

Abstract
The cyclopic and laterality phenotypes in model organisms linked to disturbances in the generation or propagation of Nodal-like signals are potential examples of similar impairments resulting in birth defects in humans. However, the types of gene mutation(s) and their pathogenetic combinations in humans are poorly understood. Here we describe a mutational analysis of the human NODAL gene in a large panel of patients with phenotypes compatible with diminished NODAL ligand function. Significant reductions in the biological activity of NODAL alleles are detected among patients with congenital heart defects (CHD), laterality anomalies (e.g. left-right mis-specification phenotypes), and only rarely holoprosencephaly (HPE). While many of these NODAL variants are typical for family-specific mutations, we also report the presence of alleles with significantly reduced activity among common population variants. We propose that some of these common variants act as modifiers and contribute to the ultimate phenotypic outcome in these patients; furthermore, we draw parallels with strain-specific modifiers in model organisms to bolster this interpretation.
Keywords: NODAL, GDF1, TOF, TGA, DORV, cardiac defects, laterality, HPE
INTRODUCTION
The basic vertebrate body plan becomes defined with the establishment of the anterior-posterior (A-P), dorsal-ventral (D-V), and left-right (L-R) axes during gastrulation. Efforts to understand these processes at a molecular level have guided a significant fraction of recent developmental biological research, since disturbances in these embryological steps can influence the development of the forebrain [1], midline structures [2], as well as the elaboration of L-R sidedness necessary for the asymmetric development of the brain, heart, lungs, gut, and abdominal organs [3,4]. While there are clear differences among distinct vertebrate species, these are often variations on a basic theme [5] that may allow for meaningful extrapolation to human biology.
Nodal is a member of the TGF-β class of secreted signaling molecules that is implicated in the establishment of the A-P axis and generation of mesoderm and definitive endoderm [6]. It is also involved in a dose-dependent cell-fate specification of midline precursor cells in the anterior primitive streak; these cells will ultimately form the prechordal plate, notochord, and floorplate [7]. The subsequent anterior migration of these axial mesendoderm cells, in turn, influences the patterning of the developing central nervous system, including the forebrain. Holoprosencephaly (HPE) is considered a “default state” of forebrain development since, in the absence of midline patterning signals from the prechordal plate signaling center, the future forebrain fails to correctly divide into left and right hemispheres and paired subcortical structures [1,2,8,9]. Furthermore, the axial midline divides the embryo into left-right compartments that can elaborate different molecular properties to affect asymmetric organogenesis [2,3]. Therefore, Nodal-like factors are crucial for both the development of midline structures and left-right organ specification.
Once the organizer (or node, in the mouse [7]) develops at the distal tip of the primitive streak, asymmetric signals (including Nodal- and Gdf1-dependent signals) [10] are subsequently transmitted to the left lateral plate mesoderm, resulting in a cascade of asymmetrically expressed genes such as Nodal itself, Lefty2 and the downstream pathway effector Pitx2 [2,3,11–13]. While the asymmetric left-sided expression of Nodal is transient, the continued asymmetric expression of the Pitx2c isoform is important for the development of the heart and other similarly asymmetrically patterned organs [12–13]. Nodal signals induce the expression of Nodal itself through an autoregulatory loop; these signals simultaneously induce Nodal’s antagonists (the Lefty genes) and, therefore, establish an intricate mechanism to control the effective level of Nodal expression [5,6,7,14–17]. Such a tight regulation of Nodal expression is important, since the effective strength or duration of Nodal signals has a direct influence on cellular response [18,19].
Nodal/TGF-β signals are also essential for the specification of the secondary heart field. The secondary heart field provides a source of myocardial precursor cells in the ventral pharyngeal mesoderm [20] that, in turn, directly contributes to the growth of the cardiac outflow tract or conotruncus [21–25]. Therefore, the impaired signaling of the Nodal pathway that has been implicated in cyclopia, craniofacial, cardiovascular, and situs phenotypes in mice [8] and other model systems can be plausibly extrapolated to similar phenotypes in humans.
While the total absence of murine Nodal is embryonic lethal [26], heterozygosity for a null allele and a normal allele is entirely compatible with normal development in the mouse strains examined [6,7,27,28]. Interestingly, a hypomorphic allele of Nodal and a null allele also causes considerable lethality and severe anterior forebrain truncations, while two copies of the same hypomorphic allele allows 50% of the animals to survive to term, but with a spectrum of craniofacial and cardiac defects [18]. Lowe et al. described a different hypomorphic allele of Nodal that also manifested a spectrum of axial, forebrain, left-right visceral and conotruncal cardiac malformations, supporting the notion of graded Nodal signaling in the mouse [15,18,29]. Heterozygosity for Nodal loss-of-function mutations also can genetically interact with other genes such as Smad2 [30], Foxa2 [31], Activin receptor A [32,33], or Gdf1 [10] to generate a similar spectrum of cyclopia and laterality findings, suggesting that compound transheterozygotes might account for some forms of these defects in humans. These results imply that a spectrum of phenotypes can be generated in the mouse when total levels of Nodal activity are between nil and 50%, and that the effective level of cumulative signaling influences which developmental events can proceed normally.
A summary of the core components of the TGF-β signaling pathway utilized by human NODAL [MIM*601265], and the Nodal-like factor GDF1 [MIM*602880], is presented in Fig. 1. Both TGF-β class proteins (see 34–35 for reviews) are structurally similar and interact with a similar array of receptors and downstream effectors. Given that we had previously characterized genetic lesions affecting CFC1, TDGF1 and GDF1 and FOXH1 in our patients [36–40] we now turned our attention to evaluating the potential role(s) of human NODAL. Here we describe new NODAL mutations and determine their activity in a new bioassay. We find functionally abnormal mutations in NODAL’s signal sequence, pro domain and mature domain. We also report on two common polymorphisms in NODAL’s pro-domain that affect NODAL activity, and note several cases where the presence of a common weak allele appears to be predictive of disease severity.
Figure 1. The essential components of Nodal signaling.

TGF-2β proteins are synthesized as pre-pro-proteins requiring cleavage of the signal peptide (black) and prodomain (orange) prior to dimerization of active ligand (red diamonds). Cells responsive to Nodal-like ligands display Type I/II Activin receptors on their cell surface with co-receptors of the EGF-CFC family required for certain ligands, such as Nodal and Gdf1. The Lefty proteins (yellow) interfere with mature ligand binding to receptors. Ligand binding triggers trans-phosphorlyation of Type I receptors by the Type II receptor serine-threonine kinase domain. Receptor-regulated R-Smads (green) become activated on the inner surface of the cell membrane through phosphorylation by Type I receptors. Activated Smad complexes include two R-Smads and the common Smad4 (black) that translocate into the nucleus to affect gene expression. Target genes typically contain binding sites for Smads and co-factors such as FoxH1 (red triangle) to achieve tissue-specific gene expression. Human mutations have been previously described in LEFTYA [MIM+601877] and LEFTYB [ref. 57; MIM*603037], ACVR2B [ref. 58; MIM+602730], CFC1 [ref. 36, 38; MIM*605194], and TDGF1 [ref. 37; MIM+187395].
MATERIALS AND METHODS
Study population
We analyzed approximately 400 HPE patients for sequence variations in NODAL under our NHGRI approved brain research protocol. In addition, our congenital heart disease (CHD) pilot consortium study consists of 375 unrelated individuals prospectively ascertained at several urban centers with a wide spectrum of defects (see both Fig. 1 and Table 1 of [40]). These congenital cardiovascular malformations include transposition of the great arteries (TGA), tetralogy of Fallot (TOF), double outlet right ventricle (DORV), atrial septal defect (ASD), common atrioventricular canal (CAVC), and interrupted aortic arch (IAA). Coded affected proband DNA samples were obtained from three consortium centers (Children’s Hospital of Philadelphia, PA; Baylor College of Medicine, Houston, TX; and Yale University School of Medicine, New Haven, CT) following the guidelines of the Institutional Review Boards (IRBs) of each institution and with the supervisory approval of the IRB of the NHGRI/NIH for patients with cardiovascular anomalies (Laterality protocol). All CHD patient samples had been collected purely for research purposes and no consent had been obtained to study family members or return results of unknown significance to the subjects. None of the patients with cardiovascular anomalies were seen at the NIH. In addition to patients with HPE or CHD, we studied 125 unrelated individual normal controls obtained as anonymous samples from the Coriell Institute for Medical Research.
Table 1.
Summary of mutational data for NODAL and GDF1.
| Gene | Disease | Classe | Variation(s)f | Activity | Domain | NODAL165 | Reference |
|---|---|---|---|---|---|---|---|
| NODAL | |||||||
| TOF | dv | C5Fa | 100% | Signal peptide |
N.D. | This study | |
| TOF | dv | C5F | 100% | Signal peptide |
N.D. | This study | |
| TOF | uniq | P7S | 85% | Signal peptide |
This study | ||
| CHD | uniq | 85% | Signal peptide |
H/H | This study | ||
| syn | (Q34Q) | N.D. | prodomain | This study | |||
| TOF | dv | A21S | 100% | Signal peptide |
N.D. | This study | |
| DORV | uniq | A22S | 65% | Signal peptide |
N.D. | This study | |
| common | cp | A22V | N.D. | Signal peptide |
This study | ||
| DORV | uniq | S46I | 100% | prodomain | N.D. | This study | |
| TGA | uniq | R49L | 100% | prodomain | N.D. | This study | |
| TOF | uniq | A55E | N.D. | prodomain | This study | ||
| TOF | dv | A55V and | 60% | prodomain | This study | ||
| dv | A21S | 100% | Signal peptide |
This study | |||
| TOF | uniq | 25% | prodomain | H/H | This study | ||
| cp | IVS1+11G>T | N.D. | intron | ||||
| TOF | uniq | S60R | N.D. | prodomain | This study | ||
| TOF | dv | S60I | 15% | prodomain | This study | ||
| TOF | dv | S60I | 15% | prodomain | This study | ||
| TGA | dv | S60I | 15% | prodomain | This study | ||
| TOF | dv | A63S | 50% | prodomain | R/R | This study | |
| DORV | dv | 50% | prodomain | This study | |||
| cp | IVS1+6C>T | N.D. | intron | ||||
| Laterality | dv | A63E | N.D. | prodomain | N.D. | Baylord | |
| HPE/CHD | cp(rs1904589) | H165R | 45% | prodomain | various | ||
| Laterality | cp(rs10999334) | E203K | 60% | prodomain | This study; Baylord |
||
| Laterality | uniq | delSJ | 15% | Cleavage site |
N.D. | Baylord | |
| Laterality | uniq | G260R | 80% | Mature domain |
N.D. | This study; Baylord |
|
| Laterality | uniq | R275C | 20% | Mature domain |
N.D. | This study; Baylord |
|
| Laterality | uniq | V284F | 60% | Mature domain |
N.D. | This study; Baylord |
|
| DORV | uniq | N292K | 70% | Mature domain |
This study | ||
| TOF | dv | R302C | 35% | Mature domain |
This study | ||
| HPE | dv | R302C | 35% | Mature domain |
R/R | This study | |
| Laterality | uniq |
 b b
|
90% | Mature domain |
N.D. | [36] (B. Casey unpublished); Baylord. |
|
| GDF1 | |||||||
| AVC | uniq |
 c c
|
N.D. | prodomain | H/H | [39] | |
| TOF | uniq | G162D | N.D. | prodomain | N.D. | [39] | |
| TGA | uniq | C227X | N.D. | truncation | [39] | ||
| AVC | uniq | G262S | reduced | Mature domain |
R/R | [39] | |
| DORV | uniq | C267Y | L.O.F. | Mature domain |
R/R | [39] | |
| TOF | uniq | S309P | L.O.F. | Mature domain |
N.D. | [39] | |
| TOF | uniq | P312T | L.O.F. | Mature domain |
[39] | ||
| TGA | uniq | A318T | L.O.F. | Mature domain |
[39] |
NODAL C5F (100%) also FOXH1  (25,90%, see [58]) and (P354P; A353A)
(25,90%, see [58]) and (P354P; A353A)
NODAL  (95%) also CFC1
(95%) also CFC1  (L.O.F.) [36]
(L.O.F.) [36]
GDF1 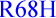 (N.D.) [39] also CFC1
(N.D.) [39] also CFC1 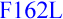 (N.D.) and
(N.D.) and 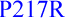 (N.D., see companion manuscript)
(N.D., see companion manuscript)
Patient samples studied at Baylor [41]; only bp changes were supplied to allow functional studies (see Materials and Methods, as well as Fig. 1 of [40]).
syn = synonymous; dv = disease variant; cp = common polymorphism; and uniq = unique variant.
Color code conforms to usage in the Figures: namely, red = <50% activity; orange = 50–99% activity; blue = not adequately tested; black = not different from the normal gene.
A separate set of samples was studied for NODAL and FOXH1 variations using independent methods [40,41]. In these cases (cited here in Table 1 with the footnote d), only information regarding the nature of the mutation and general phenotype was shared in order to allow functional analysis in zebrafish to complement their cell-based assays. Qualitatively similar conclusions were drawn, using independent methods, for the deleterious nature of mutations revealed by functional analysis used in [41] and this study.
For the association analysis (see below) study subjects were recruited prospectively from the Cardiac Center at The Children’s Hospital of Philadelphia (from 1992–2007) in accordance with a protocol approved by the Internal Review Board for the Protection of Human Subjects for studies on the genetic basis of congenital heart disease. Medical records including, when necessary, original imaging studies were reviewed to confirm the cardiac diagnosis and identify additional medical issues. Subjects with one of the five classic conotruncal defects (CTD) were included in the study cohort, including those with tetalogy of Fallot (TOF), D-transposition of the great arteries (D-TGA), double outlet right ventricle (DORV), truncus arteriosus (TA) and interrupted aortic arch (IAA). Subjects with perimembranous or posterior malalignment type ventricular septal defects, and those with isolated aortic arch anomalies were also included in this study because patients with these types of ventricular septal defects and aortic arch anomalies share a common genetic basis, namely a 22q11 deletion, with a subset of CTD [49–51]. Therefore, it is appropriate to include these additional cardiac defects in a study designed to identify genetic risk factors for conotruncal cardiac defects. All subjects were tested for a 22q11 deletion. Subjects with a recognized chromosomal alteration (including a 22q11 deletion) or genetic syndrome were excluded from the current study.
PCR amplification, mutation screening, and DNA sequencing
Oligo™ 4.1 was used to design primers for the relevant NODAL exons or regulatory elements (Suppl. Table 1). Amplification of human genomic DNA was performed in a 30 µl reaction volume [37], using 60–100 ng DNA template, 50 µM each of deoxynucleotide triphosphate, 0.25 µM of each primer, 3 µl of 10X PCR Amplification buffer (Invitrogen), 1.5 µl 10X Enhancer buffer (Invitrogen) and 0.3 µl Taq polymerase. All reactions were performed using a PTC-255 thermocycler (MJ Research, MA). Typical PCR cycling parameters were 95°C for 4 minutes followed by 30 cycles at 95°C, annealing at 62°C, extension at 72°C for 1 minute, and a final extension step of 72°C for 5 minutes. One half of the PCR product was used for denaturing high-pressure liquid chromatography (dHPLC) analysis (Trangenomics WAVE™) and the remainder was retained for direct DNA sequencing. Amplicons displaying heterozygous profiles were purified using a high pure PCR purification kit (Roche, IN) and bi-directionally sequenced using the BigDye™ version 3.1 terminator cycle sequencing kit according to the manufacturer’s protocol (Applied Biosystems, CA) on an ABI 3100 automated sequencer.
Genotyping for the A494G NODAL SNP (rs#1904589; R165H) was done in the High-Throughput Genotyping Core Laboratory at the Molecular Diagnosis and Genotyping Facility at the University of Pennsylvania, using the ABI 7900 HT Sequence Detection System (Applied Biosystems, Inc., Foster City, CA) by routine methods (see supplement for details). To validate assay performance, the primers and probes were first tested on a panel of DNA samples composed of CEPH family members (Family 1331, XC01331) obtained from Coriell Human Variation Collection (Coriell Institute, Camden, NJ). Callable genotypes and appropriate allele frequencies were obtained. The validity of the genotypes for the CEPH family was checked by validation of Mendelian inheritance. Thereafter, the study subjects were genotyped and data transferred electronically into an Excel spreadsheet for analysis.
Statistical methods and association analysis
Log-linear analyses were used to assess the association between conotruncal heart defects (CTD) and the case genotype for the NODAL H165R variant [52]. Specifically, the relative risks (and associated 95% confidence intervals) of CTD in cases with the HR and HH genotypes, as compared to cases with the RR genotype, were estimated, and the significance of the case genotype as a predictor of CTD was determined using the likelihood ratio test. These analyses were run using LEM [53], a program for log-linear analysis with missing data that allows data from triads that have not been completely genotyped to be included in the analysis.
Site-directed mutagenesis and construct design
The cDNA-derived coding region of the NODAL (H165 and E203 variant) gene was directionally cloned into pcDNA3.1 by standard methods. Site-directed mutagenesis was either performed with a minor modification of the Transformer kit (Promega, WI) by Transponics (York, PA) or by using the GeneTailer™ (InVitrogen) kit to introduce the human sequence variations.
Structural modeling
Molecular modeling of the TGF-β domain of NODAL was performed using the MODELLER package [54], as implemented within Discovery Studio (Accelrys, San Diego, CA). Modeling was based on the solved structure of BMP7, as determined by X-ray diffraction and refined to a resolution of 2 Å (pdb|1LXI) [55]. The Nodal sequence was aligned with the template sequence using Clustal W. The two sequences share 42% residue identity over the region modeled. The MODELLER program was run in a fully-automated mode with a high optimization level in order to construct an energy-minimized three-dimensional model of the aligned Nodal sequence by satisfaction of spatial restraints extracted from the template PDB (protein database) file.
NODAL luciferase assay
The activities of WT and variant NODAL alleles were determined from their relative ability to drive luciferase activity from a co-microinjected reporter vector (pGL3–3ARE) when microinjected as mRNA into zebrafish embryos, as compared to wild type [WT] NODAL mRNA. Binding of assembled phospho-Smad/FoxH1 complexes (which form in response to Nodal signals) to ARE response elements in the reporter vector causes coupled transcription/translation of a tandem firefly luciferase gene [17]. As an internal standard, embryos were also microinjected with a third element, the pRL-CMV vector (10:1 w/w ratio of pGL3-3ARE:pRL-CMV, 25 pg/embryo), which constitutively produces the independently-measurable renilla luciferase, providing an internal standard of translational efficiency. Positive control and experimental samples included 50 pg of WT or variant NODAL mRNAs and the two luciferase vectors. Negative control samples, which provided a measure of endogenous zebrafish Nodal activity levels, included the two luciferase vectors alone. After several hours of growth, embryos were lysed and lysates were measured for luciferase activity with the Dual Luciferase Reporter Assay System (Promega), as previously described [56]. Firefly luciferase (FL) values were normalized to renilla luciferase (RL) values and averaged. Experimental (EXP) values were further adjusted and normalized by subtracting the negative (NEG) control value and dividing by the adjusted WT NODAL (NOD) value. The following formula summarizes the conversion: wild type adjusted activity (WTAA) of NODALEXP = (Ave [FLEXP/RLEXP] - Ave [FLNEG/RLNEG])/(Ave [FLNOD/RLNOD] - Ave [FLNEG/RLNEG]).
RESULTS
Functional analysis of NODAL variations
We have identified numerous sequence variations of unknown significance in NODAL in patients with either holoprosencephaly or congenital heart defects (Table 1). In order to rapidly evaluate these variations, we microinjected mutated synthetic mRNAs into zebrafish so as to provide an indicator of bioactivity relative to the normal NODAL gene. As shown in Fig. 2, fifteen of the twenty individual sequence variations tested were significantly reduced in their measured ability to activate an Activin-responsive ARE-luciferase reporter construct. Since this assay measures the in vivo ability of the mRNA to be translated, the protein to be correctly processed, and the ultimate assembly of transcription factors onto the reporter gene, we reasoned that many potential categories of mutation could be captured by a single rapid test. We note that the assay is somewhat artificial, in that it requires the human gene to interact with signaling components of the zebrafish host embryo; nevertheless, the findings are readily interpretable and consistent since we measure activity relative to a wild-type control.
Figure 2.

Relative activity of human NODAL alleles. In vitro-synthesized mRNA encoding variant NODAL alleles were individually injected into zebrafish embryos together with a Nodal-reponsive luciferase vector and an internal luciferase standard, and embryos were harvested and measured for luciferase activity several hours later (see methods). Induced luciferase activity from each allele was normalized relative to wild type NODAL activity as described in the methods. The X axis in the graphical display indicates the allele tested (DSJ: deletion splice junction) and the Y axis (WT-adjusted activity [WTAA]) indicates the activity of the allele relative to WT NODAL. The horizontal solid gray line indicates 100% WT activity. Filled circles represent the average WTAA index from multiple experiments and vertical lines indicate the SEM. Values used for the graphical display and the number of independent experiments for each allele are given in the accompanying Suppl. Table 2. One patient was identified by sequencing to have two different changes p.A21S;A55V in cis and this construct was tested with both changes incorporated into the wild-type backbone. Similarly, a single patient had p.A19E; A63E changes in cis and a double mutant construct was studied. The remainder of the missense changes were analyzed individually.
A cluster of sequence variations was detected in the region of the signal peptide, as illustrated in Fig. 3A and Table 1. The first two variations, p.C5F and p.P7S, retain normal (or nearly normal) function (Fig. 2). However, residues 19 (data not shown) and 22 (Fig. 2) appear to be functionally important, based on our bioassay (residue 22) or computational sequence analysis (residue 19). The efficiency of signal peptide cleavage might be affected by these missense changes.
Figure 3.

Summary of pre-pro-domain variations in NODAL and GDF1. A), Domain architecture is shown as in Fig. 1, with arrows indicating the sites of cleavage of the signal peptide and prodomains from the canonical TGF-β ligand domain. Positions of the variant amino acids are numbered and color-coded as follows: red, severe (less than 50% activity); orange, mild (50–99% activity); black, normal activity; and blue, not tested. Note that NODAL missense changes p.H165R and p.E203K represent functionally abnormal common polymorphic variants. The leftmost panel shows a Clustal W-generated multiple sequence alignment of a putative novel domain within the NODAL prodomain associated with a cluster of functionally significant human variations between positions p.55–65. This alignment uses the Clustal W biological similarity coloring scheme (orange, identical; red, highly similar; pink, biochemically related; and black, biochemically distinct). The central panel reveals that the amino acid sequences of higher mammals can be aligned in the vicinity of the p.H165 position to allow evolutionary comparisons. Multiple sequence alignments of this region indicate that this region of the prodomain is only conserved among highly related vertebrate Nodals (data not shown). The rightmost panel contains a multiple sequence alignment of TGF-β prodomains (orange) and ligands (red) separated by the canonical RXXR consensus sequence. Numbers in italic precede the cleavage site, while those in normal font denote the amino-terminus of the mature ligand. Note that the construct delSJ (deletion of splice junction) tested in our assays is missing the consensus site. The patient with this variation has a complex in-del, with the indicated amino acids deleted and replaced in frame with heterologous aminoacids. B), The GDF1 p.C227X truncation mutation is depicted by the red triangle without fill to signify a presumed loss-of-function allele.
In addition to these potential defects in secretion, the release of active ligand requires tissue-specific convertases and the proper removal of the prodomain. Certain sequence variations in the prodomain also affect the bioactivity of NODAL (Fig. 3A). These variations are also within a novel motif we observe in the NODAL prodomain, between residues 55 and 63 (Fig. 3A). While all higher vertebrate Nodal proteins contain this motif, we find it intriguing that the residues implicated in our functional studies are completely conserved in Xenopus Nodal-related 4 and that biochemically similar motifs are also present in the zebrafish Nodals Cyc and Sqt. A working hypothesis would be that this motif could allow for interactions with processing proteins or folding necessary for such processing. The investigation of prodomain variants in GDF1 (Fig. 3B) was not possible because the assays utilized incorporated a synthetic BMP-derived prodomain in a chimeric construct.
A third line of evidence that protein processing could be important in disease pathogenesis comes from our functional analysis of a construct containing an insertion-deletion (in-del) (Fig. 3A). We analyzed the in-frame deletion version of human NODAL, which lacks the consensus cleavage site RXXR and adjacent residues. This mutation was identified in a patient with laterality findings, and cell-based transfection studies also suggest that the combined in-del variation significantly reduces NODAL bioactivity [41].
Some NODAL polymorphisms are functionally abnormal
Our functional analysis of mutations outside of the mature ligand domain also led us to discover two instances of reduced activity in common alleles. We first detected NODAL p.H165R as an extremely common sequence variant with a heterozygosity of almost 50% in healthy normal controls (our data and public databases, e.g., dbSNP). The only other significant variant among the HPE cases was a single case of p.R302C. Interestingly, both variations were substantially impaired in our bioassay (Fig. 2) as well as in their ability to induce the mesodermal markers no tail and goosecoid in zebrafish (A.F. Schier, unpublished, data not shown). The concordance among three independent measures (the induction of two different genes by in situ hybridization and by luciferase reporter activity) firmly indicates that these NODAL polymorphisms with a high prevalence in clinically normal individuals can substantially alter protein function. A second example of a functionally abnormal single nucleotide polymorphism (SNP) is NODAL p.E203K, detected in our study (Table 1), a recent independent study [41], and also annotated as a common variant in dbSNP with a lower heterozygosity (0.10 +/−0.06). There is complete consistency between the results of cell based transfection studies [41] and our estimates in zebrafish. It is therefore possible that these functionally significant polymorphisms confer a risk for disease, particularly in conjunction with additional genetic alterations in this or other genes (see Discussion).
We were curious to evaluate when the NODAL p.H165R variant had been observed in other vertebrates. As shown in Fig. 3A, the p.R165 variant is present in chimpanzees and humans, but not in other higher mammals. This comparison indicates that the p.H165 is the ancestral version and correctly used as the normal version of the gene in functional studies. The significance of the p.H165R variant is not completely understood, and there is little amino acid conservation in this portion of the prodomain to explain the differences in bioactivity that we detect.
Family based association testing of the p.H165R variant in a large, heterogeneous group of conotruncal defect-affected families detects a small independent effect
Genotyping for the functionally abnormal p.H165R variant was performed to assess the role of this variant among families with conotruncal defects. The genotyping call rate for the NODAL p.H165R variant was 95% and no discrepancies were observed among the samples that were subjected to repeat genotyping. Genotype combinations that were incompatible with Mendelian inheritance were observed in 23 (3.1%) triads. Genotype data from the members of these triads were excluded from the analyses. A valid NODAL p.H165R genotype was obtained in 1677 samples, representing 644 case-parent triads. As compared to the NODAL RR genotype, the HR and HH genotypes were associated with decreased risks of CTD (relative risk (RR) and 95% confidence interval (CI): RRRH versus RR = 0.91, 95% CI 0.72–1.15; RRHH versus RR = 0.78, 95% CI 0.55–1.10). However, neither the relative risks nor the likelihood ratio test (p=0.37) assessing the significance of the genetic effect were statistically significant. Hence, if this particular variant is truly associated with disease risk it is either a very weak risk factor, or associated with risk in only a subset of conotruncal defects.
NODAL variations in the TGF-β ligand domain
Most of the sequence variations in the ligand domain are apparently unique and found associated with a single proband. We examined the conservation and placement of the affected residues by performing multiple protein alignments (Fig. 4A) and by deriving a theoretical model for NODAL based on the solved crystal structure of bone morphogenic protein (BMP7; pdb|1LXI) (Fig. 4B). While residue conservation or localization within critical structural motifs are both valuable predictors for mutations affecting function, in the case of these NODAL mutations, they were not sufficient. For example, p.N292 is completely conserved among 14 members of the TGF-β family and is predicted to be part of an alpha helix in the core barrel domain of NODAL, yet substitution of a p.K292 residue results in only mildly altered activity in our assay (70% - Fig. 2). Similarly, p.G260 is highly conserved and is predicted to contribute to an alpha helical turn, but substitution with p.R260 has a mild effect (80% - Fig. 2).
Figure 4.


Homology within the TGF-β domain. A), Unlike the signal peptide and prodomains, there is extensive homology (blue for identity, yellow for similarity) within the ligand domain (red font). Structural features include two anti-parallel β-sheets (arrows) forming wing-like projections (termed finger 1 and finger 2), separated by the α-helical core buried between them. Two of the missense changes occur in finger 1, one in finger 2, and the remaining three occur within or near the core (barrel). Symbols to describe variations are as in legend for Fig. 3. B), The predicted three-dimensional structure of human NODAL. The alpha-helices are indicated by purple barrels, while the beta-sheets are indicated by the yellow, ribbon-like arrows. The ligand domain is modeled after the solved structure of BMP7 [55] and conforms to the typical features of the cystine-knot superfamily [59,60]. The positions of the missense changes are indicated within the model, with the wild type amino acid side chains shown in red.
Note that a patient with TOF has the same non-synonymous p.R302C alteration as the unrelated proband with HPE. The HPE patient is also homozygous for the NODAL R/R p.165 variant, implying that at least one allele has two variations in cis that may interact at the structural level, thereby contributing to the more severe condition (HPE) (see Table 1). This particular p.R302C variation was observed in different individuals with two very different extremes of the clinical spectrum. There are several possible explanations for the presence of p.R302C in these two individuals, such as unrecognized blood relationships or independent de novo changes, but we propose that this and other disease variant (dv) changes represent rare variations in the general population that are disproportionately found in affected individuals.
Loss-of-function mutations in GDF1
As described in Fig. 3B and Table 1, eight different unique GDF1 mutations were detected among the cardiac samples but not in HPE or healthy normal controls [39]. We note that two of these are putative prodomain variations. The processing of Vg1-like factors such as GDF1 is extremely difficult to detect in vivo and further studies will be needed to clarify any role for these two unanalyzed variations. Interestingly, five of these unique GDF1 variations detected in cardiac patients with typical conotruncal malformations were shown to have reduced bioactivity in zebrafish assays (Table 1) [39]. These same individuals were either heterozygous p.H165R or homozygous for NODAL p.R165. These findings raise the possibility of genetic interactions in these patients. Similarly, clear examples of synergy between Gdf1 and Nodal have been described in model organisms [10].
DISCUSSION
Congenital heart defects occur in nearly 1% of infants and approximately one third of these involve the conotruncus [42,43]. By far, the most common malformation associated with defective NODAL signaling detected in this study involves the conotruncal malformations TOF and, to a lesser extent, TGA and DORV [40]. These are particularly fascinating malformations that depend on the migration of cardiac neural crest cells and cardiac precursors in the ventral pharanx as the outflow tract grows and progressively rotates to orient the systemic and pulmonic circulations. These developmental events display significant sensitivity to Nodal signaling in animal models suggesting that similar mechanisms occur in all higher vertebrates including humans. Several defective Nodal alleles were also associated with laterality defects in humans, a link that is also supported by animal models. Several of the laterality-associated alleles we characterized (E203K, G260R, R275C, and V284F) were independently found to be loss-of-function in a recent study (41).
Studies in model organisms have suggested that the Nodal signaling pathway is crucial to the development and signaling potential of the axial midline and, consequently, is a major etiological cause of cyclopic phenotypes. Our systematic analysis of the human NODAL gene at the mutational level would suggest that defective signaling of this pathway contributes to only a minority of HPE cases (see [40]).
The acknowledged difficulty in comparing the phenotypes observed in model systems to humans (e.g. heterozygous mutations detected in patients vs. null genotypes in model organisms) could have several causes. First, the set of modifer genes important for a mouse line is often strain-specific [8]. For example, the phenotypic penetrance associated with targeted disruption of the murine HPE gene Cdo is highly strain-specific, ranging from a single central incisor to full-blown HPE [44,45]. By analogy, it is virtually certain that similar effects of modifier genes are important in out-bred human populations [9]. It is also important to recognize that 97% of HPE cases occurring in utero result in embryonic arrest and pregnancy loss [1,46,47]. Therefore, since none of these cases would be represented in our live-born patient samples, there are undoubtedly a significant proportion of cases of this type that cannot be analyzed genetically. Dysfunction of NODAL signaling could be a major undetected contributor to pregnancy loss reflecting the cumulative effects of genetic lesions. This interpretation is consistent with murine models of cyclopia where significant embryonic lethality is observed, especially in situations where the level of Nodal signaling is substantially reduced.
Collectively, our data is consistent with extremely rare family-specific mutations (e.g. in NODAL or GDF1) interacting with a limited set of common modifiers in key genes as a working model for complex traits such as CHD, laterality or HPE. Typically, these mutations can be observed in a large set of target genes within the NODAL pathway [40, 41]. Therefore, while individual gene mutations are exceedingly rare, collectively those within a pathway of interacting genes add up to a considerable genetic burden. It is this cumulative genetic burden that goes a long way towards explaining their common incidence. While simple heterozygosity for one of these loss-of-function alleles might not be considered sufficient, it can be considered necessary for the observed phenotype(s). Similarly, at the other extreme, the effects of two (or more) strong loss-of-function alleles might not be compatible with postnatal survival.
Although the p.165R variant does not act as an independent risk factor for conotruncal heart defects, its effects might still be seen in a context-dependent manner with another interacting gene. Recall that this variant is present in both chimpanzees and humans; therefore, while functionally deleterious when tested in vivo, it has not been obviously subjected to purifying selection. The presence of common modifiers with context-dependent effects in humans (such as NODAL p.H165R, or others) would help to resolve the conundrum that if defects in more than one gene are necessary to produce effects, yet individual mutations are so uncommon, how can these be some of the most common malformations in humans? The presence of functionally deficient common modifiers in human populations [48] would provide the opportunity for an individually rare deleterious sporadic mutation to manifest itself. In other words, a clinically normal mutation carrier parent would frequently have a healthy spouse with a common context-dependent modifier gene, such that offspring would be at enhanced risk for malformations. In this view, neither parent is directly affected by being a carrier of a deleterious gene; it is only the combination of alleles in the offspring that results in a recognizable clinical outcome.
Finally, these considerations argue for a procedural strategy to dissect gene associations with human disease. A systematic inventory of common variations in the general population should be undertaken for all critical genes within a given pathway. These common variations should be assessed for their potential to alter the functional flow of a signaling pathway such as that triggered by Nodal-like factors. Once this core baseline is established, it should be possible to predict the additive effect of deleterious family-specific mutations.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank the patients who participated in this research. We are also grateful to Richard Bamford and June de la Cruz for initiating this study, and A.F. Schier for permission to use unpublished data.
FUNDING
This research was supported by the Division of Intramural Research of the National Human Genome Research Institute, National Institutes of Health, and by grants from the NIH to J.A.T. and J.W.B. and by P50 HL74731 to E.G.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTARY MATERIAL
Supplementary Material is available.
CONFLICT OF INTEREST STATEMENT
None declared.
WEB RESOURCES
Accession numbers and URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
GeneBank, http://www.ncbi.nlm.nih.gov/GeneBank/ (for human NODAL [accession number NM_018055], GDF1 [accession number NM_001492],
University of California Santa Cruz Bioinformatics Site, http://genome.ucsc.edu/, (gene structure and annotation)
BLASTN, http://www.ncbi.nlm.nih.gov/blast/, (nucleotide homology searches)
SignalP3.0, http://www.cbs.dtu.dk/services/SignalP/, (for signal peptide analysis)
ClustalW, http://align.genome.jp/, (for protein alignment analysis)
REFERENCES
- 1.Muenke M, Beachy PA. Holoprosencephaly. In: Scriver CR, Beaudet AL, Valle D, Sly WS, editors. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 2001. pp. 6203–6230. [Google Scholar]
- 3.Roessler E, Muenke M. Midline and laterality defects: left and right meet in the middle. Bioessays. 2001;23:888–900. doi: 10.1002/bies.1130. [DOI] [PubMed] [Google Scholar]
- 4.Hamada H, Meno C, Watanabe D, Saijoh Y. Establishment of vertebrate left-right asymmetry. Nat. Rev. Genet. 2003;3:103–113. doi: 10.1038/nrg732. [DOI] [PubMed] [Google Scholar]
- 5.Zhu L, Belmont JW, Ware SM. Genetics of human heterotaxias. Eur. J. Hum. Genet. 2006;14:17–25. doi: 10.1038/sj.ejhg.5201506. [DOI] [PubMed] [Google Scholar]
- 6.Whitman M. Nodal signaling in early vertebrate embryos: themes and variations. Dev. Cell. 2001;1:605–617. doi: 10.1016/s1534-5807(01)00076-4. [DOI] [PubMed] [Google Scholar]
- 7.Schier AF. Nodal signaling in vertebrate development. Ann. Rev. Cell. Dev. Biol. 2003;19:589–621. doi: 10.1146/annurev.cellbio.19.041603.094522. [DOI] [PubMed] [Google Scholar]
- 8.Beddington RSP, Robertson EJ. Axis development and early asymmetry in mammals. Cell. 1999;96:195–209. doi: 10.1016/s0092-8674(00)80560-7. [DOI] [PubMed] [Google Scholar]
- 9.Krauss RS. Holoprosencephaly: new models, new insights. Expert Rev Mol Med. 2007;9:1–17. doi: 10.1017/S1462399407000440. [DOI] [PubMed] [Google Scholar]
- 10.Ming JE, Muenke M. Multiple hits during early embryonic development: digenic disease and holoprosencephaly. Am. J. Hum. Genet. 2002;71:1017–1032. doi: 10.1086/344412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Andersson O, Reissmann E, Jornvall H, Ibanez CF. Synergistic interaction between Gdf1 and Nodal during anterior axis development. Dev. Biol. 2006;293:370–381. doi: 10.1016/j.ydbio.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 12.Cheng SK, Olale F, Brivanlou AH, Schier AF. Lefty blocks a subset of TGFβ signals by antagonizing EGF-CFC coreceptors. PLoS Biology. 2004;2:215–226. doi: 10.1371/journal.pbio.0020030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Franco D, Campione M. The role of pitx2 during cardiac development. Trends Cardiovasc. Med. 2003;13:157–163. doi: 10.1016/s1050-1738(03)00039-2. [DOI] [PubMed] [Google Scholar]
- 14.Ai D, Liu W, Ma L, Dong F, Lu F-L, Wang D, Verzi MP, Cai C, Gage PJ, Evans S S, et al. Pitx2 regulates cardiac left-right asymmetry by patterning second cardiac lineage-derived myocardium. Dev. Biol. 2006;296:437–449. doi: 10.1016/j.ydbio.2006.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brennan J, Lu CC, Norris DP, Rodriguez TA, Beddington RSP, Robertson EJ. Nodal signaling in the epiblast patterns the early mouse embryo. Nature. 2001;411:965–969. doi: 10.1038/35082103. [DOI] [PubMed] [Google Scholar]
- 16.Gritsman K, Talbot WS, Schier AF. Nodal signaling patterns the organizer. Development. 2000;127:921–932. doi: 10.1242/dev.127.5.921. [DOI] [PubMed] [Google Scholar]
- 17.Norris DP, Brennan J, Bikoff EK, Robertson EJ. The Foxh1-dependent autoregulatory enhancer controls the level of Nodal signals in the mouse embryo. Development. 2002;129:3455–3468. doi: 10.1242/dev.129.14.3455. [DOI] [PubMed] [Google Scholar]
- 18.Osada S-I, Saijoh Y, Frisch A, Yeo C-Y, Adachi H, Watanabe M, Whitman M, Hamada H, Wright CVE. Activin/Nodal responsiveness and asymmetric expression of a Xenopus nodal-related gene converge on a FAST-regulated module in intron 1. Development. 2000;127:2503–2514. doi: 10.1242/dev.127.11.2503. [DOI] [PubMed] [Google Scholar]
- 19.Vincent SD, Ray Dun N, Hayashi S, Norris DP, Robertson EJ. Cell fate decisions within the mouse organizer are governed by graded Nodal signals. Genes Dev. 2003;17:1646–1652. doi: 10.1101/gad.1100503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hagos EG, Dougan ST. Time-dependent patterning of the mesoderm and endoderm by Nodal signals in zebrafish. BMC Dev. Biol. 2007;7:22. doi: 10.1186/1471-213X-7-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Buckingham M, Meilhac S, Zaffran S. Building the mammalian heart from two sources of myocardial cells. Nat. Rev. Genet. 2005;6:826–835. doi: 10.1038/nrg1710. [DOI] [PubMed] [Google Scholar]
- 22.Kelly RG, Brown NA, Buckingham ME. The arterial pole of the mouse heart forms from Fgf10-expressing cells in pharyngeal mesoderm. Dev. Cell. 2001;1:435–440. doi: 10.1016/s1534-5807(01)00040-5. [DOI] [PubMed] [Google Scholar]
- 23.Waldo KL, Kuminski DH, Wallis KT, Stadt HA, Hutson MR, Platt DH, Kirby ML. Conotruncal myocardium arises from a secondary heart field. Development. 2001;128:3179–3188. doi: 10.1242/dev.128.16.3179. [DOI] [PubMed] [Google Scholar]
- 24.Ward C, Stadt H, Hutson M, Kirby ML. Ablation of the secondary heart field leads to tetralogy of Fallot and pulmonary atresia. Dev. Biol. 2005;284:72–83. doi: 10.1016/j.ydbio.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 25.Bajolle F, Zaffran S, Kelly RG, Hadehouel J, Bonnet D, Brown NA, Buckingham ME. Rotation of the myocardial wall of the outflow tract implicated in the normal positioning of the great arteries. Circul. Res. 2006;98:421–428. doi: 10.1161/01.RES.0000202800.85341.6e. [DOI] [PubMed] [Google Scholar]
- 26.Kelly RG. Molecular inroads into the anterior heart field. Trends Card. Med. 2005;15:51–56. doi: 10.1016/j.tcm.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 12.Zhou X, Sasaki H, Lowe L, Hogan BL, Kuehn MR. Nodal is a novel TGF-β–like gene expressed in the mouse node during gastrulation. Nature. 1993;361:543–547. doi: 10.1038/361543a0. [DOI] [PubMed] [Google Scholar]
- 27.Collignon J, Varlet I, Robertson EJ. Relationship between asymmetric nodal expression and the direction of embryonic turning. Nature. 1996;381:155–158. doi: 10.1038/381155a0. [DOI] [PubMed] [Google Scholar]
- 28.Varlet, Collingnon J, Robertson EJ. Nodal expression in the primitive endoderm is required for specification of the anterior axis during mouse gastrulation. Development. 1997;124:1033–1044. doi: 10.1242/dev.124.5.1033. [DOI] [PubMed] [Google Scholar]
- 29.Lowe LA, Yamada S, Kuehn MA. Genetic dissection of nodal function in patterning the mouse embryo. Development. 2001;128:1831–1843. doi: 10.1242/dev.128.10.1831. [DOI] [PubMed] [Google Scholar]
- 30.Nomura M, Li E. Smad2 role in mesoderm formation, left-right patterning and craniofacial development. Nature. 1998;393:786–789. doi: 10.1038/31693. [DOI] [PubMed] [Google Scholar]
- 31.Varlet, Colingnon J, Norris DP, Robertson EJ. Nodal signaling and axis formation in the mouse. Cold Spring Harb Symp Quant Biol. 1997;62:105–113. [PubMed] [Google Scholar]
- 32.Oh SP, Li E. The signaling pathway mediated by the type IIB activin receptor controls axial patterning and lateral asymmetry in the mouse. Genes Dev. 1997;11:1812–1826. doi: 10.1101/gad.11.14.1812. [DOI] [PubMed] [Google Scholar]
- 33.Song J, Oh SP, Schrewe H, Nomura M, Lei H, Okano M, Gridley T, Li E. The type II activin receptors are essential for egg cylinder growth, gastrulation, and rostral head development in mice. Dev. Biol. 1999;213:157–169. doi: 10.1006/dbio.1999.9370. [DOI] [PubMed] [Google Scholar]
- 34.Massagué J. TFGβ signal transduction. Ann. Rev. Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- 35.Shi Y, Massagué J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 36.Bamford RN, Roessler E, Burdine RD, Saplakoglu U, de la Cruz J, Splitt M, Towbin J, Bowers P, Marino B, Schier AF, et al. Loss-of-function mutations in the EGF-CFC gene CFC1 are associated with human left-right laterality defects. Nat. Genet. 2000;26:365–369. doi: 10.1038/81695. [DOI] [PubMed] [Google Scholar]
- 37.de la Cruz J, Bamford RN, Burdine RD, Roessler E, Donnai D, Schier AF, Muenke M. A loss of function mutation in the CFC domain of TDGF-1 is associated with human forebrain defects. Hum. Genet. 2002;110:422–428. doi: 10.1007/s00439-002-0709-3. [DOI] [PubMed] [Google Scholar]
- 38.Goldmuntz E, Bamford RN, Karkera JD, dela Cruz J, Roessler E, Muenke M. CFC1 mutations in patients with Transposition of the Great Arteries and Double Outlet Right Ventricle. Am. J. Hum. Genet. 2002;70:776–780. doi: 10.1086/339079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Karkera JD, Lee JS, Roessler E, Banerjee-Basu S, Ouspenskaia MV, Mez J, Goldmuntz E, Bowers P, Towbin J, Belmont JW, Baxevanis AD, Schier AF, Muenke M. Loss-of-function mutations in Growth Differentiation Factor - 1 (GDF1) are associated with congenital heart defects in humans. Am. J. Hum. Genet. 2007;81:987–994. doi: 10.1086/522890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roessler E, Ouspenskaia MV, Karkera JD, Vélez JI, Kantipong A, Lacbawan F, Bowers P, Belmont JW, Towbin JA, Goldmuntz E, Feldman B, Muenke M. Reduced NODAL signaling strength via mutation of several pathway members including FOXHI is linked to human heart defects and holoprosencephaly. Am. J. Hum. Genet. 2008;83:18–29. doi: 10.1016/j.ajhg.2008.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mohapatra B, Casey B, Li H, Ho-Dawson T, Smith L, Fernbach SD, Molinari L, Niesh SR, Jefferies JL, Craigen WJ, Towbin JA, Belmont JW, Ware SM. Identification and functional characterization of NODAL rare variants in heterotaxy and isolated cardiovascular malformations. Hum. Mol. Genet. 2009;18:861–871. doi: 10.1093/hmg/ddn411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Samaneck M. Congenital heart malformations: prevalence, severity, survival and quality of life. Cardiol. Young. 2000;10:179–185. doi: 10.1017/s1047951100009082. [DOI] [PubMed] [Google Scholar]
- 43.Hoffman H, Kaplan S. The incidence of congenital heart disease. J. Am. Coll. Cardiol. 2002;39:1890–1900. doi: 10.1016/s0735-1097(02)01886-7. [DOI] [PubMed] [Google Scholar]
- 44.Cole F, Krauss RS. Microform holoprosencephaly in mice that lack the Ig superfamily member. Cdon. Cur. Biol. 2003;13:411–415. doi: 10.1016/s0960-9822(03)00088-5. [DOI] [PubMed] [Google Scholar]
- 45.Zhang W, Kang JS, Cole F, Yi MJ, Krauss RS. Cdo functions at multiple points in the Sonic Hedgehog pathway, and Cdo-deficient mice accurately model human holoprosencephaly. Dev. Cell. 2006;10:657–665. doi: 10.1016/j.devcel.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 46.Matsunaga E, Shiota K. Holoprosencephaly in human embryos: epidemiological studies on 150 cases. Teratology. 1997;16:261–272. doi: 10.1002/tera.1420160304. [DOI] [PubMed] [Google Scholar]
- 47.Yamada S, Uwabe C, Fujii S, Shiota K K. Phenotypic variability in human embryonic holoprosencephaly in the Kyoto Collection. Birth Defects Res Part A Clin Mol Teratol. 2004;70:495–508. doi: 10.1002/bdra.20048. [DOI] [PubMed] [Google Scholar]
- 48.Krykov GV, Pennacchio LA, Sunyaev SR. Most rare missense alleles are deleterious in humans: implications for complex disease and association studies. Am. J. Hum. Genet. 2007;80:727–739. doi: 10.1086/513473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goldmuntz E, Clark BJ, Mitchell LE, Jawad AF, Cuneo BF, Reed L, McDonald-McGinn D, Chien P, Feuer J, Zackai EH, Emanuel BS, Driscoll DA. Frequency of 22q11 deletions in patients with conotruncal defects. J. Am. Coll. Cardiol. 1998;32:492–498. doi: 10.1016/s0735-1097(98)00259-9. [DOI] [PubMed] [Google Scholar]
- 50.McElhinney DB, Clark BJ, Weinberg PM, Kenton ML, McDonald-McGinn D, Driscoll DA, Zackai EH, Goldmuntz E. Association of chromosome 22q11 deletion with isolated anomalies of aortic arch laterality and branching. J. Am. Coll. Cardiol. 2001;37:2114–2119. doi: 10.1016/s0735-1097(01)01286-4. [DOI] [PubMed] [Google Scholar]
- 51.McElhinney DB, Driscol DA, Levin ER, Jawad AF, Emanuel BS, Goldmuntz E. Chromosome 22q11 deletion in patients with ventricular septal defect: frequency and associated cardiovascular anomalies. Pediatrics. 2003;112:e472. doi: 10.1542/peds.112.6.e472. [DOI] [PubMed] [Google Scholar]
- 52.Weinberg CR, Wilcox AJ, Lie RT. A log-linear approach to case-parent-triad data: assessing effects of disease genes that act either directly or through maternal effects and that may be subject to parental imprinting. Am. J. Hum. Genet. 1998;62:969–978. doi: 10.1086/301802. 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vermunt JK. LEM: a general program for the analysis of categorical data. Tilberg University. 1997 [Google Scholar]
- 54.Sali L, Potterton L, Yuan F, van Vlijmen H, Karplus M. Evaluation of comparative protein modeling by MODELLER. Proteins. 1995;23:318–326. doi: 10.1002/prot.340230306. [DOI] [PubMed] [Google Scholar]
- 55.Greenwald J, Groppe J, Gray P, Wlater E, Kwiatkowski W, Vale W, Choe S. The BMP7/ActRII extracellular domain complex provides new insights into the cooperative nature of receptor assembly. Mol. Cell. 2003;11:605–617. doi: 10.1016/s1097-2765(03)00094-7. [DOI] [PubMed] [Google Scholar]
- 56.Pei W, Noushmehr H, Ouspenskaia MV, Elkahloun AG, Feldman B. An early requirement for maternal FoxH1 during zebrafish gastrulation. Dev. Biol. 2007;310:10–22. doi: 10.1016/j.ydbio.2007.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kosaki K, Bassi MT, Kosaki R, Lewin M, Belmont J, Schauer G, Casey B. Characterization and mutation analysis of human LEFTYA and LEFTYB, homologues of murine genes implicated in left-right axis development. Am. J. Hum. Genet. 1999;64:712–721. doi: 10.1086/302289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kosaki B, Gebbia M, Kosaki K, Lewin M, Bowers P, Towbin JA, Casey B. Left-right axis malformations associated with mutations in ACVR2B. Am J Med Genet. 1999;82:70–76. doi: 10.1002/(sici)1096-8628(19990101)82:1<70::aid-ajmg14>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 59.Isaacs NW. Cystine knots. Curr. Opin. Struct. Biol. 1995;5:391–395. doi: 10.1016/0959-440x(95)80102-2. [DOI] [PubMed] [Google Scholar]
- 60.MacDonald NO, Hendrickson WA. A structural superfamily of growth factors containing a cystine knot motif. Cell. 1993;73:421–424. doi: 10.1016/0092-8674(93)90127-c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.