg/ml BSA and fixed by incubating in 4% paraformaldehyde in HBBS for 5 min. Cells were then permeabilized in 100% methanol for 5 min and stained with rat polyclonal ING2 antibody for 1 hr at 37° C. Cells were subsequently stained with Alexa Fluor 555 goat anti-rat IgG-conjugated secondary antibody (Invitrogen, Carlsbad, CA). DNA was counterstained with DAPI. Photomicrographs were recorded using a Nikon fluorescence microscope (TE S2000) equipped with CCD camera.
Results
Dose- and time-dependent induction of ING2 in response to MNNG treatment
ING2, a member of the inhibitor of growth (ING) family has been implicated in mediating cellular responses to UV-induced DNA damage [44, 45]. To evaluate its role in alkylator-induced responses we first monitored intracellular ING2 protein levels in MNNG-treated cells. Anti-ING2 immunoblotting of the lysates formed from HeLa cells at 0, 3, 6, 12 and 24 h after MNNG (10 μM) showed a time-dependent increase in ING2 protein with optimal induction observed at 6 h post-treatment (Fig. 1A, top panel). Levels of ING2, however, decreased slightly at 24 h post-MNNG in all the cell lines examined. Similar time-dependent increases in ING2 protein were also observed after MNNG treatment in U2OS, HEK293 and HCT116+Ch3 (H3) (middle and bottom panels). Induction of ING2 was dose-dependent as exposure to increasing concentrations of MNNG (5, 10, 25 and 50 μM) caused a proportionate increase in ING2 (~1.4, 2.3, 4.1 and 4.5-fold) at the optimal induction time point of 6 h (Fig. 1B). When ING2 induction was assessed in the presence of, O6-benzylguanine (BQ), a methyl-guanine methyl transferase (MGMT) inhibitor that increases O6-MeG lesion load in DNA, we observed higher levels than observed with MNNG alone (Fig. 1C, compare lane 4 to 2 top panel). This result showed that alkylator-induced DNA lesion signals ING2 protein accumulation.
Figure 1. Time- and dose-dependent ING2 upregulation in response to MNNG treatment.
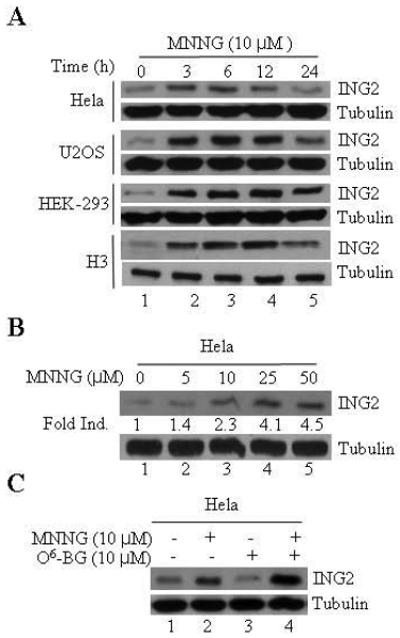
A. HeLa, U2OS, HEK-293 and H3 cells were exposed to 10 μM of MNNG for 1 h and collected 0, 3, 6, 12 and 24 h post-treatment. Lysates were formed, adjusted for equal protein content using Bio-Rad protein dye assay and resolved on a 4-12% SDS-PAGE. The proteins were transferred onto Immobilon-P and subjected to immunoblotting with rat polyclonal ING2 antibody. An aliquot of the lysate was also subjected to immunoblotting with β-tubulin antibody to assure equal protein loading. B. Dose-dependent ING2 induction in HeLa cells at 6 h post-MNNG. C. MNNG (10 μM; 1h)-induced ING2 upregulation in HeLa cells in the presence of MGMT inhibitor, O6-benzylguanine (10 μM).
ING2 is required for MNNG-induced cell death
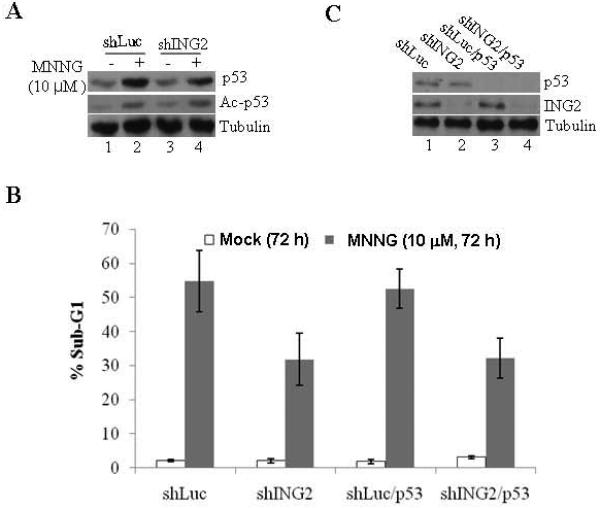
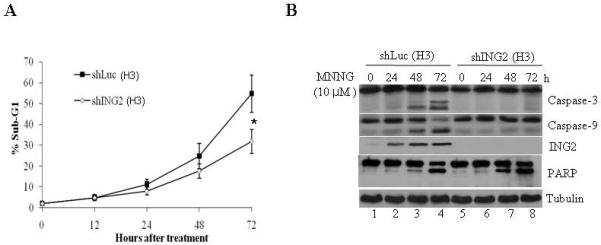
To assess the role of ING2 in cell-cycle arrest/ cell death responses induced by MNNG, we suppressed ING2 expression in mismatch repair-proficient human colorectal cancer cells (HCT116+Ch3;H3) using shRNA as described in methods. As control, luciferase shRNA was used. After confirming efficient suppression of ING2 protein levels in ING2-Knockdown (shING2) cells (see Fig. 3C, middle panel; compare lane 2 to 1), these cells, along with control Luciferase Knockdown population (shLuc), were exposed to MNNG (10 μM) and % G2/M population was assessed by flow cytometric analysis. Comparable G2 blockade in shLuc and shING2 cells at 24 and 48 h after treatment demonstrated that ING2 is dispensable for cell-cycle arrest induced by MNNG (Suppl. Fig. S1). We next assessed % sub-G1 DNA containing cells representing apoptotic population by flow cytometric analysis after MNNG treatment. Low levels of comparable cell death were observed in both shLuc and shING2 cells at 12 and 24 h post-MNNG; however, a clear difference was noted at 48 and 72 h post-treatment between these two cell lines (Fig. 2A). Specifically, at 72 h, shING2 cells showed approximately two-fold reduced cell death than similarly-treated shLuc cells (~27.9% vs. ~54.8%) and measured a statistically significant difference (p < 0.01) between these two cell lines. To independently assess the differential cell death induction, extracts were formed from MNNG-treated shLuc and shING2 cells and subjected to immunoblotting against anti-caspase-3 and -9 antibodies. MNNG-treated shING2 cells showed decreased caspase-3 and -9 cleavages compared to similarly-treated shLuc cells at 48 and 72 h after treatment (Fig. 2B, compare lanes 7 and 8 to lanes 3 and 4), consistent with attenuation of cell death following suppression of ING2 expression. Reprobing of the membrane with anti-ING2 antibody confirmed the presence of ING2 protein in shLuc cells at 24, 48 and 72 h time points. Interestingly, immunoblotting with anti-PARP antibody revealed no quantitative difference in PARP cleavage between shLuc and shING2 cells following MNNG treatment (Fig. 2B). Since MNNG induces non-apoptotic cell death such as necrosis in mouse fibroblasts [46] and caspase-independent cleavage of PARP has been noted during necrosis induced by MNNG [47] we dually stained cells with annexin V and PI. Costaining with annexin V and PI allows segregation of viable, apoptotic, and necrotic cell populations based on characteristics and integrity of the extracellular membrane to these dyes. Using this approach, we analyzed shLuc (H3) and shING2 (H3) cells treated with 10 μM MNNG at 72 h after drug exposure. In MNNG-treated shLuc cells, ~33.58 % of the cells was in either late or early apoptosis at this time point (Fig. 3C; upper right and lower quadrant). Less than 2% of the cells showed necrotic cell death (PI-positive/annexin V-negative, upper left quadrant). On the other hand, shING2 cells showed ~17.91% apoptotic and >3% nonapoptotic cell death at this time point. Collectively, this analysis demonstrated that, at the concentration employed, MNNG induces predominantly apoptotic cell death in these cell types and that ING2 is required for optimal apoptotic cell death induction. The PARP cleavage observed in these cells may represent other forms of cell death such as autophagy. To that end, additional studies are required to disseminate the role of PARP in the MNNG-induced cell killing.
Figure. 3. ING2 is dispensable for MNNG-induced p53 acetylation.
A. Mock- (DMSO) and MNNG (10 μM; 1h at 37 C)-treated shLuc and shING2 cells were collected at 6 h post-MNNG and extracts were prepared and adjusted for equal protein concentration. Proteins were resolved and subjected to immunoblotting with monoclonal anti-p53 (DO1) antibody. An aliquot of the lysate was also probed with site specific anti-acetyl p53 (K373/K382) antibody. Anti-β-tubulin immunoblotting ensured equal protein loading. B. p53 is dispensable for MNNG-induced cell death. shLuc, shING2, shp53, and shING2/p53 cells were either mock (DMSO)-treated or exposed to MNNG (10 μM; 1h) and collected 72 h later. Cells were stained with propidium iodide and % apoptosis (sub-G1) were determined by flow cytometry. Graphed are the mean ± SEM of three independent experimental determinations. C. Extracts formed from shLuc, shING2, shLuc/p53, and shING2/p53 cells were analyzed for suppression of p53 (top panel) and ING2 (bottom panel) expression by immunoblotting with anti-p53 (DO1) and anti-ING2 antibody, respectively. Equal protein loading was confirmed by anti-β-tubulin antibody.
Figure 2. Suppression of ING2 attenuates MNNG-induced cell death.
A. Luciferase (shLuc) and ING2-knocked down (shING2) cells were exposed to MNNG (10 μM; 1h) and apoptosis (% sub-G1) was assessed at 0, 12, 24, 48 and 72 h after treatment by flow cytometry following propidium iodide staining. Results are mean ± SEM of three independent determinations. *P<0.01 compared with the control group. B. MNNG-treated shLuc and shING2 cell lysates prepared at 0, 24, 48 and 72 h post-treatment were subjected to immunoblotting with caspase-3 (top panel), and caspase-9 (middle panel), PARP and ING2 antibodies. Equal protein content was confirmed by anti-β-tubulin immunoblotting. C. Mock- and MNNG (10 μM; 1 h)-treated cells were dually stained with annexin V and PI, and subsequently analyzed by flow cytometry. % cells showing PI-positive/annexin V-positive (upper right quadrant), PI-negative/annexin V-positive (lower right quadrant), and PI-positive/annexin V-negative (upper left quadrant) staining is indicated.
ING2 is dispensable for p53 induction/ acetylation induced by MNNG
MNNG treatment increases cellular levels of p53 [17] and in some cell lines apoptosis, induced by this alkylator proceeds in a p53-dependent manner [19]. Thus, in an effort to elucidate the mechanism of ING2-mediated cell death, we first examined the p53 upregulation in ING2-knock cells after MNNG treatment. Immunoblotting of the extracts prepared from shLuc and shING2 cells at 6 h post-MNNG with anti-p53 antibody revealed comparable p53 upregulation in both the cell lines (Fig. 3A, top panel). Since ING2 is linked to p53 acetylation during onset of replicative senescence [48] we also examined the acetylation status by reprobing the membrane with a site-specific anti-acetyl p53 (K373/K382) antibody. Comparable p53 acetylation in MNNG-treated shING2 cells and shLuc cells (middle panel, compare lane 1 to 2) revealed that ING2 is dispensable for p53 acetylation induced by MNNG. To explore a possible link between ING2 and p53 in MNNG-induced lethality, we compared % apoptotic cell population in cells knocked down either for p53 (shLuc/p53) or both p53 and ING2 (shING/p53) that were obtained by transfection of p53 siRNA into luciferase and ING2-knocked down H3 cells (described in Methods). After confirming suppression of p53 expression in shLuc/p53 and shING2/p53 cells by immunoblotting (Fig. 3B), the cells were exposed to MNNG (10 μM; 1h) and % sub-G1 cell population was assessed at 72 h post-treatment by flow cytometry. As anticipated, ING2-knocked down cells showed reduced cell death (~33.2% sub-G1; Fig. 3B) compared to Luciferase-knocked down cells (~51.2% sub-G1). Cells knocked down for p53, however, showed a cell death response similar to Luciferase-suppressed cells (~51.2% and ~53.7% sub-G1 cells in shp53 and shLuc, respectively). Shp53/ING2 cells and shING2 cells displayed similar levels of cell death (~31.2% and ~33.8%, respectively). Together, the data demonstrated that p53 is dispensable for cell death induced by the SN1 alkylator, MNNG.
ING2 regulates p73α induction/ acetylation in response to MNNG
MNNG modestly elevates cellular levels of p73α and its functional abrogation reduces MMR-dependent apoptosis induced by this alkylator [18]. Additionally, in response to IR treatment p73α undergoes acetylation modification that is correlated with activation of its transcriptional activity against pro-apoptotic genes [49]. Given this, we examined the requirement of ING2 in MNNG-induced p73α upregulation/ acetylation. In agreement with a previous report [18], a slight increase in p73α level was observed in MNNG-treated shLuc cells at 6, 12 and 24 h (Fig. 4A, lane 2, top panel). Levels of p73α in shING2 cells, however, remained unchanged at similar time points after treatment. Examination of p73α acetylation status by immunoprecipitation followed by immunoblotting with anti-acetyl lysine antibody showed a clear ING2 dependency in p73α acetylation induced by MNNG (middle panel). We also overexpressed ING2 in 293 cells by lipofectamine-mediated transfection of Ing2 cDNA and observed that the transgene expression alone increased p73α acetylation (Fig. 4C, top panel, compare lane 1 to 3). MNNG treatment further enhanced p73α acetylation (compare lane 2 to 4). In an inverse experiment, we examined ING2 induction in cells suppressed for p73α and observed that suppression of p73α had no impact on ING2 upregulation induced by MNNG (Fig. 4B, middle panel). The result clearly supports the notion that ING2 is a required upstream element in the MNNG-induced p73α signaling.
Figure. 4. ING2 is required for p73 stabilization/ acetylation induced by MNNG.
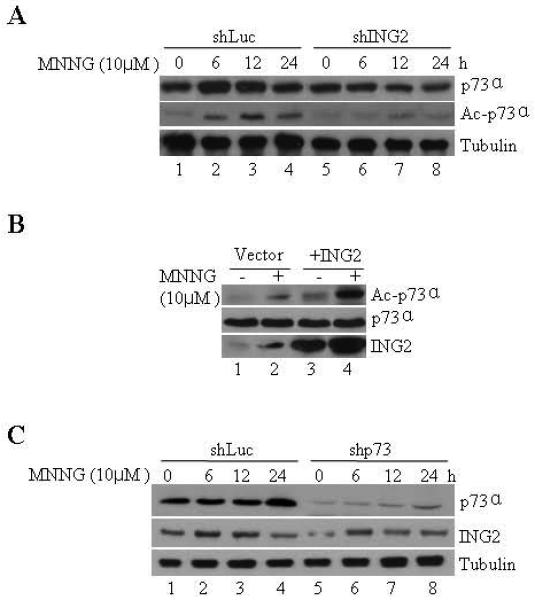
A. ShLuc and shING2 cells were treated with MNNG (10 μM; 1h) and collected at 0, 6, 12, and 24 h later. Extracts were formed, adjusted for equal protein concentration, and subjected to electrophoresis followed by immunoblotting with monoclonal anti-p73α antibody (top panel). Anti-β-tubulin immunoblotting ensured equal protein loading (bottom panel). B. An aliquot of the lysate was also subjected to immunoprecipitation with anti-p73α antibody. The immune-complexes were collected on Protein A beads and resolved on a 10% SDS-PAGE. The proteins were transferred onto Immobilon-P and immunoblotted against anti-acetyl lysine (top panel) or anti-p73α antibody (bottom panel). C. 293 cells were transfected with either vector (pcDNA3) alone or containing Ing2 cDNA using lipofectamine. At 36 h post-transfection, cells were exposed to MNNG (10 μM; 1h) and lysates were prepared 4 h later, adjusted for equal protein concentration and p73α induction/ acetylation was assessed by immunoprecipitation followed by immunoblotting with anti-p73α or anti-acetyl antibody. Anti-ING2 immunoblotting confirmed ING2 expression in Ing2 cDNA transfected cells (bottom panel, lanes 3 and 4).
MNNG promotes ING2/p73α complex formation in the nucleus
To test whether ING2, as an adaptor protein, interacts with p73α to facilitate acetylation induced by MNNG we performed a co-immunoprecipitation assay. Anti-ING2 immune-complexes prepared from mock-treated lysates showed low levels of p73α that was greatly enhanced after MNNG treatment (Fig. 5A, middle panel, compare lane 2 to 1). Reprobing of the membrane with pan specific anti-acetyl lysine antibody showed that ING2-complexed p73α is acetylated as well (middle panel). Presumably, MNNG treatment leads to enhanced binding and acetylation of p73α by ING2. Anti-ING2 immunoblotting confirmed the presence of ING2 in mock and MNNG-treated cells (bottom panel). Since ING2 resides mostly in the nucleus with 74% in the chromatin/nuclear matrix and 9% in the nucleoplasm in HT1080 fibrosarcoma cells ING2 [38], we examined alterations in endogenous ING2 localization after MNNG treatment. Immunofluorescence analysis of untreated H3 cells stained with rat polyclonal anti-ING2 antibody showed faint, diffuse, peri-nuclear and cytoplamic ING2 localization (Fig. 5B). Corroborating well with heightened expression observed by immunoblot analyses cells exposed to MNNG showed increased ING2 staining both in the nucleus and in the cytoplasm (Fig. 1A). Since p73α is localized both in the nucleus and cytoplasm [50], we assessed ING2/p73α complex formation in nuclear and cytoplasmic extracts formed from mock- and MNNG (10 μM; 1h)-treated H3 cells. Subsequent to confirming the integrity of cytoplasmic and nuclear fractions (described in Methods) by immunoblotting with anti-tubulin and anti-lamin A/C antibody (Fig. 5C), p73α was immunoprecipitated and the immune-complex was resolved and immunoblotted against anti-ING2 antibody. Nuclear extracts formed from mock-treated cells also showed very little ING2-p73α interaction, but the abundance of this complex increased following MNNG treatment (Fig. 5D; compare lane 3 to 4). In contrast, MNNG-treated cytoplasmic fraction showed decreased levels of p73α-associated ING2 compared to mock-treated cytoplasmic extracts. Together, the results support the notion that MNNG treatment leads to increased ING2-p73α complex formation in the nucleus.
Figure. 5. MNNG-induced ING2 localization and ING2/p73α complex formation.
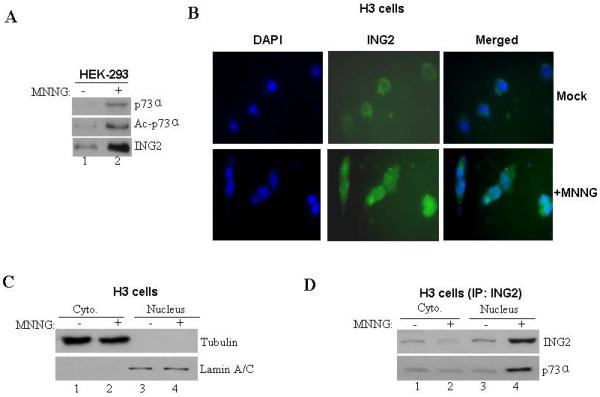
A. Mock- (DMSO) and MNNG- (10 μM; 1h)-treated HEK-293 cells were collected 6 h later. Lysates were formed, adjusted for equal protein content and incubated with anti-ING2 antibody and Protein A Sepharose. The immune-complexes were resolved on a 4-12% gradient SDS-PAGE and analyzed by immunoblotting with anti-73α antibody. The membrane was stripped (described in methods) and re-probed with anti-acetyl lysine antibody. An aliquot of the lysate was also probed with anti-ING2 antibody. B. Mock- and MNNG (10 μM; 1h)-treated HCT116+Ch3 (H3) cells were collected 6 h later cells, fixed and stained with anti-ING2 antibody and subsequently with a FITC-conjugated secondary antibody. Cells were also counterstained with DAPI and visualized under immunofluorescence microscope. C. Mock- and MNNG-treated cells were subjected to hypotonic lysis and nuclei isolated by sucrose cushion centrifugation. Following this, nuclei were lysed and both cytoplasmic (lanes 1 and 2) and nuclear fractions (lanes 3 and 4) were normalized for equal protein content and immunoblotted against tubulin (top panel) and nuclear protein lamin A/C (bottom panel) antibody to confirm the integrity of fractionation. D. Lysates of both cytoplasmic and nuclear fractions from mock- and MNNG-treated cells were used for immunoprecipitation reactions by incubation with anti-p73α antibody. Subsequently, immune-complexes were resolved and immunoblotted with anti-ING2 (top panel) and anti-p73α (bottom panel) antibody.
Both ING2 and p73α are required for MNNG-induced cell death
To evaluate the functional significance of ING2/ p73α interaction in MNNG responses we compared cell death induction in cells suppressed for p73α, ING2 or both. ShLuc/p73α and shING2/p73α cells were obtained by transfecting p73α siRNA into Luciferase and ING2-knocked down cells using lipofectamine as described in Methods. After confirming suppression of p73α protein expression by immunoblotting (Fig. 6A), the transfectants were either mock treated or exposed to MNNG (10 μM) and % apoptosis (sub-G1) was assessed by flow cytometry. At 72 h post-treatment, cells suppressed for either p73α or ING2 showed reduced apoptosis (Fig. 6B). However, a comparison of cell death between these two cell lines showed that suppression of p73α had a greater impact on cell death as demonstrated by ~18.7% apoptosis in shp73α cells versus ~31.8% apoptosis in shING2 cells. Cell death induction in shING2/p73α cells were similar to that observed in shp73α cells (~17.4% and ~18.7%). Corroborating well with these results, decreased caspase-3 cleavage was observed in MNNG-treated shp73α cells compared to similarly-treated shING2 cells (Fig. 6C, compare lane 4 to 6). Again, MNNG-treated shp73α and shING2/shp73α cells showed much reduced caspase-3 cleavage (compare lane 4 to 8) than shING2 cells. We reconcile these results by concluding that ING2 promotes cell death, in part, through p73α and that p73α plays a prominent ING2-independent role in MNNG-induced cell death.
Figure 6. Effect of ING2, p73α and ING2/p73α suppression on MNNG-induced cell death.
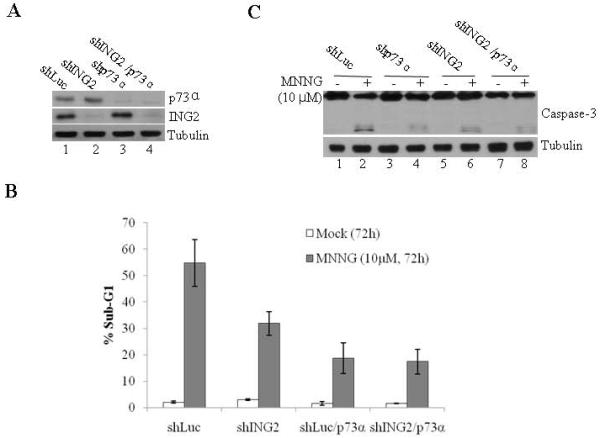
A. Cell extracts formed from shLuc, shING2, shLuc/p73α, and shING2/p73α cells were adjusted for equal protein content and immunoblotted against anti-p73α (top panel), ING2 (middle panel) and tubulin (bottom panel) antibody. B. ShLuc, shp73, shLuc/ING2 and shING2/p73α cells were either mock-treated or exposed to MNNG (10 μM; 1h) and collected 72 h later. Cells were stained with propidium iodide and % cell death (sub-G1) was assessed by flow cytometry. Mean of three experiments with S.D is given. C. Mock- and MNNG-treated cells, described in Panel B, were evaluated for caspase-3 cleavage by immunoblotting the lysates with anti-caspase-3 antibody (top panel). Equal protein loading was confirmed by anti-β-tubulin antibody immunoblotting (bottom panel).
MMR/c-Abl-dependency of ING2 signaling induced by MNNG
The proto-oncogene kinase, c-Abl is activated by MNNG [22]. In addition, a recent study demonstrated that both c-Abl and p73α are required for MNNG-induced G2 arrest, apoptosis and lethality [18, 62]. Given this, we investigated the role of c-Abl in MNNG-induced ING2 upregulation and p73α acetylation using c-Abl tyrosine kinase inhibitor, STI571. Co-treatment of HEK293 cells with MNNG (10 μM; 1h) and STI571 (10 μM) abrogated ING2 upregulation (Fig. 7A; compare lane 2 to 4, top panel). In addition, STI571 treatment completely abrogated p73α acetylation induced by MNNG (middle panel, compare lane 2 to 4). Inhibition of c-Abl kinase by STI571 was confirmed by immunoprecipitation of c-Abl followed by in vitro kinase assay using GST-CTD of RNAP II as substrate (Suppl. Fig S2). Subsequent assessment of the impact of STI571 on MNNG-induced cell death in shLuc and shING2 cells showed that the inhibitor reduced MNNG-induced cell death as demonstrated by ~18% in the presence Vs ~51.3% sub-G1 cells in the non-treated shLuc cells (Fig. 7B). Since Gleevec has other targets, we examined cells suppressed for c-Abl expression (Fig. 7C, top panel) for ING2 induction, p73 stabilization/acetylation induced by MNNG. As anticipated, c-Abl-knocked down cells showed attenuated ING2 induction and p73α stabilization/acetylation compared to Luciferase-knock down cells in response to MNNG treatment (Fig. 7C, middle panels). Annexin V staining of MNNG-treated shLuc and shAbl cells followed by flow cytometric analysis revealed that suppression of c-Abl expression reduced cell death (~48.9% sub-G1) compared to control knock down cells (~16.2% sub-G1) (Fig. 7D). Together, the results established that c-Abl is a required upstream component in the ING2>p73α apoptotic signaling pathway activated by MNNG.
Fig. 7. STI571 blocks ING2 upregulation/ p73α acetylation induced by MNNG.
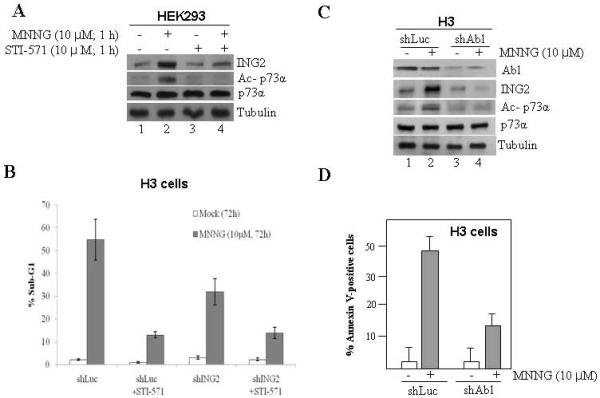
A. HEK-293 cells were incubated with MNNG (10 μM) in the presence or absence of STI571 (10 μM) for 1 h at 37° C and ING2 protein induction was assessed by immunoblotting with anti-ING2 antibody (top panel). Lysates were also used to determine p73α acetylation by immunoprecipitation followed by immunoblotting with anti-acetyl lysine or anti-p73α antibody (middle panels) as described in Methods. Tubulin immunoblotting determined equal loading (bottom panel). B. ShLuc and shING2 cells were treated with MNNG (10 μM) ± STI571 (10 μM) and % apoptosis (sub-G1) was assessed at 72 h post-treatment by flow cytometry. Graphed are the mean ± SEM of three independent experimental determinations. C. ShLuc and shAbl cells were either mock treated or exposed to MNNG (10 μM; 1h) and collected at 6 h post-treatment. Extracts were formed, adjusted for equal protein concentration and then subjected to immunoblotting with anti-c-Abl and ING2 antibody. Lysates were also subjected to immunoblotting with anti-p73α antibody and the immune-complex was resolved and probed with anti-acetyl lysine antibody. Anti-tubulin immunoblotting confirmed equal protein loading. Mock and MNNG-treated shLuc and shAbl cells were harvested at 72 h after treatment and stained with Annexin V. Cells were subjected to flow cytometric analysis and % Annexin V-positive cells are given. Mean of three experiments with S.D is given.
We also evaluated the requirement of MMR in ING2 signaling activated by MNNG. Treatment of isogenic MLH1-deficient (HCT116) and -proficient cells (HCT116/MLH1+) with MNNG resulted in ING2 induction at 3, 4 and 6 h post-treatment in MLH1+ cells (Fig. 8A, top panel). Under the same condition, MLH1-deficeint cells failed to upregulate ING2. Similarly, MNNG-induced p73α acetylation was observed in MMR-positive cells (Fig. 8A, middle panel). MNNG-treated MMR-deficient cells failed to display p73α acetylation. To independently confirm the MMR requirement, MLH1 expression was suppressed in HCT116+Ch3 (H3) cells by shRNA and ING2 induction and p73α acetylation, induced by MNNG, was assessed. Result showing attenuated ING2 induction and p73α acetylation in MLH1-suppressed cells confirmed MMR requirement (Fig. 7C). Anti-MLH1 immunoblotting shows suppression of MLH1 expression in the knocked down cell populations. Collectively, all these findings demonstrated that MMR and c-Abl are required component in the ING2>p73α signaling event activated by MNNG.
Fig. 8. MMR function is required for MNNG-induced ING2 upregulation and p73 acetylation.
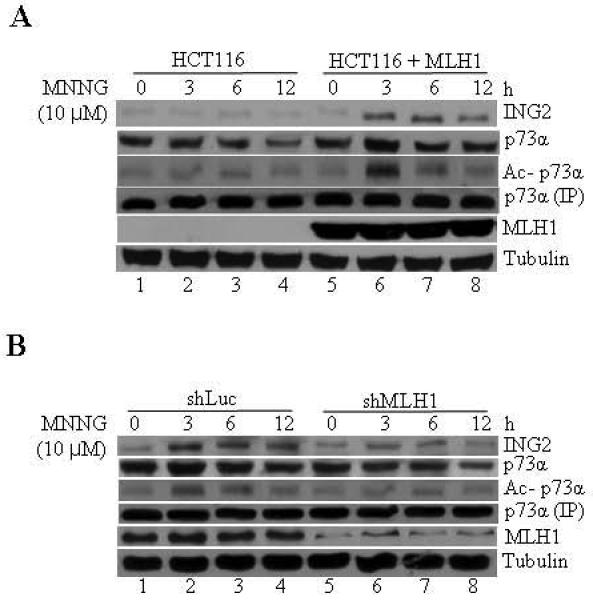
A. HCT116 and HCT116 cells reconstituted with Mlh1 (HCT116+MLH1) were exposed to MNNG (10 μM; 1h) and lysates formed at 0, 3, 6 and 12 h later were subjected to immunoblotting with anti-ING2 or anti-p73α antibody. An aliquot of the lysate was used to immunoprecipitate p73α and its acetylation status was assessed by immunoblotting with anti-acetyl lysine antibody. MLH1 expression in reconstituted cells was confirmed by immunoblotting with anti-MLH1 antibody. Anti-β-tubulin immunoblotting shows equal protein loading. B. MNNG-induced ING2 induction and p73α acetylation in shLuc and shMLH1 cells. Suppression of MLH1 expression was confirmed by immunoblotting with anti-MLH1 antibody. Anti-β-tubulin immunoblotting determined equal protein loading.
Discussion
Although members of the ING family have been implicated in cellular responses elicited by UV and etoposide [45, 51], their involvement in alkylator-induced responses is not known. The work outlined in this report has demonstrated an involvement of ING2 in mediating cell death activated by SN1-methylating agent, MNNG. Briefly, activation of this pathway leads to heightened expression of ING2 protein that in turn promotes cell death, in part, through acetylation and activation of the pro-apoptotic function of p73α. In support of this view, we have observed that suppression of ING2 expression by shRNA reduced MNNG-induced cell death by ~2 fold compared to control Luciferase-knocked down cells (Fig. 4). In addition, ING2-Knockdown cells failed to display p73α upregulation and acetylation induced by MNNG (Fig. 5). The presence of ING2-p73α complex in the nuclear fractions of MNNG-treated cells and the augmented p73α acetylation observed upon ING2 overexpression indicate a direct role for ING2 in promoting p73α induction and acetylation. Interestingly, p73α suppressed cells showed much reduced cell death than ING2-depleted cells leading us to conclude that p73α plays an additional ING2-independent role in eliciting the cell death response to MNNG. On the other hand, p53 acetylation/stabilization induced by MNNG does not require ING2. Moreover, consistent with the report of Li et al [18], in these colorectal cancer cells, cell death activated by this alkylator does not require p53. Using genetic and pharmacological approaches, we further established that MMR and c-Abl are required upstream elements in the activation of ING2>p73α signaling induced by MNNG. Finally, the augmented ING2 upregulation by MNNG in the presence of O6-benzylguanine that raises the O6MeG DNA lesion load, lead us to conclude that alkylator-induced DNA damage activates the ING2>p73α signaling pathway.
Although our findings underscore a potential role for ING2/p73α signaling in MNNG-induced apoptosis, we are unsure of how ING2 exerts control over cell death. As an adaptor protein, ING2 facilitates p300 HAT-mediated p53 acetylation during the onset of replicative senescence [48]. Given that acetylation modification activates pro-apoptotic proteins, we propose that ING2 promotes apoptosis, induced by MNNG, through HAT-mediated acetylation and activation of key apoptotic molecules such as p53 and p73α. Our study determined that p53 is dispensable for MNNG-induced apoptosis. On the other hand, we found that p73α is required for apoptosis induced by MNNG. As a transcriptional factor, p73α mediates IR-induced apoptosis through induction of apoptotic genes such as puma and bax and [57]. Moreover, the transcriptional activity of p73α is regulated by acetylation. We observed that MNNG-induced p73α acetylation requires ING2 and that alkylator treatment enhances interaction between ING2 and p73α. Conceivably, ING2-mediated cell death response to MNNG involves recruitment of p73α by ING2 to HAT consequently promoting acetylation and activation of the transcriptional function of p73α. Since p73α principally controls the onset of apoptosis by triggering the transcription of several pro-apoptotic genes, the delay we have observed between ING2 induction and the onset of apoptosis likely corresponds to the time necessary for p73-responsive genes to achieve the cellular levels required to trigger cell death.
Despite the demonstration that MMR and c-Abl are required for ING2 upregulation, the molecular events leading to ING2 induction remains elusive. Hickman and Samson found that the DNA mismatch recognition complex, MutSα (MSH2/MSH6 heterodimer) is required for signaling apoptosis in response to MNNG and that MMR status, rather than p53 status, is a strong indicator of the susceptibility of cells to alkylation-induced apoptosis [52]. Previous studies from our laboratory support the notion that MMR complex assembly at the site of DNA damage lesion signals activation of Chk2 and ATM kinase activity in response to MNNG [26]. Others have proposed that MMR-mediated processing of O6-methyl lesions leads to formation of DNA strand breaks that perhaps signals apoptosis. Clearly, several DNA strand break responsive molecules including ATM and c-Abl are activated by MNNG in MMR-dependent manner [22, 23]. Since ING2 is upregulated by DSB-inducing agent such as neocarzinostatin [48], it is possible that DSB generated during MMR-mediated processing of O6-MeG lesion signals ING2 upregulation. In addition to MMR, the c-Abl kinase function is also required for ING2>p73α signaling event activated by MNNG since inhibition of c-Abl kinase by STI571 blocked MNNG-induced ING2 upregulation. Again, c-Abl is activated and associates with MLH1 following MNNG treatment [22]. C-Abl-mediated targeting of proteins through the ubiquitin-mediated proteosome pathway has also been reported [58]. Possibly, recruitment of c-Abl to the O6-methyl lesion is required for ING2 upregulation. Alternatively, c-Abl may prevent ING2 degradation consequently leading to its upregulation. Of note, a recent study showed that MMR-dependent G(2) arrest responses triggered by MNNG are dependent on a human MLH1/c-Abl/GADD45alpha signaling pathway and activity (62). Given this, we postulate that although MLH1/c-Abl complex regulates both apoptotic and G2 blockade triggered by MNNG, this complex dependent induction of ING2 participates in the cell death response but dispensable for cell cycle arrest induced by this alkylator.
ING genes are rarely mutated but their transcript levels are often suppressed in many cancer cells [59,60]. Methylation of the ING promoter has been reported [61]. Therefore, the lack of ING2 upregulation due to Ing2 promoter silencing may also confer an inability of these cells to activate ING2-mediated signaling events. Clearly, further study is required to fully elucidate the role of MMR and c-Abl in activating ING2 signaling in response to MNNG.
In sum, the principal advance stemming from this investigation is the demonstration that ING2, previously implicated in other genotoxin-induced responses, regulates MNNG-induced cell death. We further demonstrated that ING2-regulated cell death response is mediated, in part, through acetylation and activation of p73α function. We also observed a deficiency in ING2 in the MMR-proficient human colorectal cancer cells reduced sensitivity to MNNG. This is reminiscent of the “alkylation tolerant” phenotype observed in cells deficient in MMR function. The lack of ING2>p73α signaling in cells compromised for MMR function suggests that an inactivation of this signaling event could contribute to alkylation tolerance observed in MMR-deficient colorectal cells.
Supplementary Material
Acknowledgements
We thank Dr. Or Gozani at Stanford University for rat ING2 antibody and Dr. Curtis Harris at NIH for ING2 cDNA. This work was supported by NIH grant (GM60945) to R.B.
Abbreviations
- MNNG
N-methyl-N’-nitro-N-nitrosoguanidine
- ING
inhibitor of growth
- MMR
mismatch repair
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hartwell LH, Kastan KB. Cell cycle control and cancer. Science. 1994;266:1821–1828. doi: 10.1126/science.7997877. [DOI] [PubMed] [Google Scholar]
- 2.Loeb LA, Loeb KR, Anderson JP. Multiple mutations and cancer. Proc Natl Acad Sci USA. 2003;100:776–781. doi: 10.1073/pnas.0334858100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eadie JS, Conrad MD, Toorchen D, Topal MD. Mechanism of mutagenesis by O6-methylguanine. Nature. 1984;308:201–203. doi: 10.1038/308201a0. [DOI] [PubMed] [Google Scholar]
- 4.Loveless A. Possible relevance of O-6 alkylation of deoxyguanosine to the mutagenicity and carcinogenicity of nitrosamines and nitrosamides. Nature. 1969;223:206–207. doi: 10.1038/223206a0. [DOI] [PubMed] [Google Scholar]
- 5.Wyatt MD, Pittman DL. Methylating agents and DNA repair responses: Methylated bases and sources of strand breaks. Chem Res Toxicol. 2006;19:1580–1594. doi: 10.1021/tx060164e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Foote RS, Mitra S, Pal BC. Demethylation of O6-methylguanine in a synthetic DNA polymer by an inducible activity in Escherichia coli. Biochem Biophys Res Commun. 1980;97:654–659. doi: 10.1016/0006-291x(80)90314-9. [DOI] [PubMed] [Google Scholar]
- 7.Olsson M, Lindahl T. Repair of alkylated DNA in Escherichia coli. Methyl group transfer from O6-methylguanine to a protein cysteine residue. J Biol Chem. 1980;255:10569–10571. [PubMed] [Google Scholar]
- 8.Bobola MS, Berger MS, Silber JR. Contribution of O6-methylguanine-DNA methyltransferase to resistance to 1,3-(2-chloroethyl)-1-nitrosourea in human brain tumor-derived cell lines. Mol Carcinog. 1995;13:81–88. doi: 10.1002/mc.2940130204. [DOI] [PubMed] [Google Scholar]
- 9.Kalamegham R, Warmels-Rodenhiser S, MacDonald H, Ebisuzaki K. O6- methylguanine-DNA methyltransferase-defective human cell mutant: O6- methylguanine, DNA strand breaks and cytotoxicity. Carcinogenesis. 1988;9:1749–1753. doi: 10.1093/carcin/9.10.1749. [DOI] [PubMed] [Google Scholar]
- 10.Tominaga Y, Tsuzuki T, Shiraishi A, Kawate H, Sekiguchi M. Alkylation- induced apoptosis of embryonic stem cells in which the gene for DNA-repair, methyltransferase, had been disrupted by gene targeting. Carcinogenesis. 1997;18:889–896. doi: 10.1093/carcin/18.5.889. [DOI] [PubMed] [Google Scholar]
- 11.Duckett DR, Drummond JT, Murchie AI, Reardon JT, Sancar A, Lilley DM, Modrich P. Human MutSalpha recognizes damaged DNA base pairs containing O6-methylguanine, O4-methylthymine, or the cisplatin-d(GpG) adduct. Proc Natl Acad Sci U S A. 1996;93:6443–6447. doi: 10.1073/pnas.93.13.6443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Griffin S, Branch P, Xu YZ, Karran P. DNA mismatch binding and incision at modified guanine bases by extracts of mammalian cells: implications for tolerance to DNA methylation damage. Biochemistry. 1994;33:4787–4793. doi: 10.1021/bi00182a006. [DOI] [PubMed] [Google Scholar]
- 13.Modrich P, Lahue R. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu Rev Biochem. 1996;65:101–33. doi: 10.1146/annurev.bi.65.070196.000533. [DOI] [PubMed] [Google Scholar]
- 14.Meyers M, Wagner MW, Hwang HS, Kinsella TJ, Boothman DA. Role of the hMLH1 DNA mismatch repair protein in fluoropyrimidine-mediated cell death and cell cycle responses. Cancer Res. 2001;61:5193–5201. [PubMed] [Google Scholar]
- 15.Stojic L, Brun R, Jiricny J. Mismatch repair and DNA damage signaling. DNA Repair (Amst) 2004;3:1091–101. doi: 10.1016/j.dnarep.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 16.Meikrantz W, Bergom MA, Memisoglu A, Samson L. O6-alkylguanine DNA lesions trigger apoptosis. Carcinogenesis. 1998;19:369–372. doi: 10.1093/carcin/19.2.369. [DOI] [PubMed] [Google Scholar]
- 17.Duckett DR, Bronstein SM, Taya Y, Modrich P. hMutSalpha- and hMutLalpha-dependent phosphorylation of p53 in response to DNA methylator damage. Proc Natl Acad Sci U S A. 1999;96:12384–12388. doi: 10.1073/pnas.96.22.12384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li LS, Morales JC, Hwang A, Wagner MW, Boothman DA. DNA mismatch repair-dependent activation of c-Abl/p73alpha/GADD45alpha-mediated apoptosis. J Biol Chem. 2008;283:21394–21403. doi: 10.1074/jbc.M709954200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim WJ, Beardsley DL, Adamson AW, Brown KD. The monofunctional alkylating agent N-methyl-N’-nitro-N-nitrosoguanidine triggers apoptosis through p53-dependent and -independent pathways. Toxicol Appl Pharmacol. 2005;202:84–98. doi: 10.1016/j.taap.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 20.Roos WP, Batista LF, Naumann SC, Wick W, Weller M, Menck CF, Kaina B. Apoptosis in malignant glioma cells triggered by the temozolomide-induced DNA lesion O6-methylguanine. Oncogene. 2007;26:186–197. doi: 10.1038/sj.onc.1209785. [DOI] [PubMed] [Google Scholar]
- 21.Dunkern T, Roos W, Kaina B. Apoptosis induced by MNNG in human TK6 lymphoblastoid cells is p53 and Fas/CD95/Apo-1 related. Mutat Res. 2003;544:167–72. doi: 10.1016/j.mrrev.2003.06.005. [DOI] [PubMed] [Google Scholar]
- 22.Kim WJ, Rajasekaran B, Brown KD. MLH1- and ATM-dependent MAPK signaling is activated through c-Abl in response to the alkylator N-methyl-N’-nitro- N’-nitrosoguanidine. J Biol Chem. 2007;2:32021–32031. doi: 10.1074/jbc.M701451200. [DOI] [PubMed] [Google Scholar]
- 23.Adamson AW, Kim WJ, Shangary S, Baskaran R, Brown KD. ATM is activated in response to N-methyl-N’-nitro-N-nitrosoguanidine-induced DNA alkylation. J. Biol. Chem. 2002;277:38222–38229. doi: 10.1074/jbc.M204409200. [DOI] [PubMed] [Google Scholar]
- 24.Adamson AW, Beardsley DI, Kim WJ, Gao Y, Baskaran R, Brown KD. Methylator-induced, mismatch repair-dependent G2 arrest is activated through Chk1 and Chk2. Mol Biol Cell. 2005;16:1513–1526. doi: 10.1091/mbc.E04-02-0089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stojic L, Mojas N, Cejka P, Di Pietro M, Ferrari S, Marra G, Jiricny J. Mismatch repair-dependent G2 checkpoint induced by low doses of SN1 type methylating agents requires the ATR kinase. Genes Dev. 2004;18:1331–1344. doi: 10.1101/gad.294404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brown KD, Rathi A, Kamath R, Beardsley DI, Zhan Q, Mannino JL, Baskaran R. The mismtach repair system is required for S phase checkpoint. Nat Genet. 2003;33:80–84. doi: 10.1038/ng1052. [DOI] [PubMed] [Google Scholar]
- 27.Goldmacher VS, Cuzick RA, Jr, Thilly WG. Isolation and partial characterization of human cell mutants differing in sensitivity to killing and mutation by methylnitrosourea and N-methyl-N’-nitro-N-nitrosoguanidine. J Biol Chem. 1986;261:12462–12471. [PubMed] [Google Scholar]
- 28.Branch P, Aquilina G, Bignami M, Karran P. Defective mismatch binding and a mutator phenotype in cells tolerant to DNA damage. Nature. 1993;362:652–654. doi: 10.1038/362652a0. [DOI] [PubMed] [Google Scholar]
- 29.Campos EI, Chin MY, Kuo WH, Li G. Biological functions of the ING family tumor suppressors. Cell. & Mol. Life Sci. 2004;61:2597–2613. doi: 10.1007/s00018-004-4199-4. [DOI] [PubMed] [Google Scholar]
- 30.Howe L, Kusch T, Muster N, Chaterji R, Yates JR, 3rd, Workman JL. Yng1p modulates the activity of Sas3p as a component of the yeast NuA3 Hhistone acetyltransferase complex. Mol Cell Biol. 2002;22:5047–5053. doi: 10.1128/MCB.22.14.5047-5053.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nourani A, Doyon Y, Utley RT, Allard S, Lane WS, Cote J. Role of an ING1 growth regulator in transcriptional activation and targeted histone acetylation by the NuA4 complex. Mol Cell Biol. 2001;21:7629–7640. doi: 10.1128/MCB.21.22.7629-7640.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nourani A, Howe L, Pray-Grant MG, Workman JL, Grant PA, Cote J. Opposite role of yeast ING family members in p53-dependent transcriptional activation. J Biol Chem. 2003;278:19171–19175. doi: 10.1074/jbc.C300036200. [DOI] [PubMed] [Google Scholar]
- 33.Kuzmichev A, Zhang Y, Erdjument-Bromage H, Tempst P, Reinberg D. Role of the Sin3-histone deacetylase complex in growth regulation by the candidate tumor suppressor p33(ING1) Mol Cell Biol. 2002;22:835–848. doi: 10.1128/MCB.22.3.835-848.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vieyra D, Loewith R, Scott M, Bonnefin P, Boisvert FM, Cheema P, Pastyryeva S, Meijer M, Johnston RN, Bazett-Jones DP, McMahon S, Cole MD, Young D, Riabowol K. Human ING1 proteins differentially regulate histone acetylation. J Biol Chem. 2002;277:29832–29839. doi: 10.1074/jbc.M200197200. [DOI] [PubMed] [Google Scholar]
- 35.Doyon Y, Selleck W, Lane WS, Tan S, Cote J. Structural and functional conservation of the NuA4 histone acetyltransferase complex from yeast to humans. Mol Cell Biol. 2004;24:1884–1896. doi: 10.1128/MCB.24.5.1884-1896.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Doyon Y, Cayrou C, Ullah M, Landry AJ, Cote V, Selleck W, Lane WS, Tan S, Yang XJ, Cote J. ING tumor suppressor proteins are critical regulators of chromatin acetylation required for genome expression and perpetuation. Mol. Cell. 2006;21:51–64. doi: 10.1016/j.molcel.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 37.Aasland R, Gibson TJ, Stewart AF. The PHD finger: implications for chromatin-mediated transcriptional regulation. Trends Biochem Sci. 1995;20:56–59. doi: 10.1016/s0968-0004(00)88957-4. [DOI] [PubMed] [Google Scholar]
- 38.Gozani O, Karuman P, Jones DR, Ivanov D, Cha J, Lugovskoy AA, Baird CL, Zhu H, Field SJ, Lessnick SL, Villasenor J, Mehrotra B, Chen J, Rao VR, Brugge JS, Ferguson CG, Payrastre B, Myszka DG, Cantley LC, Wagner G, Divecha N, Prestwich GD, Yuan J. The PHD finger of the chromatin-associated protein ING2 functions as a nuclear phosphoinositide receptor. Cell. 2003;114:99–111. doi: 10.1016/s0092-8674(03)00480-x. [DOI] [PubMed] [Google Scholar]
- 39.Martin DG, Baetz K, Shi X, Walter KL, MacDonald VE, Wlodarski MJ, Gozani O, Hieter P, Howe L. The Yng1p plant homeodomain finger is a methylhistone binding module that recognizes lysine 4-methylated histone H3. Mol Cell Biol. 2006;26:7871–7879. doi: 10.1128/MCB.00573-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pena PV, Davrazou F, Shi X, Walter KL, Verkhusha VV, Gozani O, Zhao R, Kutateladze TG. Molecular mechanism of histone H3K4me3 recognition by plant homeodomain of ING2. Nature. 2006;442:100–103. doi: 10.1038/nature04814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shi X, Hong T, Walter KL, Michishita E, Hung T, Carney D, Peña P, Lan F, Kaadige MR, Lacoste N, Cayrou C, Davrazou F, Saha A, Cairns BR, Ayer DE, Kutateladze TG, Shi Y, Côté J, Chua KF, Gozani O. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature. 2006;442:96–99. doi: 10.1038/nature04835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang J, Chin MY, Li G. The novel tumor suppressor p33ING2 enhances nucleotide excision repair via inducement of histone H4 acetylation and chromatin relaxation. Cancer Res. 2006;66:1906–1911. doi: 10.1158/0008-5472.CAN-05-3444. [DOI] [PubMed] [Google Scholar]
- 43.Koi M, Umar A, Chauhan DP, Cherian SP, Carethers JM, Kunkel TA, Boland CR. Human chromosome 3 corrects mismatch repair deficiency and microsatellite instability and reduces N-methyl-N’-nitro-N-nitrosoguanidine tolerance in colon tumor cells with homozygous hMLH1 mutation. Cancer Res. 1994;54:4308–4312. [PubMed] [Google Scholar]
- 44.Wang Y, Wang J, Li G. Leucine zipper-like domain is required for tumor suppressor ING2-mediated nucleotide excision repair and apoptosis. FEBS Lett. 2006;580:3787–3793. doi: 10.1016/j.febslet.2006.05.065. [DOI] [PubMed] [Google Scholar]
- 45.Chin MY, Ng KC, Li G. The novel tumor suppressor p33ING2 enhances UVB-induced apoptosis in human melanoma cells. Exp Cell Res. 2005;304:531–543. doi: 10.1016/j.yexcr.2004.11.023. [DOI] [PubMed] [Google Scholar]
- 46.Wsierska-Gadek J, Gueorguieva M, Wojciechowski J. MNNG induced dramatic DNA damage and non-apoptotic changes in cervical HeLa cells. Ann N Y Acad Sci. 2003;1010:278–282. doi: 10.1196/annals.1299.048. [DOI] [PubMed] [Google Scholar]
- 47.Wesierska-Gadek J, Gueorguieva M, Schloffer D, Uhl M, Wojciechowski J. Non-apoptogenic killing of HeLa cells after short exposure to the alkylating agent N-Methyl-N-’-nitro-N-nitrosoguanidine (MNNG) J Cell Biochem. 2003;89:1222–34. doi: 10.1002/jcb.10586. [DOI] [PubMed] [Google Scholar]
- 48.Pedeux R, Sengupta S, Shen JC, Demidov ON, Saito S, Onogi H, Kumamoto K, Wincovitch S, Garfield SH, McMenamin M, Nagashima M, Grossman SR, Appella E, Harris CC. ING2 regulates the onset of replicative senescence by induction of p300-dependent p53 acetylation. Mol Cell Biol. 2005;25:6639–6648. doi: 10.1128/MCB.25.15.6639-6648.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Costanzo A, Merlo P, Pediconi N, Fulco M, Sartorelli V, Cole PA, Fontemaggi G, Fanciulli M, Schiltz L, Blandino G, Balsano C, Levrero M. DNA damage-dependent acetylation of p73 dictates the selective activation of apoptotic target genes. Molecular Cell. 2002;9:175–186. doi: 10.1016/s1097-2765(02)00431-8. [DOI] [PubMed] [Google Scholar]
- 50.Inoue T, Stuart J, Leno R, Maki CG. Nuclear import and export signals in control of the p53-related protein p73. J Biol Chem. 2002;277:15053–15060. doi: 10.1074/jbc.M200248200. [DOI] [PubMed] [Google Scholar]
- 51.Nagashima M, Shiseki M, Miura K. DNA damage-inducible gene p33ING2 negatively regulates cell proliferation through acetylation of p53. Proc Natl Acad Sci U S A. 2001;98:9671–9676. doi: 10.1073/pnas.161151798. K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hickman MJ, Samson LD. Apoptotic signaling in response to a singly type of DNA lesion, O(6)-methylguanine. Mol Cell. 2004:105–116. doi: 10.1016/s1097-2765(04)00162-5. [DOI] [PubMed] [Google Scholar]
- 53.Li GM. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008:85–98. doi: 10.1038/cr.2007.115. [DOI] [PubMed] [Google Scholar]
- 54.Tang Y, Luo J, Zhang W, Gu W. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell. 2006;24:827–839. doi: 10.1016/j.molcel.2006.11.021. [DOI] [PubMed] [Google Scholar]
- 55.Sykes SM, Mellert HS, Holbert MA, Li K, Marmorstein R, Lane WS, McMahon SB. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol Cell. 2006;24:841–851. doi: 10.1016/j.molcel.2006.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ivanov GS, Ivanova T, Kurash J, Ivanov A, Chuikov S, Gizatullin F, Herrera-Medina EM, Rauscher F, 3rd, Reinberg D, Barlev NA. Methylation-acetylation interplay activates p53 in response to DNA damage. Mol Cell Biol. 2007:6756–6769. doi: 10.1128/MCB.00460-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Melino G, Bernassola F, Ranalli M, Yee K, Zong WX, Corazzari M, Knight RA, Green DR, Thompson C, Vousden KH. p73 Induces apoptosis via PUMA transactivation and Bax mitochondrial translocation. J Biol Chem. 2004;279:8076–8083. doi: 10.1074/jbc.M307469200. KH. [DOI] [PubMed] [Google Scholar]
- 58.Gao B, Lee SM, Fang D. The tyrosine kinase c-Abl protects c-Jun from ubiquitination-mediated degradation in T cells. J Biol Chem. 2006;281:29711–29718. doi: 10.1074/jbc.M604596200. D. [DOI] [PubMed] [Google Scholar]
- 59.Gunduz M, Gunduz E, Rivera RS, Nagatsuka H. The inhibitor of growth (ING) gene family: potential role in cancer therapy. Curr Cancer Drug Targets. 2008;8:275–284. doi: 10.2174/156800908784533454. H. [DOI] [PubMed] [Google Scholar]
- 60.Nouman GS, Anderson JJ, Lunec J, Angus B. The role of the tumour suppressor p33 ING1b in human neoplasia. J Clin Pathol. 2003;56:491–496. doi: 10.1136/jcp.56.7.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gunduz M, Ouchida M, Fukushima K, Hanafusa H, Etani T, Nishioka S, Nishizaki K, Shimizu K. Genomic structure of the human ING1 gene and tumor-specific mutations detected in head and neck squamous cell carcinomas. Cancer Res. 2000;60:3143–3146. K. [PubMed] [Google Scholar]
- 62.Wagner MW, Li LS, Morales JC, Galindo CL, Garner HR, Bornmann WG, Boothman DA. Role of c-Abl kinase in DNA mismatch repair-dependent G2 cell cycle checkpoint arrest responses. J Biol Chem. 2008;283:21382–93. doi: 10.1074/jbc.M709953200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.