

Abstract
Promiscuous 6-aminohexanoate-linear dimer (Ald)-hydrolytic activity originally obtained in a carboxylesterase with a β-lactamase fold was enhanced about 80-fold by directed evolution using error-prone PCR and DNA shuffling. Kinetic studies of the mutant enzyme (Hyb-S4M94) demonstrated that the enzyme had acquired an increased affinity (Km = 15 mM) and turnover (kcat = 3.1 s−1) for Ald, and that a catalytic center suitable for nylon-6 byproduct hydrolysis had been generated. Construction of various mutant enzymes revealed that the enhanced activity in the newly evolved enzyme is due to the substitutions R187S/F264C/D370Y. Crystal structures of Hyb-S4M94 with bound substrate suggested that catalytic function for Ald was improved by hydrogen-bonding/hydrophobic interactions between the Ald—COOH and Tyr370, a hydrogen-bonding network from Ser187 to 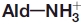 , and interaction between and Gln27-Oɛ derived from another subunit in the homo-dimeric structure. In wild-type Ald-hydrolase (NylB), Ald-hydrolytic activity is thought to be optimized by the substitutions G181D/H266N, which improve an electrostatic interaction with (Kawashima et al., FEBS J 2009; 276:2547–2556). We propose here that there exist at least two alternative modes for optimizing the Ald-hydrolytic activity of a carboxylesterase with a β-lactamase fold.
, and interaction between and Gln27-Oɛ derived from another subunit in the homo-dimeric structure. In wild-type Ald-hydrolase (NylB), Ald-hydrolytic activity is thought to be optimized by the substitutions G181D/H266N, which improve an electrostatic interaction with (Kawashima et al., FEBS J 2009; 276:2547–2556). We propose here that there exist at least two alternative modes for optimizing the Ald-hydrolytic activity of a carboxylesterase with a β-lactamase fold.
Keywords: 6-aminohexanoate-dimer hydrolase, nylon oligomer, β-lactamase, esterase, DD-peptidase, directed evolution, X-ray crystallography
Introduction
Biodegradation of man-made compounds provides a good system for studying how microorganisms have evolved specific enzymes that degrade synthetic compounds. We have studied the degradation of a byproduct from the manufacture of nylon-6,6-aminohexanoate (Ahx)-oligomer (or nylon oligomer), by Arthrobacter sp.1,2 We found that the Ahx-linear dimer (Ald) is hydrolyzed by Ald hydrolase (NylB) according to the following reaction.
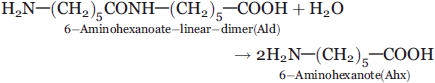 |
NylB specifically hydrolyzes amide compounds containing Ahx as the N-terminal residue, but it has no detectable activity towards 60 kinds of natural amide compounds tested.3 Therefore, we sought to identify the structural factors responsible for this unique substrate specificity. Southern hybridization and nucleotide sequencing revealed that NylB and a carboxylesterase (NylB′) that has 88% amino acid sequence identity to NylB are encoded on a plasmid in a strain of Arthrobacter.4,5 NylB′ possesses promiscuous catalytic activity towards Ald (ca. 0.5% of the activity of NylB).6 A NylB/NylB′ hybrid protein (designated as Hyb-24) containing five amino acid substitutions (T3A, P4R, T5S, S8Q, and D15G) in the NylB′ protein possesses similar activity levels to NylB′ and yields crystals suitable for X-ray crystallographic analysis (Fig. 1).7 Hyb-24 generated a two-domain structure (α and α/β) that is similar to the folds of the penicillin-recognizing family of serine-reactive hydrolases. This two-domain structure is particularly similar to that of d-alanyl-d-alanine carboxypeptidase (DD-peptidase) from Streptomyces and carboxylesterase (EstB) from Burkholderia.8,9
Figure 1.
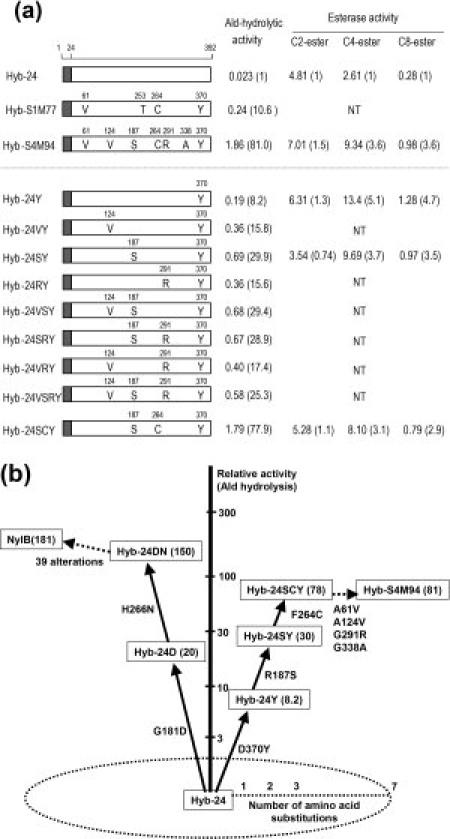
Relationship between the amino acid alterations in Hyb-24 and enzyme activity. (a) Hyb-24 is a NylB/NylB′ hybrid protein constructed from conserved PvuII sites located 24 amino acid residues downstream of the initiation codons and contains five amino acid replacements (T3A, P4R, T5S, S8Q, and D15G) in the NylB′ protein. His-tagged proteins were purified to homogeneity, and the specific activity (μmol/min (U)/mg protein) of each enzyme was estimated using 10 mM Ald under standard assay conditions. Esterase activities were assayed using 0.2 mM p-nitrophenylacetate (C2-ester), 0.2 mM p-nitrophenylbutyrate (C4-ester), and p-nitrophenyloctanoate (C8-ester) as substrates. The number in parentheses indicates the activity relative to the specific activity of parental Hyb-24. NT means “not tested.” Open boxes and shaded boxes represent the region of NylB′ and NylB, respectively. Amino acid residues substituted in the mutant enzymes are shown as one-letter codes in the open boxes. (b) The cumulative effect of typical amino acid alterations on Ald-hydrolytic activity is illustrated. Activity is expressed relative to the specific activity of parental Hyb-24 (0.023 U/mg).
Esterase/lipases are classified into eight families (I–VIII) based on amino acid sequence and biological properties.10,11 Most esterases belong to the α/β hydrolase superfamily and possess the short consensus sequence Gly-x-Ser-x-Gly (the esterase motif). The central Ser is a catalytic residue constituting the Ser-His-Asp triad. Although NylB′ is classified as a carboxylesterase due to its high activity for carboxylesters with short acyl chains, multiple three-dimensional alignment has revealed that the putative esterase motif sequence is altered to Trp-Arg-Thr-Arg-Arg in Hyb-24, demonstrating that the motif sequence is not conserved in Hyb-24.8 The specificity of Hyb-24 for carboxylesters seems to be similar to that of EstB carboxylesterase.12 However, EstB shows no β-lactamase or peptidase activity.13 In addition, enzymes having promiscuous amidase activity are rather rare among carboxylesterases,14 and esterase activity is also rare in ordinary Class A β-lactamases and Class C β-lactamases.15,16 Thus, despite the functional divergence between amidases and esterases, a single substitution (G181D) in a carboxylesterase (Hyb-24D) results in a 20-fold increase in Ald-hydrolytic activity, and an additional substitution (H266N, Hyb-24DN) results in the same level of Ald-hydrolytic activity as the parental NylB enzyme [Fig. 1(b)].9,17
In Hyb-24 and Hyb-24DN, Ser112, Lys115, and Tyr215 are catalytic residues; that is (i) Tyr215-Oηfunctions as a general base to increase the nucleophilicity of Ser112, (ii) Ser112-Oγ performs a nucleophilic attack on amide compounds, and (iii) the positively charged Lys115-Nζ stabilizes the anion.9 However, in Hyb-24DN, additional amino acid residues (Tyr170, Asp181, and Asn266) that are unnecessary for the esterolytic activities are required to confer a NylB-level of hydrolytic activity toward Ald. Asp181-COO− stabilizes substrate binding by electrostatic interactions with Ald—NH3+, while Asn266 makes suitable contacts with Ald and improves the electrostatic environment at the N-terminal region of Ald cooperatively with Asp181.18 In addition, the nylon oligomer hydrolase exhibited unique structural alterations induced by Ald, specifically, a movement of the loop region and the flip flop of Tyr170. On the basis of these findings, we proposed that amino acid replacements in the catalytic cleft of a pre-existing esterase with a β-lactamase fold resulted in the evolution of the nylon-oligomer hydrolase, and that catalysis proceeds according to the following steps: (i) Ald-induced transition from the open (substrate-unbound) to the closed (substrate bound) form, (ii) nucleophilic attack by Ser112 on Ald to form a tetrahedral intermediate, (iii) formation of an acyl enzyme and transition to the open form, and (iv) deacylation.8,9
In a PCR-induced random mutagenesis experiment using the parental Hyb-24 gene followed by selection for enhanced Ald-hydrolase activity, the G181D substitution was preferentially selected for in two independent experiments (pQ1M series and pR1M series) [Fig. 2(a)].17 However, a D370Y substitution occurring on the opposite side of the catalytic cleft from Gly181 (17 Å apart from Tyr370 at Cα position) was selected for a separate experiment (pS1M series). Hyb-24Y, containing the D370Y substitution, exhibited eight-fold enhanced activity relative to the parental Hyb-24 [0.023 μmol/min (U)/mg protein] (Fig. 1). Recently, we found that the D370Y substitution in Hyb-24DN improves the kcat/Km value five-fold by stable binding at both the N-terminal and C-terminal regions of Ald.18 These observations raise questions regarding what sort of structural alterations would be required for a catalytic cleft optimized as an esterase to evolve a new activity toward a nylon oligomer.
Figure 2.
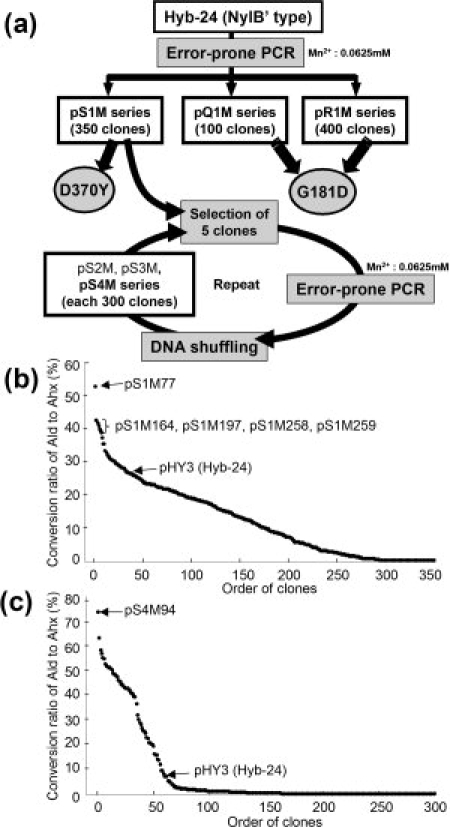
Directed evolution of nylon oligomer hydrolase from carboxylesterase. (a) Scheme for constructing the mutant library from the parental carboxylesterase (Hyb-24) gene (see Fig. 1). The Ald-hydrolytic activities of Hyb-24 were enhanced by PCR-induced random mutation and DNA shuffling. Details are described in “Materials and Methods.” (b) Distribution pattern of the conversion ratio of Ald to Ahx in random mutant populations in the first-cycle library. The conversion ratios after the 6 h reaction are arranged from highest to lowest. The positions of the Hyb-24 parent (average value of 28 experiments) and those of the mutant enzymes used for the present study are shown. (c) The distribution pattern of the conversion ratio of Ald to Ahx in random mutant populations in the fourth-cycle library. The conversion ratios after 30 min reactions are arranged from highest to lowest. The positions of the Hyb-24 parent (average value of 16 experiments) and those of the mutant enzymes produced in clones harboring pS4M94 are shown.
In this article, we have directly evolved Ald-hydrolytic activity from clones with a D370Y substitution. We identified the three-dimensional structures of the mutant enzymes and analyzed the structural requirements essential for generating the catalytic centers responsible for nylon oligomer hydrolase activity.
Results
Isolation of Hyb-24-mutant enzymes with increased Ald-hydrolytic activity
The strategy used to isolate clones with enhanced Ald-hydrolytic activity is illustrated in Figure 2(a). Plasmid pS1M77 was found to encode the enzyme (Hyb-24 A61V/A253T/F264C/D370Y) (designated as Hyb-S1M77) with the highest Ald-hydrolytic activity among the 350 clones in the first-cycle pS1M mutant library [Fig. 2(b)].17 The plasmids pS1M164, pS1M197, pS1M258, and pS1M259 were found to produce enzymes having the second to fifth highest activity. Therefore, a mixture of the five pS1M plasmids was used for the present experiment. To increase the divergence of the mutant library, we performed mutagenic PCR coupled with DNA shuffling.19 To select clones with enhanced Ald-hydrolytic activity, the enzyme reactions were performed at 30°C for 0.5–6 h with 10 mM Ald (pH 7.3). The enzyme activity was semiquantitatively assayed using TLC, and among the 300 independent clones (pS2M series), the five clones with the highest Ald-hydrolytic activity were selected on the basis of their conversion ratio of Ald to Ahx (Supporting Information Fig. S2). These clones were used for successive mutation cycles combined with DNA shuffling.
A clone (pS4M94) obtained from the fourth-cycle mutant library exhibited the maximum Ald-hydrolytic activity by semiquantitative TLC assay [Fig. 2(c)]. Therefore, this clone was selected for further study. Nucleotide sequencing of the 1.4 kb EcoRI/HindIII-fragment encoding the nylB/nylB′ region in pS4M94 revealed that the mutant enzyme (Hyb-S4M94) had six amino acid alterations (A61V, A124V, R187S, F264C, G291R, and G338A) in addition to the D370Y mutation. To characterize Hyb-S4M94 and several other Hyb-24 mutant enzymes, we fused a His-tagged linker to the N-terminal region of the mutant gene and purified the enzyme to homogeneity (Supporting Information Fig. S3). We have found that the His-tagged region barely affects the enzyme activity.8 The specific activity of Hyb-S4M94 was found to be 1.86 U/mg, which is 81 times the value obtained for Hyb-24 (0.023 U/mg; Fig. 1).
Kinetic studies showed that the kcat/Km of Hyb-S4M94 increased 13-fold relative to the value of Hyb-24Y and reached up to 61% of the level of Hyb-24DN18 (Fig. 3). We have not succeeded in estimating the Km and Vm values of parental Hyb-24 (Gly181-enzyme) because of its low activity toward Ald. However, we have found that D181N and D181E substitutions in the NylB-type protein (Hyb-2; Asp181-enzyme) decrease the enzyme's affinity for Ald [Km = 80 mM (Asn181-enzyme); Km = 220 mM (Glu181-enzyme)], and that D181K and D181H substitutions result in a drastic decrease in Ald-hydrolytic activity (<0.01% of the level of Asp181-enzyme).8,9,20 In contrast, the kinetic studies of Hyb-S4M94 (Gly181-enzyme) demonstrate that the enzyme acquired a high affinity (Km = 15 mM) and turnover (kcat = 3.1 s−1) for Ald, generating a catalytic center suitable for nylon-oligomer hydrolysis.
Figure 3.
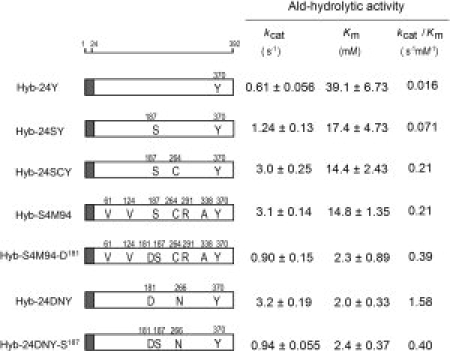
Kinetic parameters of Hyb-24 mutant enzymes. Ald-hydrolytic activity was assayed using the His-tagged enzymes under standard assay conditions, except that various concentrations of Ald were used. Kinetic parameters (kcat and Km values) were evaluated by directly fitting the Michaelis–Menten equation to the data using GraphPad prism, version 5.01 (GraphPad, San Diego, CA). The kcat values were expressed as turnover numbers per subunit (Mr of the subunit: 42,000). The kinetic parameters of Hyb-24Y and Hyb-24DNY reported previously (18) are shown for comparison. Shaded box, polypeptide regions from NylB; open box, region from NylB′. Amino acid alterations are shown as one-letter codes in the box.
Amino acid alterations essential for increasing Ald-hydrolytic activity in Hyb-S4M94
To identify amino acid substitutions responsible for increasing Ald-hydrolytic activity among the seven alterations introduced into Hyb-S4M94, we examined the combined effects of the substitutions by site-directed mutagenesis [Fig. 1(a)]. The D370Y substitution in Hyb-S1M77 was found to possess the primary activity-enhancing effect.17 The activity of His-tagged purified Hyb-24Y (0.19 U/mg) was enhanced 3.6-fold by the R187S substitution (Hyb-24SY). The addition of the substitutions A124V and G291R to Hyb-24Y increased its activity two-fold (see Hyb-24VY, Hyb-24RY), whereas neither A124V, G291R nor an A124V/G291R double substitution in Hyb-24SY resulted in any further increase in activity (see Hyb-24VSY, Hyb-24SRY, and Hyb-24VSRY). By contrast, the triple substitution R187S/F264C/D370Y in Hyb-24 (Hyb-24SCY) was enough to enhance the activity to the level of Hyb-S4M94. In addition, kinetic studies revealed that the kcat and Km of Hyb-24SCY are almost identical to those of Hyb-S4M94 (Fig. 3). These results demonstrate that the enhancement of the Ald-hydrolytic activity can be explained by the combined effects of R187S/F264C/D370Y. As Hyb-S1M77 contains the F264C/D370Y substitutions, its high activity is attributable to the combined effects of F264C/D370Y introduced in the first mutation cycle and R187S introduced in the successive mutation cycles. To simplify the cumulative effects for enhancing the activity, the effects are represented in the order of D370Y, R187S, and F264C in Figure 1(b).
X-ray crystallographic analysis
To analyze the structural basis for the increased activities, we determined the substrate-bound and unbound crystal structures of Hyb-24Y and Hyb-S4M94 by X-ray crystallographic analysis at resolutions of 1.50–1.58 Å (Supporting Information Table S3) and compared the structures with that of Hyb-24DN, which had been solved previously.9 In the three-dimensional structure model of Hyb-S4M94, three of the substitutions (R187S/F264C/D370Y) are located in the catalytic cleft, whereas three others (A61V/G291R/G338A) are located on the surface, and the remaining one (A124V) is located on the inside of the protein molecule [Fig. 4(a)]. Superimposition of the substrate-unbound structures of Hyb-24, Hyb-24DN, Hyb-24Y, and Hyb-S4M94 revealed that the overall structures share almost identical folding patterns with root mean square deviations (rmsd) smaller than 0.3 Å. The effects of the R187S/F264C/D370Y substitutions on catalytic function are discussed later.
Figure 4.
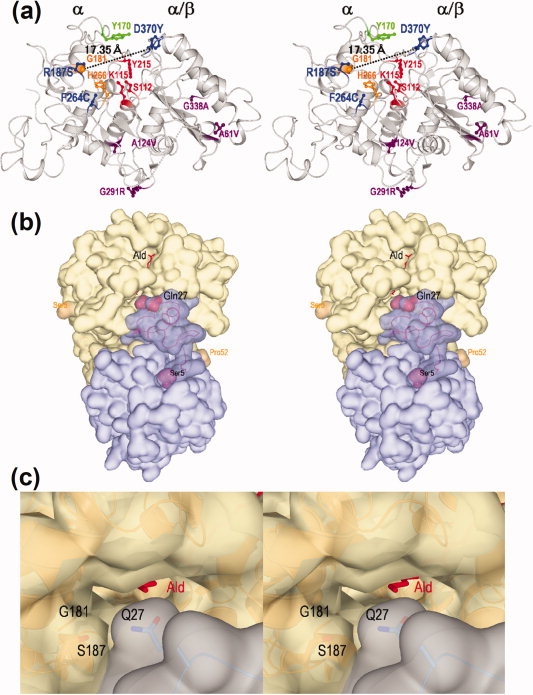
Stereo views of the three-dimensional structure of nylon oligomer hydrolase. (a) Overall structure of a monomer molecule of Hyb-S4M94 shown as a ribbon diagram. The left portion of the molecule is assigned as an α domain and the right portion as an α/β domain. The main chain folding is shown as a gray ribbon diagram, and the side-chains of the following amino acid residues are shown as a stick diagram. Proposed amino acid residues constituting the catalytic centers (S112/K115/Y215) are shown in red. Tyr170 is essential for Ald-hydrolytic activity is shown in green. Two alterations (G181D/H266N) increasing the Ald-hydrolytic activity in Hyb-24DN are shown in orange. Among the seven alterations differing between Hyb-24 and Hyb-S4M94, three (R187S/F264C/D370Y) were able to effectively increase the Ald-hydrolytic activity and are shown in blue, whereas the remaining four (A61V, A124V, G291R, and G338A) are shown in purple. (b) A homo-dimeric molecule of Hyb-S4M94 and interactions with substrate Ald. Surface structures of each monomeric unit are colored blue and gray, and substrate Ald is shown as a stick diagram colored red. Regions 1–4 and 53–57 were not traced because of poor electron density maps. The Ser5-Pro52 region located at the interface with another subunit is shown as semitransparent dark blue. Gln27, responsible for substrate binding, and Ser5/Pro52 are represented as a space-filing model. (c) Surface structure of the entrance of the catalytic cleft of the Hyb-S4M94-A112/Ald complex (closed form). The positions of Gln27, Gly181, and Ser187 are shown.
Induced fit motion by Ald-binding
In Hyb-24Y, the 169Asp-Ala174 region had poor electron density, and therefore, Tyr170 could not be identified in the three-dimensional models. This suggests that the region constitutes a flexible loop. A similar flexible structure has been found in the region 169Asp-Ala174 in parental Hyb-24.8 In contrast, in substrate-unbound Hyb-S4M94 and Ald-bound forms (Hyb-24Y-A112/Ald and Hyb-S4M94-A112/Ald complexes), catalytic/binding residues and Ald in the catalytic cleft had clear electron density distributions for which structural models could be determined (Supporting Information Fig. S4). Hyb-S4M94 exhibits movement of the loop region (5.1 Å at Val171-Cα) and a flip flop of Tyr170 upon substrate-binding, resulting in the formation of hydrogen bonds with Ald (amide—N) (distance = ca. 3 Å). Through these combined effects, Tyr170-Oη moves ∼10 Å, and these combined structural alterations also cover the active site to generate a closed form (Supporting Information Fig. S5). The surface structure around the flexible loop region in Hyb-S4M94 was basically similar to that of Hyb-24DN. However, it should be noted that the cleft of Hyb-S4M94 is more exposed to solvent, as the side chains of Gly181/Ser187 (Hyb-S4M94) are more compact than those of Asp181/Arg187 (Hyb-24DN) [Fig. 4(c)]. Actually, both Hyb-S4M94 and Hyb-24Y maintained additional water molecules around the N-terminal region of Ald in the catalytic cleft [Fig. 5(c,d)]. Substrate binding and the roles of hydrogen bonding networks and water molecules are discussed later.
Figure 5.
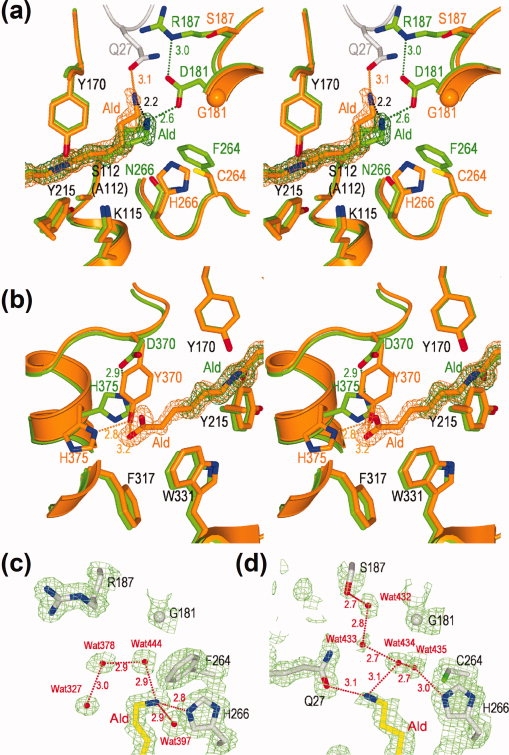
Interaction of Hyb-24 mutant enzymes with substrate Ald. (a,b) Stereo views of the superimposition of the Hyb-S4M94-A112/Ald complex (orange) with the Hyb-24DN-A112/Ald complex (green) at the amino-terminal portion (a) and carboxyl-terminal portion (b) of the substrate. The side-chains of catalytic/binding residues and substrate Ald with the 2Fo-Fc electron density maps contoured at 1.0σ are shown. (c,d) 2Fo−Fc electron density maps of the Hyb-24Y-A112/Ald complex (c) and the Hyb-S4M94-A112/Ald complex (d) contoured at 1.0σ. The side-chains of some residues [Gly181, Arg187 (Ser187), Phe264 (Cys264), His266, substrate Ald, and water molecules (Wat327, Wat378, Wat397, Wat432, Wat433, Wat434, Wat435, and Wat444) are shown. Hydrogen bonds between two atoms in the enzyme/substrate complex are indicated as dotted lines with the distance in angstroms.
Enzyme-substrate interaction
On the basis of the three-dimensional structures of the Hyb-24-mutant enzymes complexed with Ald, we estimated the following enzyme/substrate interactions in the catalytic cleft of the nylon-oligomer hydrolases.
Catalytic residues
Superimposition of the substrate-bound and unbound structures of Hyb-24Y, Hyb-S4M94, and Hyb-24DN revealed that Ser112 and Lys115 are conserved at the original position even after the binding of Ald. Upon substrate-binding, Tyr215 rotates its side chain 11–22° around the Cβ—Cγ bond, and the phenolic oxygen moves 0.53–0.60 Å in Gly181-enzymes (Hyb-24Y, Hyb-S4M94), whereas the Asp181-enzyme (Hyb-24DN) exhibits a larger movement of Tyr215, either in the rotation around Cβ—Cγ bond (33°) or in the position of the phenolic oxygen (0.81 Å). These results suggest that stable binding of Ald by electrostatic interaction with Asp181—COO− causes a larger movement of Tyr215 suitable for the positioning of the enzyme/substrate complex (Supporting Information Fig. S5).
Interactions at the N-terminal region of Ald
In parental Hyb-24, Gly181 provides no possible interactions that could stabilize the binding of Ald. This conclusion is supported by the observation that the distance between Ald—N and Gly—Cα in Hyb-24 fluctuates during 1 ns of molecular dynamic simulation.9 Unstable substrate binding at the N-terminal region of Ald is also estimated for the Hyb-24Y mutant. Actually, x-ray crystallographic analysis of the Hyb-24Y-A112/Ald complex revealed that the complex holds three water molecules (Wat327, Wat378, and Wat444) around the N-terminal region [Fig. 5(c)]. These observations suggest that the N-terminal region of Ald is exposed to solvent and that effective substrate binding is absent around the N-terminal region of Ald.
However, in the Hyb-S4M94-A112/Ald complex, we found a possible interaction that could be responsible for Ald binding. We suggest that (i) the R187S substitution forms a new hydrogen-bonding network starting from Ser187 to Ald—NH3+ via Wat432-Wat433-Wat434 [Fig. 5(d)] and (ii) the N-terminal half of Ald is bent at a maximum 2.2 Å (at the amino nitrogen) compared to that of Hyb-24DN/Ald complex [Fig. 5(a)].
Molecular modeling of the homodimeric structures of Hyb-S4M94 demonstrates that the N-terminal region (Ser5 to Pro52) is located at the interface between two subunits, and this region is thought to be related to domain swapping [Fig. 4(b,c)]. The Ald—NH3+ is located 3.1 Å apart from Gln27—Oɛ derived from another subunit [Fig. 5(a)]. As described later, we estimate that stable binding of the C-terminal region and the absence of electrostatic interactions with Asp181 eventually position Ald close to the alternative polar residue, Gln27 derived from another subunit. From these findings, we now conclude that a hydrogen-bonding network including Ser187 and interaction with Gln27 at the N-terminal region of Ald contributes to the increased substrate binding of Hyb-S4M94. Kinetic studies also revealed that the R187S substitution in Hyb-24Y improved both kcat and Ald-binding, resulting in four-fold higher kcat/Km values than those of Hyb-24Y (see Fig. 3, Hyb-24SY). However, it should be noted that the effect was lower than that of the G181D substitution (electrostatic stabilization), because of the G181D substitution in Hyb-24Y improved both kcat and Km more significantly (20-fold increase in kcat/Km values) (17). Moreover, the F264C substitution in Hyb-24SY increases the kcat value 2.4-fold, but barely improves the affinity (see Fig. 3, Hyb-24SCY). We found that the H266N substitution in Hyb-24D, spatially close to the F264C substitution, also enhances the kcat 4.6-fold without significantly affecting the Km (Hyb-24DN).18 This demonstrates that the F264C and H266N alterations, close to the Ser112-Lys115-Tyr215 catalytic triad, significantly increase the turnover of the enzyme reaction (Fig. 3).
Interactions at the C-terminal region of Ald
In Hyb-24DN, significant movement of Asp370/His375 was not observed, even after substrate binding (Supporting Information Fig. S5). In addition, the electron density map was poor for the C-terminal half of Ald [Fig. 5(b)].9 In contrast, in the Hyb-S4M94-A112/Ald and Hyb-24Y-A112/Ald complexes, the entire structure of Ald in the catalytic cleft had clear electron density distribution from which structural models could be determined, suggesting that Ald-binding is improved by the D370Y substitution [Fig. 5(b)]. Interestingly, Ald-binding induces the movement of Tyr370 (located at the border of Helix 18 and the flexible loop) and His375 (located at Helix 18). Specifically, the His375 side-chain rotates ∼100° around Cα—Cβ and ∼170° around Cβ—Cγ, and it flips the imidazole ring. Through this cooperative effect, Tyr370 moves the aromatic ring (ca. 7 Å at Tyr—Oη) into close contact with Ald [Fig. 5(b), Supporting Information Fig. S5]. We have found that Ald-binding induces similar movements of Tyr370/His375 in Hyb-24DNY (Hyb-24 having G181D/H266N/D370Y mutations).18 These results suggest that the interaction of Tyr370 with the C-terminal region of Ald plays an important role in substrate binding in the Tyr370 enzymes. Moreover, the carboxyl-half of the substrate Ald is surrounded by hydrophobic residues such as Trp331, Phe317, and Ile343 [Fig. 5(b), Supporting Information Fig. S4], and the D370Y substitution should make the environment of the catalytic cleft even more hydrophobic, resulting in improved substrate binding.
Combined effects of amino acid substitutions originating from alternative optimization routes
To examine whether or not mutations accumulated in the NylB-type enzyme (Hyb-24DN) and the directly evolved enzyme (Hyb-S4M94) are additive, we constructed Hyb-S4M94-D181 (Hyb-S4M94 having G181D substitution) and Hyb-24DNY-S187 (Hyb-24DNY having R187S substitution). These two mutants have similar kcat (0.90–0.94 s−1) Km (2.3–2.4 mM) for Ald (Fig. 3). However, it should be noted that the G181D substitution in Hyb-S4M94 decreases kcat to 30% of that of Hyb-S4M94. Similarly, R187S substitution in Hyb-24DNY also decreased kcat and kcat/Km (ca. 30% that of Hyb-24DNY), without affecting the Km value. These results suggest that coexistence of Asp181 and Ser187 is rather inhibitory for turnover of the enzyme reaction. Combined effects of amino acid substitutions on enzyme activity are discussed later on the basis of the three-dimensional structures.
Discussion
The penicillin-recognizing family of serine-reactive hydrolases possesses diverse catalytic functions, including β-lactam hydrolysis, DD-peptide hydrolysis, DD-transpeptidation, and carboxylester hydrolysis. Despite the similarity of its protein fold, NylB specifically recognizes nylon-6 related compounds. The spatial location of the Ser/Lys/Tyr catalytic triad of nylon oligomer hydrolase is similar to that of DD-peptidase, EstB carboxylesterase, and Class C β-lactamase (Fig. 6). However, the dual coordination of tyrosine (Tyr170 and Tyr215) to amide—N in the enzyme/Ald complex is unique for Ald-hydrolase among the serine-reactive hydrolases (Fig. 5).
Figure 6.
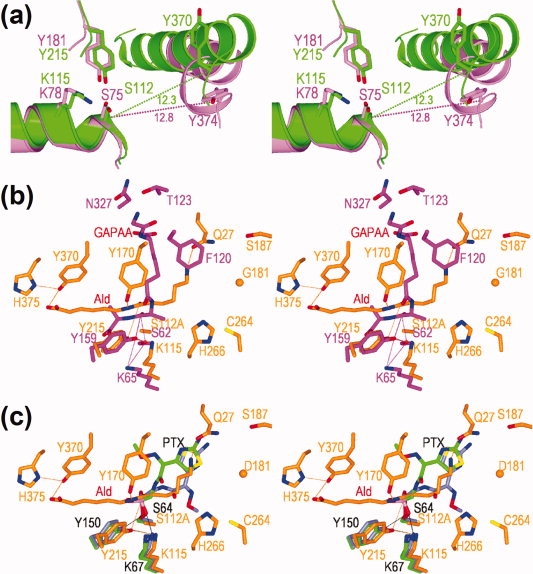
Comparison of catalytic/binding residues among nylon oligomer hydrolase, EstB carboxylesterase, DD-peptidase, and Class C β-lactamase. (a) Superimposition of Hyb-24Y (green) and EstB carboxylesterase (PDB ID code: 1CI8) (pink). The main chain folding (ribbon diagram), side-chain (stick diagram) of catalytic/binding residues are shown. (b) Superimposition of the Hyb-S4M94-A112/Ald complex (orange) with the R61 DD-peptidase/glycyl-l-α-amino-ɛ-pimelyl-d-alanyl-d-alanine (designated as GAPAA) complex (PDB ID code: 1IKG) (pink). (c) Superimposition of the Hyb-S4M94-A112/Ald complex (orange) with wild-type Class C β-lactamase/m-nitrophenyl-2-(2-aminothiazol-4-yl)-2-[(Z)-methoxyimino]acetylaminomethylphosphonate) (designated as PTX) complex (PDB ID code: 1RGY) (light green) and extended spectrum Class C β-lactamase/PTX complex (PDB ID code: 1RGZ) (navy).
In substrate-free structures, the positions of Tyr170 differ among Hyb-24 mutants having different levels of Ald-hydrolytic activity. In Hyb-24Y, the position of Tyr170 should be fluctuating, since the loop (169Asp-Ala174) region gave a poor electron density. In Asp181 enzymes (Hyb-24DN and Hyb-24DNY), Tyr170 forms a hydrogen bond with Asp181, and this interaction stably maintained the structure in an opened form9 (Supporting Information Fig. S5). In contrast, in directly evolved Hyb-S4M94, Tyr170 was oriented in the opposite direction and exposed to solvent. We estimate that fixation of Tyr170 in Hyb-S4M94 is due to a hydrogen-bonding network including Tyr170-Oη, Wat79, Tyr370-N (to the backbone), and Tyr370-O (to the carbonyl) (Supporting Information Fig. S5). However, despite these differences in the original positions, the loop region including Tyr170 was located at spatially equivalent positions in the Ald-bound structures. These results demonstrate that the movement of the loop and the flip flop of Tyr170 play an essential role in generating the catalytic centers of nylon-oligomer hydrolases. We have proposed that the primary role of Tyr170 is the stable binding of Ald, but we would also like to suggest the possibility that Tyr170-OηH cooperatively functions with Tyr215-OηH as a proton donor for the breakdown of the tetrahedral intermediate to the acyl enzyme.9
In substrate-unbound Hyb-24DN, Arg187 is present in a free form (Supporting Information Fig. S6). However, upon substrate binding, Arg187 rotates its side chains 130° around Cγ—Cδ, and 176° around Cδ—Nɛ, whereas Tyr170 flips its side-chain towards Ald (Supporting Information Fig. S6). Thus, it is likely that the dynamic motion of Tyr170 is stimulated by the cooperative movement of Arg187. In addition, as Arg187—Nɛ is 3.0 Å apart from Asp181—COO− in the Ald-bound structures [Fig. 5(a)], it is possible that a positively charged Arg187-guanidium ion promotes the ionization of Asp181—COO−, contributing to the catalytic function.
G181D substitution in Hyb-S4M94 improves its Km for Ald from 14.8 mM to 2.3 mM (see Fig. 3, Hyb-S4M94-D181). This increased affinity can be explained by the additive effect of electrostatic stabilization between Asp181—COO− and Ald—NH3+. However, it should be noted that the G181D substitution decreased kcat to about 30% that of Hyb-S4M94 (Fig. 3). Similarly, R187S substitution in Hyb-24DNY decreased its kcat and kcat/Km (ca. 30% of Hyb-24DNY), without affecting the Km value (Fig. 3). This indicates that the coexistence of Asp181 and Ser187 around N-terminal region of Ald is rather inhibitory for turnover of the catalytic reaction. As Ser187 in Hyb-S4M94 exhibits no significant movement even after Ald binding, it is likely that Ser187 provides no stimulating effect for the motion of Tyr170 bound to Asp181. In contrast, Tyr170, exposed to solvent in Hyb-S4M94, can exhibit induced fit motion toward Ald-amide-N without cooperative movement of residue at position 187.
We have suggested that steric hindrance to Ald binding by the bulky residues Phe264/His266 is suppressed by the H266N substitution in Hyb-24DNY, and Asn266 makes suitable contacts with Ald.18 Thus, Asp181/Arg187/Asn266 is responsible for catalytic function in NylB-type enzymes. In Hyb-S4M94, the steric hindrance by Phe264/His266 should be diminished by F264C substitution, and Ser187 becomes a better counterpart functioning with Cys264. Thus, selected Ser187/Cys264 increases Ald-hydrolytic activity in Hyb-S4M94 [Fig. 5(a)]. On the basis of these findings, we propose that there exist at least two alternative modes for optimizing the Ald-hydrolytic activity of a carboxylesterase with a β-lactamase fold.
Even after Ald-hydrolytic activity was enhanced 10–80-fold over that of parental Hyb-24 through accumulated amino acid substitutions, esterase activity towards C2-ester was not severely affected [Fig. 1(a)]. Ald-hydrolase activity requires strict substrate binding achieved by induced fit motion, whereas the enzyme performs the carboxylesterase catalytic function in a more relaxed open form. This model is consistent with our finding that Ald-hydrolytic activity is significantly affected by amino acid substitutions at positions 170, 181, 187, 264, 266, and 370 responsible for Ald-binding, whereas amino acid substitutions at these positions have less effect on the esterase activity.
The multiple three-dimensional alignment shows that Asp370 in Hyb-24 (Tyr370 in Hyb-24Y and Hyb-S4M94) is aligned with Tyr374 of EstB esterase. Interestingly, Tyr370 (Hyb-24Y) and Tyr374 (EstB) are located at the border of the Helix and loop regions conserved in the overall folds, although the side chain atoms spatially share different positions [Fig. 6(a)]. In addition, Asp370 in Hyb-24 is aligned with Asn327 in R61 DD-peptidase, which has been identified as a substrate binding site in an R61 DD-peptidase/substrate (glycyl-l-α-amino-ɛ-pimelyl-d-alanyl-d-alanine) complex (PDB ID code: 1IKG) [Fig. 6(b)].21 In Hyb-24DN, substrate Ald is extended straight across the catalytic cleft, and Asp181 interacts with the terminal ɛ-amino-group in Ald (Fig. 5, Supporting Information Fig. S5). In contrast, in DD-peptidase, the substrate (glycyl-l-α-amino-ɛ-pimelyl-d-alanyl-d-alanine) is bent at the junction of the pimelyl and d-alanyl groups, and its amino-terminal region (glycyl-l-α-amino-ɛ-pimelyl group) spatially shares positions occupied by Tyr170 in Hyb-24DN [Fig. 6(b)]. Thus, Ald adopts a position that is different from that of DD-peptidase. In wild-type Class C β-lactamase (PDB ID code: 1RGY),22 the large side chains of the thiazole ring in the cefotaxime analogue (m-nitrophenyl-2-(2-aminothiazol-4-yl)-2-[(Z)-methoxyimino]acetylaminomethylphosphonate) spatially overlapped with the position of Tyr170 [Fig. 6(c)]. In contrast, in extended-spectrum (ES) Class C β-lactamase (PDB ID code: 1RGZ),22 a mutated Ω-loop has a unique three-residue repeat insertion and adopts an alternate conformation, allowing a about 180° flip around the CO—C bond of the phosphonate in the cefotaxime analogue. Interestingly, in the Hyb-S4M94/Ald complex, the terminal amino group of Ald is bent toward Gln27, and through this effect, the position of Ald at the N-terminal region is more readily superimposable with the alternative position of the cefotaxime analogue in ES Class C β-lactamase [Fig. 6(c)].
It should be noted that the R187S substitution, which is responsible for increasing the activity of Hyb-S4M94, is located at the second position corresponding to the Gly-x-Ser-x-Gly esterase motif sequence, although the sequence is altered to Trp-Arg(Ser)-Thr-Arg-Arg in the multiple three-dimensional alignment of Hyb-24.8 Amino acid replacements required for specific substrate binding should occur at a suitable position in the catalytic cleft. Therefore, it is likely that the selected amino acid residues (Tyr370 and Ser187) in Hyb-S4M94 eventually coincide with the conserved residue (Tyr370) or share positions in the motif sequences (Ser187) with the other enzyme family members. In contrast, Cys264 in Hyb-S4M94 is less conserved, and it is substituted with Thr227 in DD-peptidase and Ser272 in EstB esterase.
From these analyses, we now conclude that nylon-oligomer hydrolase possesses unique substrate-binding residues responsible for Ald-hydrolysis, and that effective binding is achieved by induced-fit motion, including interconversion between open and closed forms during each catalytic cycle. However, we also suggest that the enzyme interacts with substrate at positions that are spatially similar to the substrate binding sites in other members of the penicillin-recognizing family of serine reactive hydrolases. Moreover, the presence of high esterolytic activity in the natural and evolved enzymes indicates that promiscuous amidase activities can evolve without severely compromising the original esterolytic activity. The present findings demonstrate that new enzymes can diverge from ancestral proteins having promiscuous activities to create families and superfamilies composed of highly specialized enzymes.
Materials and Methods
Strategy for the isolation of clones with enhanced Ald-hydrolytic activity
Ald-hydrolytic activity enhanced mutants were isolated according to the strategy illustrated in Figure 2(a). The 1.5 kb fragment containing the nylB24 mutant gene from five clones [pS1M77, pS1M164, pS1M197, pS1M258, and pS1M259, see Fig. 2(b)] was individually amplified by PCR in the presence of 0.0625 mM MnCl2 using the following primers: [FpKP1 (5′-GAGCGGATAACAATTTCACACAGG-3′) and RpKP1 (5′-AGGCTGAAAATCTTCTCTCATCCG-3′)]. After phenol/chloroform extraction followed by ethanol precipitation, primers were removed using SUPREC™-02 (Takara). The amplified 1.5 kb fragment (ca. 1 μg) (0.1 mL) was digested with DNase I (0.014 U) for 4 min at 25°C in Tris-HCl (pH 5.0) containing 1 mM MnCl2. The reactions were stopped by heating at 80°C for 15 min, and then the proteins in the reaction mixture were removed by phenol/chloroform extraction. The digested DNA fragments were fractionated on agarose and 50–150 bp fragments were recovered from the gel using SUPREC™-01 (Takara). The recovered DNA fragments were reassembled by an initial PCR (without primers). After verifying the increase in the fragment sizes by agarose gel electrophoresis, the 1.5 kb fragments corresponding to the original size were amplified by a second PCR using the primers: [NYL3 (5′-GCCGAGGCCATGGGCTACATCGATCTC-3′) and EII-2 (5′-GGCGGTCGGGTTTCGACTCGTCGC-3′)]. The 1.1 kb PvuII-AatII fragment was then prepared. To reconstruct the plasmid containing the entire nylB/nylB′ and vector regions, plasmid pHY3,7 was also digested with PvuII and AatII, and the 2.9 kb fragment was recovered. The 2.9 kb and 1.1 kb PvuII/AatII fragments obtained as described earlier were combined by ligation, followed by transformation of Escherichia coli KP3998,9 and the mutant library was obtained. To confirm the expression level among the cells in the mutant library, cell extract was adjusted to A280 = 10, and 20 μL of the sample was separated by SDS-polyacrylamide gel electrophoresis. We observed similar levels of protein bands corresponding to the NylB24 regions, indicating that the expression levels were maintained among the cells in library (Supporting Information Fig. S1). To select clones with enhanced Ald-hydrolytic activity, the enzyme reactions were performed at 30°C with 10 mM Ald in 20 mM potassium phosphate buffer containing 10% glycerol (pH 7.3). The reaction mixtures were fractionated by TLC and compounds having amino groups were detected by ninhydrin reaction (Supporting Information Fig. S2).6 The spots on the TLC plates were analyzed by image-analyzing programs (http://rsb.info.nih.gov/nih-image/). As two molecules of Ahx were obtained by hydrolysis of one molecule of Ald, the conversion ratios were calculated from the following equation:17
 |
Among the 300 independent clones (pS2M series), the five clones with the highest Ald-hydrolytic activity were selected on the basis of their conversion ratio of Ald to Ahx. These clones were used for successive mutation cycles combined with DNA-shuffling. After the 1.1 kb PvuII-AatII fragments were ligated with the vector (the 2.9 kb PvuII-AatII fragment from pHY3), followed by transformation into E. coli, a mutant library including 300 clones (pS3M series) was obtained. Five clones were selected on the basis of their high conversion ratio of Ald to Ahx, and mixtures of the DNA were mutagenized and shuffled, followed by ligation with the vector and transformation into E. coli, resulting in the mutant library (pS4M series). Three hundred clones were obtained. A clone containing plasmid pS4M94 obtained from the pS4M series exhibited the maximum conversion ratio [Fig. 2(c)].
Enzyme purification and assays
For x-ray diffraction experiments, the native enzymes were expressed in E. coli KP3998 using pKP1500 as a vector and purified.7 To analyze the specific activity of various mutant enzymes, a His-tag was fused to the N-terminus of each mutant enzyme (see Supporting Information). Enzyme activities were assayed at 30°C using 10 mM Ald as a substrate in 20 mM potassium phosphate buffer, pH 7.3, containing 10% glycerol (standard assay conditions). For kinetic studies, the activities were assayed under standard assay conditions, except that different Ald concentrations were used. Carboxylesterase activities were assayed using 0.2 mM p-nitrophenylacetate (C2-ester), 0.2 mM p-nitrophenylbutyrate (C4-ester) and 0.2 mM p-nitrophenyloctanoate (C8-ester). Diagrams of the molecular structures of the proteins were generated with MolFeat (ver. 3.6, FiatLux).
Coordinates
The atomic coordinates and structure factors for Hyb-24Y (PDB ID code: 2ZLY), Hyb-S4M94 (PDB ID code: 2ZM2), Hyb-24Y-A112/Ald complex (PDB ID code: 2ZM8), Hyb-S4M94-A112/Ald complex (PDB ID code: 2ZM9) have been deposited in the Protein Data Bank (http://www.rcsb.org/). The structure of Hyb-24 (PDB ID code: 1WYB),8 Hyb-24DN (PDB ID code: 1WYC)9 and Hyb-24DN-A112/Ald complex (PDB ID code: 2DCF)9 were previously reported.
Glossary
Abbreviations:
- Ahx
6-aminohexanoate
- Ald
6-aminohexanoate-linear dimer
- PCR
polymerase chain reaction
- rmsd
root mean square deviations.
References
- 1.Negoro S. Biodegradation of nylon oligomers. Appl Microbiol Biotechnol. 2000;54:461–466. doi: 10.1007/s002530000434. [DOI] [PubMed] [Google Scholar]
- 2.Negoro S. Biodegradation of nylon and other synthetic polyamides. Biopolymers. 2002;9:395–415. [Google Scholar]
- 3.Kinoshita S, Terada T, Taniguchi T, Takene Y, Masuda S, Matsunaga N, Okada H. Purification and characterization of 6-aminohexanoic acid oligomer hydrolase of Flavobacterium sp. KI72. Eur J Biochem. 1981;116:547–551. doi: 10.1111/j.1432-1033.1981.tb05371.x. [DOI] [PubMed] [Google Scholar]
- 4.Kato K, Ohtsuki K, Koda Y, Maekawa T, Yomo T, Negoro S, Urabe I. A plasmid encoding enzymes for nylon oligomer degradation: nucleotide sequence and analysis of pOAD2. Microbiology. 1995;141:2585–2590. doi: 10.1099/13500872-141-10-2585. [DOI] [PubMed] [Google Scholar]
- 5.Okada H, Negoro S, Kimura H, Nakamura S. Evolutionary adaptation of plasmid-encoded enzymes for degrading nylon oligomers. Nature. 1983;306:203–206. doi: 10.1038/306203a0. [DOI] [PubMed] [Google Scholar]
- 6.Kato K, Fujiyama K, Hatanaka HS, Prijambada ID, Negoro S, Urabe I, Okada H. Amino acid alterations essential for increasing the catalytic activity of the nylon-oligomer degradation enzyme of Flavobacterium sp. Eur J Biochem. 1991;200:165–169. doi: 10.1111/j.1432-1033.1991.tb21063.x. [DOI] [PubMed] [Google Scholar]
- 7.Ohki T, Mizuno N, Shibata N, Takeo M, Negoro S, Higuchi Y. Crystallization and X-ray diffraction analysis of 6-aminohexanoate-dimer hydrolase from Arthrobacter sp. KI72. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2005;61:928–930. doi: 10.1107/S1744309105028812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Negoro S, Ohki T, Shibata N, Mizuno N, Wakitani Y, Tsurukame J, Matsumoto K, Kawamoto I, Takeo M, Higuchi Y. X-ray crystallographic analysis of 6-aminohexanoate-dimer hydrolase: molecular basis for the birth of a nylon oligomer degrading enzyme. J Biol Chem. 2005;280:39644–39652. doi: 10.1074/jbc.M505946200. [DOI] [PubMed] [Google Scholar]
- 9.Negoro S, Ohki T, Shibata N, Sasa K, Hayashi H, Nakano H, Yasuhira K, Kato D, Takeo M, Higuchi Y. Nylon-oligomer degrading enzyme/substrate complex: catalytic mechanism of 6-aminohexanoate-dimer hydrolase. J Mol Biol. 2007;370:142–156. doi: 10.1016/j.jmb.2007.04.043. [DOI] [PubMed] [Google Scholar]
- 10.Arpigny JL, Jaeger KE. Bacterial lipolytic enzymes: classification and properties. Biochemistry J. 1999;343:177–183. [PMC free article] [PubMed] [Google Scholar]
- 11.Bornscheuer UT. Microbial carboxyl esterases: classification, properties and application in biocatalysis. FEMS Microbiol Rev. 2002;26:73–81. doi: 10.1111/j.1574-6976.2002.tb00599.x. [DOI] [PubMed] [Google Scholar]
- 12.Wagner UG, Petersen EI, Schwab H, Kratky C. EstB from Burkholderia gladioli: a novel esterase with a β-lactamase fold reveals steric factors to discriminate between esterolytic and β-lactam cleaving activity. Protein Sci. 2002;11:467–478. doi: 10.1110/ps.33002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Petersen EI, Valinger G, Soelkner B, Stubenrauch G, Schwab H. A novel esterase from Burkholderia gladioli which shows high deacetylation activity on cephalosporins is related to β-lactamases and DD-peptidases. J Biotechnol. 2001;8:11–25. doi: 10.1016/s0168-1656(01)00284-x. [DOI] [PubMed] [Google Scholar]
- 14.Kourist R, Bartsch S, Fransson L, Hult K, Bornscheuer UT. Understanding promiscuous amidase activity of an esterase from Bacillus subtilis. Chem Bio Chem. 2008;9:67–69. doi: 10.1002/cbic.200700521. [DOI] [PubMed] [Google Scholar]
- 15.Bornscheuer UT, Kazlauskas RJ. Catalytic promiscuity in biocatalysis: using old enzymes to form new bonds and follow new pathways. Angew Chem Int Ed Engl. 2004;42:6032–6040. doi: 10.1002/anie.200460416. [DOI] [PubMed] [Google Scholar]
- 16.Hult K, Berglund P. Enzyme promiscuity: mechanism and applications. Trends Biotechnol. 2007;25:231–238. doi: 10.1016/j.tibtech.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 17.Ohki T, Wakitani Y, Takeo M, Yasuhira K, Shibata N, Higuchi Y, Negoro S. Mutational analysis of 6-aminohexanoate-dimer hydrolase: relationship between nylon oligomer hydrolytic and esterolytic activities. FEBS Lett. 2006;580:5054–5058. doi: 10.1016/j.febslet.2006.08.031. [DOI] [PubMed] [Google Scholar]
- 18.Kawashima Y, Ohki T, Shibata N, Higuchi Y, Wakitani Y, Matsuura Y, Nakata Y, Takeo M, Kato D, Negoro S. Molecular design of a nylon-6 byproduct-degrading enzyme from a carboxylesterase with a β-lactamase fold. FEBS J. 2009;276:2547–2556. doi: 10.1111/j.1742-4658.2009.06978.x. [DOI] [PubMed] [Google Scholar]
- 19.Stemmer WP. DNA shuffling by random fragmentation and reassembly: in vitro recombination for molecular evolution. Proc Natl Acad Sci USA. 1994;91:10747–10751. doi: 10.1073/pnas.91.22.10747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hatanaka HS, Fujiyama K, Negoro S, Urabe I, Okada H. Alteration of catalytic function of 6-aminohexanoate-dimer hydrolase by site-directed mutagenesis. J Ferment Bioeng. 1991;71:191–193. [Google Scholar]
- 21.McDonough MA, Anderson JW, Silvaggi NR, Pratt RF, Knox JR, Kelly JA. Structures of two kinetic intermediates reveal species specificity of penicillin-binding proteins. J Mol Biol. 2002;322:111–122. doi: 10.1016/s0022-2836(02)00742-8. [DOI] [PubMed] [Google Scholar]
- 22.Nukaga M, Kumar S, Nukaga K, Pratt RF, Knox JR. Hydrolysis of third-generation cephalosporins by class C β-lactamases: structures of a transition state analog of cefotaxime in wild-type and extended spectrum enzymes. J Biol Chem. 2004;279:9344–9352. doi: 10.1074/jbc.M312356200. [DOI] [PubMed] [Google Scholar]