

Abstract
The structural basis for the T cell response to glycolipid antigens (Ags) remains poorly understood. T lymphocytes autoreactive for mouse CD1 (mCD1.1) or reactive for the glycosphingolipid αgalactosylceramide (α-GalCer) presented by mCD1.1 have been described previously. In this paper it is shown that mutations at the top of the α helices and in the bottom of the Ag-binding groove can disrupt both mCD1.1 autoreactivity and α-GalCer recognition. The locations of the positions that affect T cell responses indicate that recognition of mCD1.1 is not likely to be unconventional or superantigen-like. Furthermore, the effects of the bottom of the pocket mutation suggest that the autoreactive response could require an autologous ligand, and they indicate that α-GalCer binds to the groove of mCD1.1, most likely with the shorter 18-carbon hydrophobic chain in the A′ pocket. Natural killer T cell hybridomas with identical T cell antigen receptor (TCR) α chains and different β chains respond differently to α-GalCer presented by mCD1.1 mutants. This finding indicates a role for TCR β in defining natural killer T cell specificity, despite the more restricted diversity of the α chains in these cells. Overall, the data are consistent with a mode of lipoglycan recognition similar to that proposed for glycopeptides, in which the TCR α and β chains survey a surface composed of both mCD1.1 and the carbohydrate portion of α-GalCer.
CD1 molecules are cell surface proteins expressed as a 45- to 55-kDa heavy chain noncovalently associated with β2-microglobulin (1). Several features discriminate CD1 from the major histocompatibility complex (MHC)-encoded class I and class II molecules, most notably the ability of CD1 molecules to present lipid antigens (Ags) (1, 2). The mouse CD1 (mCD1.1) molecule has a groove typical of the MHC-encoded Ag-presenting molecules (3), but this groove is very hydrophobic, it is deeper than those in class I and class II proteins, and it contains only two pockets, termed A′ and F′.
Two subgroups of CD1 molecules have been defined on the basis of their amino acid sequence (1, 4). Considering mice and humans, group 2 molecules include the closely related mCD1.1 and mCD1.2 glycoproteins, as well as human CD1d (hCD1d). The majority of T cells reactive to group 2 CD1 molecules are natural killer (NK) T cells. NK T cells are a specialized subset of predominantly double-negative or CD4+ T lymphocytes (5, 6). They may be a regulatory cell type that plays a pivotal role between innate and adaptive immunity (2, 7). Mouse NK T cells have a limited TCR repertoire, including a Vα14–Jα4281 rearrangement with a conserved junction, and many of them are mCD1.1 autoreactive (5–7). A synthetic glycolipid, α-galactosylceramide (α-GalCer), has been identified as an Ag presented by mCD1.1 to NK T cells (8, 9). α-GalCer can bind to mCD1.1 (10), and it can greatly stimulate the mCD1.1 reactivity of T cells that express a Vα14 TCR (9). Human CD1d-autoreactive NK T cells express an α chain homologous to mouse Vα14, and they are responsive to α-GalCer in an hCD1d-dependent fashion (11–13).
Analysis of the ligands presented by the various CD1 molecules indicates that most of them have two hydrocarbon chains or a single branched chain (1, 2). It therefore has been proposed that the Ag-binding groove accommodates one chain each in the A′ and F′ pockets, while the hydrophilic head group of the Ag is exposed for TCR recognition (3, 14–16). Consistent with this view, we have recently demonstrated that binding of α-GalCer to mCD1.1 requires neither the carbohydrate nor the hydroxyls on the sphingosine, suggesting that contacts with the hydrocarbon chains are of primary importance for mCD1.1 binding (10, 17).
Despite some progress, the molecular interactions between CD1, lipid Ag, and the TCR remain largely uncharacterized. To understand better the structural basis for T cell reactivity to lipids plus CD1, we have generated transfectants expressing mutated mCD1.1 molecules. mCD1.1-autoreactive hybridomas have been tested for their ability to recognize these mutants, either in the absence of Ag to analyze autoreactivity, or in the presence of α-GalCer and related compounds.
Materials and Methods
Reagents.
The synthesis, purification, and purity of the lipid Ags have been described previously (18). All of the compounds were dissolved in 100% DMSO and sonicated before use.
The anti-IL-2 monoclonal antibodies (mAbs) used for ELISAs, JES6-N37–1A12 and biotinylated JES6–5H4, and recombinant IL-2 cytokine standard (107 units/mg) were purchased from PharMingen (San Diego). The following mAbs used for phenotypic analysis of mouse T cell hybridomas were purchased from PharMingen: anti-CD16/32 (for Fc receptor blocking), 1B1 anti-mCD1.1, anti-β TCR, anti-Vβ8.3, anti-Vβ8.1–Vβ.8.2, anti-Vβ7, and anti-Vβ10 mAbs, isotype-matched mAbs, and anti-hamster IgG mixture G70-204 and G94-90.5. Chung-Ryu Wang (University of Chicago) kindly provided the hamster 5C6 anti-mCD1 mAb.
Sequencing of TCR β Chain Complementarity-Determining Region (CDR)3s.
mRNA from T cell hybridomas was extracted and reverse transcribed by following standard procedures. Vβ fragments were amplified by PCR using specific Vβ primers and a consensus Cβ primer (19), and final PCR products were purified by gel electrophoresis and ligated into the TOPO TA cloning vector (Invitrogen, Carlsbad, CA). Clones were then sequenced with the dye primer cycle sequencing kit with the AmpliTaq DNA polymerase FS (Perkin–Elmer/Cetus, Branchburg, NJ).
Site-Directed Mutagenesis.
Wild-type mCD1.1 transfected A20 cells and A20 transfected controls, which have been obtained by transfection with the wild-type mCD1.1 cDNA in the wrong orientation, were obtained and cultured as previously described (20). The mCD1.1 mutant constructs have been generated by using the altered sites II in vitro mutagenesis system, according to the manufacturer's protocol (Promega, Madison, WI). For each mutation reaction, five to eight clones of JM109 bacteria, which were ampicillin resistant but tetracycline sensitive, were sequenced by using the dye primer cycle sequencing kit with the AmpliTaq DNA polymerase FS. The sequences of the mutagenic primers are as follows (5′ → 3′, with mutated bases in boldface): F10A, CAAAAGAATTACACCGCCCGCTGCCTGCAGATG; C12G, TACACCTTCCGCGGCCTGCAGATGTC; Y73F, CAGCATATGTTTCAAGTCTTTCGAGTCAGCTTTACCAGG; S76G, CAAGTCTATCGAGTCGGCTTTACCAGGGACATACAG; R79E, CGAGTCAGCTTTACCGAGGACATACAGGAATTAGTC; D153Y, GTGCTCAACGCTTATCAAGGGACAAGTGCA; T156I, AACGCTGATCAAGGGATAAGTGCAACCGTGCAG; F171Y, GACACCTGCCCCCTATATGTCCGTGGTCTCCTAGAG; RD79/80EA, CGAGTCAGCTTTACCGAGGCCATACAGGAATTAGTC; and LD150/153IY, GACTTGCCCATCAAAGTGATCAACGCTTATCAAGGGACAAGTGCA.
Gene Cloning and Transfection.
The mutated mCD1.1 constructs were inserted into the unique BamHI and SalI sites of the mammalian expression vector pHβAprneo. Transfections were performed as previously described (20). For each mutant, 10–15 stable lines of A20 transfectants were stained with the 1B1 or 5C6 mAbs to detect mCD1.1 surface expression, and cell lines expressing an adequate surface level of mCD1.1 were selected. At least two different lines of each mutant have been tested in the experiments described in this study, and similar results were obtained for both lines in each case.
T Cell Hybridomas.
The derivation and characterization of the mCD1.1-autoreactive T cell hybridomas have been described previously (9). Hybridomas 68 and 24 were kindly provided by S. Cardell (Lund University, Sweden). Hybridomas 1.4 and 1.2, generously provided by M. Bix (University of California, San Francisco), and hybridomas 2C12, 3C3, 2D5, 2H4 and 1A12 (M.G., unpublished results) were all derived from sorted C57BL/6 NK1.1+ thymocytes. T cell hybridomas were cultured in the presence of antigen-presenting cells (APCs) pulsed with glycolipids or their respective controls as indicated in the figure legends. The cytokine levels in cell culture supernatants were detected after 16 h of stimulation by using standard sandwich ELISAs. Cytokine levels are expressed as mean ± SD of culture triplicates.
Results and Discussion
Diverse β Chain CDR3s of Vα14+ T Cell Hybridomas.
The properties of the thymus-derived Vα14+ NK T cell hybridomas that were used in this study are presented in Table 1. Four of the six hybrids show some degree of reactivity for mCD1.1 transfectants of A20 cells in the absence of exogenous Ag. Two of the hybridomas, 2D5 and 2H4, were not autoreactive to mCD1.1+ A20 APCs (9), whereas the responses of the autoreactive ones ranged from 5 to 80 units/ml IL-2. Three of these four autoreactive T cells express Vβ8.2. All six Vα14+ cells are reactive to α-GalCer presented by mCD1.1, with a moderate (>1.5 times) to strong (>50 times) enhancement of the mCD1.1 response by the four Vα14+ cells that showed autoreactivity.
Table 1.
Properties of Vα14+ NK T cell hybridomas
| Hybridoma | mCD1.1 autoreactivity | α-GalCer reactivity, SI | Vβ | CDR3 | Jβ start | Jβ |
|---|---|---|---|---|---|---|
| 1.2 | Intermediate | 9–73 | 8.2 | GAPSGGDYAEQ | FFG | 2.1 |
| 2C12 | High | 1.5–3 | 8.2 | GDEGYTQ | YFG | 2.5 |
| 3C3 | Intermediate | 12–55 | 8.2 | GDQGITGQL | YFG | 2.2 |
| 2D5 | None | ∞ | 8.3 | SEPGTGAFYAEQ | FFG | 2.1 |
| 1.4 | Intermediate | 10–42 | 10 | SPGLENTGQL | YFG | 2.2 |
| 2H4 | None | ∞ | 7 | LRGQNTL | YFG | 2.4 |
The levels of mCD1.1 autoreactivity are indicated (intermediate = 5–50 units/ml; high > 50 units/ml). The α-GalCer reactivity is expressed as a stimulation index (SI), comparing IL-2 release induced by α-GalCer-pulsed APC versus the vehicle-pulsed APCs. ∞ = α-GalCer SI of hybridomas that are not mCD1.1 autoreactive. CDR3 is defined as beginning after the conserved serine at the end of Vβ and ending just before the conserved F/YFG motif in Jβ. N–D–N region amino acids are in boldface.
Nucleotide sequence analysis demonstrated that all six Vα14+ hybridomas express the invariant Vα14Jα281 rearrangement that is typical of NK T cells (data not shown). The sequences of the TCR β chains from these cells also are shown in Table 1. We do not find evidence for a conserved length or sequence motif in the CDR3β regions of α-GalCer-reactive T cells. In agreement with the results from previous studies of Vα14+ T cells (19, 21), however, there is an over-representation of Jβ2 segments. Furthermore, the CDR3s are enriched for the presence of amino acids that are negatively charged, polar, or have small side chains. Collectively, there are a total of eight negative charges and only one positive charge in the CDR3βs, with five of the six hybridomas having at least one negatively charged amino acid. Polar amino acids serine, threonine, glutamine, and asparagine also are highly represented. Likewise, the number of glycines is greater than expected (13 of 56 amino acids). The majority of these are in the N–D–N region of CDR3, however, and a glycine bias may be introduced by the guanosine-rich sequences of the germ-line Dβ segments. Interestingly, the Vβ11 sequences of human T cells that were expanded in α-GalCer share some of these sequence features (22).
X-ray crystallographic analyses of several carbohydrate-binding proteins have revealed that the most important interactions with sugars involve hydrogen bonds, with side chains having acidic (aspartate or glutamate) or amide (asparagine or glutamine) groups prevalent in forming these interactions with carbohydrate (23). Five of the hybridomas have three amino acids in their CDR3β that fit into these two categories, whereas 2H4 has two amino acids with amide side chains (Table 1). On the basis of these sequence patterns, it is possible that CDR3β amino acids of the NK T cells could be contacting the carbohydrate of α-GalCer. The data from the TCR β chains from mouse and human NK T cells are in marked contrast with the data obtained for a set of five human T cells reactive to diverse lipid Ags and presented by group 1 CD1 molecules, which contain many basic amino acids (16). The difference in CDR3β sequence patterns could reflect differences in the types of Ags recognized; several of the human T cells analyzed recognize Ags that do not contain carbohydrate.
Expression of mCD1.1 Mutants.
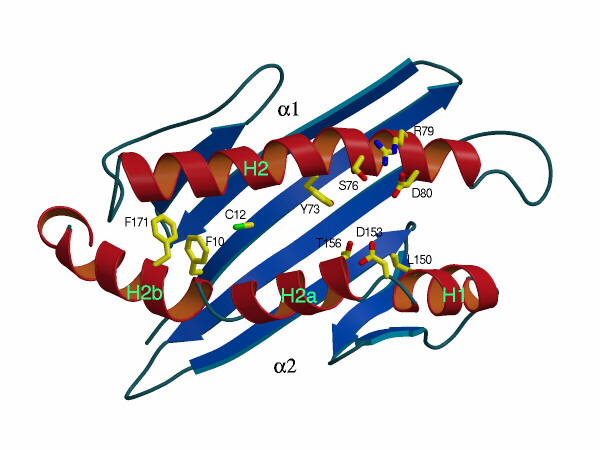
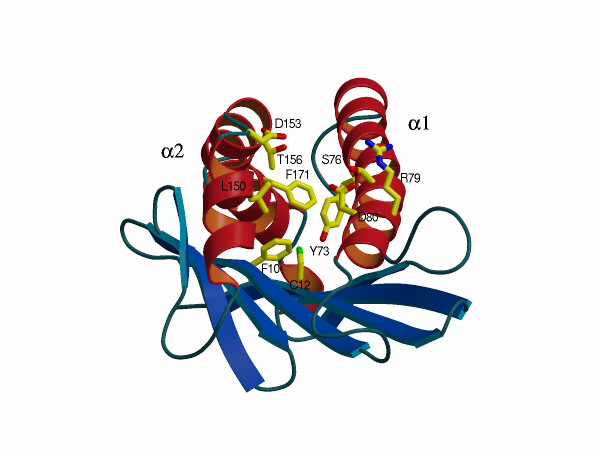
We have generated stable transfectants expressing one of eight single or two different double mCD1.1 mutations (summarized in Table 2). These α1 and α2 domain mutations have been designed on the basis of several criteria, including visual inspection of the three-dimensional structure of the mCD1.1 Ag binding groove. We also have relied on a preliminary theoretical model of the interaction between mCD1.1 and α-GalCer (H.I., unpublished results). Because of the conserved T cell recognition of mCD1.1 and hCD1d (11), positions conserved between these molecules were targeted as potentially important, while avoiding drastic changes in charge or hydrophobicity that might interfere with surface expression. In 5/10 positions, 76, 150, 153, 156, and 171, mCD1.1 amino acids were replaced with amino acids found in the same positions in human CD1b. Human CD1b can bind to α-GalCer (M.K., unpublished data), but it cannot present α-GalCer to NK T cells (12). At three other positions, we replaced mCD1.1 amino acids with amino acids having smaller side chains (F10A, C12G, and D80A). Finally, we reasoned that multiple hydrophobic interactions in the groove probably stabilize ligand binding, and that this would be difficult to disrupt, short of introducing potentially disruptive charged amino acids. We therefore focused attention on the few conserved hydrophilic amino acids, which could be important for interaction with the TCR or the hydrophilic portion of the Ag. The location of the mutated amino acid side chains is shown in a supplementary figure (www.pnas.org). These mutations fall into four categories: (i) F10A, C12G, and F171Y are located either in (positions 10 and 12) or close to (position 171) the A′ pocket. (ii) Y73F and L150I point into the F′ pocket. (iii) S76G, R79E, and D80A alter hydrophilic amino acids pointing upward near the end of the long H2 α helix encoded by the α1 domain. Amino acid positions in this group could be interacting with the TCR, and possibly also with the more external, hydrophilic portions of the mCD1.1-bound ligand. (iv) The D153Y and T156I mutations affect side chains pointing horizontally across the F′ pocket of the groove. These changes affect the character of the F′ pocket, and the side chains of these amino acids could be involved in binding to the more exposed portions of the Ag or to the TCR.
Table 2.
Summary of responses to mCD1.1 mutants
| Mutant | Expression, mfi | Location | Orientation | Reduced autoreactivity
|
Reduced α-GalCer
|
|
|---|---|---|---|---|---|---|
| Vα14− (3) | Vα14+ (4) | Vα14+ (5) | ||||
| LD150-153IY | 264* | α2 helix | 150 ↓ F′, 153 → F′ | All | All | All |
| F171Y | 1,066 | α2 helix | → A′ | None | None | None |
| T156I | 416 | α2 helix | → F′ | All | 2 (1.2, 3C3) | All |
| D153Y | 899 | α2 helix | → F′ | 2 (1A12, 24) | All | All |
| RD79-80EA | 508 | α1 helix | ↑ F′ | All | All | All |
| R79E | 1,917 | α1 helix | ↑ F′ | All | 2 (1.2, 3C3) | 3 (1.2, 3C3, 2D5) |
| S76G | 1,612 | α1 helix | ↑ F′ | 1 (24) | 1 (3C3) | 1 (2D5) |
| Y73F | 1,630 | α1 helix | ↓ F′ | None | None | None |
| C12G | 398 | A′ ↑ | 1 (1A12) | None | None | |
| F10A | 488 | A′ ↑ | 2 (1A12, 68) | None | None | |
Substitutions are shown in the left column with CD1b replacements underlined. The cell surface expression in mean fluorescence intensity (mfi) when the 1B1 anti-mCD1.1 mAb is used is shown. The location and orientation of the mutated side chains are shown: ↓, into the groove; →, across the groove; ↑, up towards the TCR. Autoreactive responses of the Vα14− cells (24, 68, 1A12) and Vα14+ hybridomas (1.2, 2C12, 3C3, 1.4), and Vα14+ α-GalCer responses (1.2, 3C3, 2D5, 1.4, 2H5) are tabulated separately; the number of hybridomas tested in each category is shown in parentheses. “None” indicates that no hybridoma was affected by the mutation, whereas “All” indicates that every tested hybridoma did not react to the mutant. Otherwise, the numbers of affected hybridomas (names in parentheses) are indicated. Effected responses were either undetectable or, in few cases, were at least 10-fold reduced. Data from at least three experiments in every case were used.
No reactivity with 1B1, mfi is for 5C6 mAb.
Two wild-type mCD1.1 transfectants were selected for this study, clone 1 with an mfi with the 1B1 anti-mCD1.1 mAb of 1,903, and clone 8 with an mfi of 444. Untransfected A20 cells do not express detectable mCD1.1 (mfi = 6). Stable transfectants that express each of the mCD1.1 mutants were generated, and those that express surface mCD1.1 levels that fell close to or between those of the two wild-type mCD1.1 transfectants were used. The mfi for the mutants ranged from 398 to 1,917 (Table 2) except for the L150I D153Y double mutant, which was not recognized by 1B1 (Table 2). 5C6, a second anti-mCD1 mAb, revealed surface expression of L150I D153Y mCD1.1. The mfi of 264 for the L150I D153Y mutant was slightly below that of clone 8 with this mAb (mfi 345).
Fine Specificity of Autoreactive T Lymphocytes for mCD1.1 Mutants.
Transfectants expressing the mutants were tested, in the absence of exogenous Ag, for their ability to stimulate seven mCD1.1-autoreactive hybridomas (Table 2, Fig. 1A). In addition to the four Vα14+ autoreactive hybridomas described above, three mCD1.1 autoreactive hybridomas that do not express Vα14 were tested. Hybridomas 24 and 68 are derived from the remaining CD4+ T cells in the spleen of MHC class II−/− mice (20), and 1A12 expresses a Vα5/Vβ14 TCR that is atypical for the NK1+ thymocyte population it originated from. Representative response data from a Vα14− and a Vα14+ hybridomas are shown in Fig. 1A. Six mutations gave similar effects on at least 6/7 hybridomas (Table 2). The F171Y and Y73F mutations generally did not reduce autoreactivity. The C12G mutation located on a β strand on the floor of the A′ pocket affected the response of 1A12 only, decreasing it to the background level. Note that the relevant comparison is with the response to the clone 8 wild-type mCD1.1 transfectant, as the expression level of the C12G mutant is only slightly less than the clone 8 transfectant (Table 2). By contrast, the D153Y mutant reduced autoreactivity to the undetectable level for every hybridoma except 68, and the two double mutations reduced it to an undetectable level in every case. The effect of the remaining mutations varied more, depending upon the cell tested. The responses to the T156I mutation, which points across the F′ pocket from the α2 helix, and the R79E mutation, pointing upward from the α1 helix above the F′ pocket, were similar. The reactivity of the 2C12 and 1.4 Vα14+ hybridomas was unaffected by these mutations, while the mCD1.1 autoreactivity of the remaining five hybridomas was eliminated by either change. The S76G mutant, which like R79E alters an amino acid pointing upward from the top of the α helix, showed a different pattern. This change completely eliminated the autoreactivity of hybridoma 24 and the Vα14+ hybridoma 3C3. It had little effect on the remaining cells, including those shown in Fig. 1A. The F10A mutation on the floor of the A′ pocket abrogated the responses of 1A12 and 68 only, with the other cells showing little or no effect compared with the relevant clone 8 wild-type control.
Figure 1.

T cell response to mCD1.1 mutants. (A) Diverse patterns of mCD1.1 autoreactivity. The responses of representative Vα14− T cells and Vα14+ NK T cells are shown. The TCR V regions expressed by these cells are indicated. mCD1.1 autoreactive T cell hybridomas were cultured in the presence of 3 × 105 APCs per well and untransfected A20 cells (A20), wild-type mCD1.1 transfectants, or the indicated mCD1.1 mutants. Mutants are arranged by location, from top to bottom: α2 helix (LD150–153IY, F171Y, T156I, D153Y), α1 helix (RD79–80EA, R79E, S76G, Y73F), and A′ pocket (C12G, F10A). Cell-free supernatants were tested by ELISA for IL-2 content as described. One representative experiment of three is shown. (B) Pattern of α-GalCer reactivity of Vα14+ NK T cells. NK T cell hybridomas were cultured as described with the indicated A20 APCs. APCs were prepulsed with 100 ng/ml α-GalCer. Cell-free supernatants were tested by ELISA for IL-2 content after culture. One representative experiment of four is shown.
Several inferences can be drawn from this analysis of mCD1.1 autoreactivity. First, the mutant mCD1.1 molecules are not likely to have a grossly altered conformation. Outside of the two double mutants, in every case there was at least one hybridoma that had an undiminished response to a particular mCD1.1 mutant. This is consistent with the ability of most of the mutants to react with two conformation-sensitive anti-mCD1.1 mAbs. Second, the mCD1.1 autoreactive T cells are heterogeneous in their response to different mutants. This heterogeneity may reflect the different TCRs expressed by the autoreactive hybridomas, and the importance of particular contacts between these TCRs and mutated mCD1.1 amino acids. Alternatively, this could reflect the recognition of different autologous ligands by the autoreactive T cells, as has been suggested previously (20, 24). Third, the CDR3 region of the β chain is important for autoreactivity. The Vα14+ hybridomas 1.2, 3C3, and 2C12 all express Vβ8.2, but only 3C3 is incapable of responding to the S76G mutant, while both 1.2 and 3C3 cannot respond to R79E. Fourth, the amino acid substitutions that affect mCD1.1-autoreactive T cells imply that the interaction with the TCR is conventional, as opposed to superantigen-like. Hydrophilic amino acids at the top of the groove, such as those at positions 76, 79, 80, 153, and 156, participate in T cell stimulation. The important hydrophilic amino acids are good candidates for those that make contacts with the TCR, although interactions with hydrophilic portions of mCD1.1-bound autologous ligands also are possible. Furthermore, amino acids at the bottom of the A′ pocket also participate in stimulating autoreactivity. The most straightforward interpretation of the effect of the A′ pocket mutations is that the autoreactive hybridomas respond to an autologous ligand(s) bound to the mCD1 groove. The alternative explanation, which is not ruled out, is that the A′ pocket mutations cause significant changes in the α helices that are detected by some of the autoreactive TCRs.
Presentation of α-GalCer by Mutant mCD1.1 Molecules.
We tested the ability of the mCD1.1 mutants to present α-GalCer to five Vα14+ hybridomas. Data from hybridoma 2C12 were not included in the analysis, because this highly autoreactive T cell exhibits a weak (approximately 2-fold) and variable stimulation with α-GalCer (Table 1). The other T cells exhibited optimal α-GalCer responses of 60–400 units/ml of IL-2 release, well above the level of mCD1.1 autoreactivity. The responses of two Vα14+ hybridomas to the mutants are presented in Fig. 1B, and the results are summarized in Table 2.
There was less diversity in the responses of various Vα14+ T cells to a defined Ag, compared with the mCD1.1-autoreactive response. A similar pattern of α-GalCer responses by all five T cells was obtained with 8 of the 10 mCD1.1 mutants. There were no α-GalCer responses to the 2 double mutants or to either the D153Y or T156I single mCD1.1 mutants, whereas all 5 Vα14+ cells responded to the 2 bottom of the A′ pocket mutants, F10A and C12G, as well as to the Y73F and F171Y mutants. Two mutations differentially affected the α-GalCer responses by Vα14+ T cells. S76G eliminated the α-GalCer reactivity of 2D5, decreasing the response from more than 100 units/ml of IL-2 release to less than 1 unit/ml. The R79E mCD1.1 mutation completely eliminated the reactivity of 1.2 and 3C3 as well as 2D5 (Fig. 1B and Table 2), whereas the other Vα14+ cells were not affected. It is noteworthy that the three cells unresponsive to α-GalCer presented by the R79E mCD1.1 mutation express a Vβ8 TCR, whereas those expressing Vβ7 and Vβ10 can respond. This finding demonstrates that the Vβ region is important for the α-GalCer response. Because the mutated mCD1.1 amino acid side chains for both positions 76 and 79 are pointing upwards, these positions represent possible mCD1.1 contact points for TCR β.
There is a high degree of similarity in the pattern of responses of the Vα14+ hybridomas to the mCD1.1 mutants in the presence or in the absence of α-GalCer, suggesting that the mode of recognition of autologous Ags and α-GalCer could be similar. There are two exceptions to this highly similar response pattern. First, the mutation S76G abolished the mCD1.1 autoreactivity of the 3C3 hybridoma, decreasing it from more than 30 units/ml of IL-2 to less than 1 unit/ml. When pulsed with α-GalCer, however, this mutant was able to achieve a stimulation of 3C3 comparable to wild-type CD1.1 transfectants (>200 units/ml of IL-2 in each case). Second, the T156I mutation did not decrease the mCD1.1 autoreactivity by the 1.4 hybridoma compared with reactivity to wild-type mCD1.1 (approximately 10 units/ml IL-2 release). However, no additional stimulation of 1.4 was achieved by adding α-GalCer to T156I-expressing APCs (Fig. 1B). APCs expressing this mCD1.1 mutation also could stimulate autoreactivity by two other T cells, 24 and 2C12, but in no case could T156I present α-GalCer. It is possible, therefore, that isoleucine at this position is compatible with the presentation of some autologous ligands but not with α-GalCer presentation.
Presentation of α-GalCer Analogs by Mutant mCD1.1 Molecules.
Three analogs that were inactive with wild-type mCD1.1, including ceramide, β-galactosylceramide, and α-mannosylceramide (8, 12, 15), likewise were unable to stimulate any of the T cell hybridomas when tested with the panel of APCs expressing mutant mCD1.1 molecules (data not shown). We also used an analog of α-GalCer, AGL 587, whose acyl chain is reduced to only 2 carbons (Fig. 2). This Ag presumably binds to mCD1.1 with the sphingosine, its only remaining hydrophobic structure. Two of the six Vα14+ hybridomas, 2C12 and 2D5, were not stimulated by AGL 587 presented by either wild-type mCD1.1 or any of the mCD1.1 mutants (data not shown). However, consistent with a previous report (15), the remaining four Vα14+ hybridomas could recognize AGL 587. The magnitude of the responses to wild-type mCD1.1 induced by 100 ng/ml AGL 587 generally were 2- to 10-fold weaker than the responses to α-GalCer tested in parallel. Although AGL 587 responses are weaker, eight of the mCD1.1 mutations were highly similar in their effects upon the responses to both α-GalCer and AGL 587(data not shown). By contrast, the responses to the bottom of the A′ pocket mutations F10A and C12G exhibit striking differences when AGL 587 and α-GalCer are compared. As shown in Fig. 2, the mutants F10A and C12G were able to support a level of α-GalCer stimulation of Vα14+ hybridomas close or equal to that observed with the clone 8 wild-type transfectant. The F10A mutant, however, was almost completely unable to present the truncated AGL 587 analog to these four hybridomas. Moreover, despite a slightly lower level of mCD1.1 expression compared with clone 8, the C12G mutant stimulated the AGL 587-responsive Vα14+ T cell hybridomas as well as the wild type, or more strongly (2- to 10-fold increase) in some cases.
Figure 2.

Glycosphingolipid Ag responses are affected by mutations at the bottom of the A′ pocket. (A) Structure of α-GalCer. AGL 587 is an α-GalCer analog with the 26-carbon acyl chain reduced to 2 carbons in length. (B) APCs expressing low levels of wild-type mCD1.1 (A20 wt CD1/8) or A′ pocket mutant mCD1.1 molecules were pulsed with 100 ng/ml either α-GalCer or AGL 587 before being cultured with indicated hybridomas. Cell-free supernatants were tested by ELISA for IL-2 content. One representative experiment of three is shown.
The opposite effects of the A′ pocket F10A and C12G mutations were further explored in dose–response experiments with the four AGL 587-responsive T cells (data not shown). The response to α-GalCer presented by the F10A mCD1.1 molecule was reduced compared with the wild-type clone 8, with an approximately 10-fold shift at lower Ag concentrations. The F10A mutant is unable to present AGL 587, however, even at the highest doses (300 ng/ml) that can be added to the tissue culture without causing toxicity due to the vehicle. The data suggest that the F10A A′ pocket mutation reduces the ability of the mCD1.1 molecule to interact with the sphingosine of AGL 587, placing the likely location of the sphingosine binding to the A′ pocket. The F10A mutation causes a more modest reduction in the reactivity to α-GalCer, however, presumably because the interaction of the mCD1.1 F′ pocket with the acyl chain partially compensates for the loss of binding energy in sphingosine binding to the mutant A′ pocket.
Conclusions.
The presentation of glycosphingolipids by mCD1.1 to NK T cells is a useful model for studying the molecular basis for lipid Ag recognition by T lymphocytes, because of the availability of both analogs of α-GalCer and mCD1.1-reactive cells with very closely related TCRs, and because the structure of mCD1.1 is known. The data presented here provide an outline of how the trimolecular complex of TCR, lipid, and mCD1.1 is formed. Regarding the TCR, we find preferential use of acidic and polar amino acids in the CDR3 region of the β chain of α-GalCer-reactive Vα14+ T cells, suggesting that some of these amino acids could be involved in hydrogen bond formation with the galactose moiety of the Ag. Although the α chain is most highly conserved, we find evidence for the involvement of TCR β in both mCD1.1 autoreactivity and α-GalCer plus mCD1.1 responses. Recent studies (25, 26) demonstrate that glycopeptides with mono- or disaccharides attached to the central region of the peptide allow for a conventional type of T cell recognition of carbohydrate plus α helical amino acids of the Ag-presenting molecule. We propose that the mechanism of contact by the NK T cell TCR is similar to that for glycopeptides, involving interactions of the TCR with both the mCD1.1 α helices and the galactose of α-GalCer. Possible contact points identified for TCR β at positions 76 and 79 located at the end of the mCD1.1 α1 helix are at least consistent with the diagonal orientation found for peptide plus MHC class I reactive TCRs (27). It remains to be determined, however, whether the strong conservation of the Vα–Jα junction in α-GalCer-reactive NK T cells is due to selection for interaction with a bound Ag.
Because mutations along the top of the helix and the bottom of the groove affect both autoreactivity and α-GalCer recognition, these processes are not likely to be similar to superantigen-type interactions. We also provide evidence that mCD1.1 autoreactive cells are heterogeneous, and by demonstrating that some of the autoreactive cells are affected by mutations in the bottom of the A′ pocket, the data suggest recognition of an mCD1.1-bound autologous ligand by these T cells. Analysis of mutations in the A′ pocket also indicates that α-GalCer binds to the groove of mCD1.1, and that it is most likely to do so with the sphingosine in the larger A′ pocket and the acyl chain in the narrower F′ pocket. If this were the case, then the entire 18-carbon sphingosine could be accommodated in the larger A′ pocket. The 26-carbon acyl chain is too long to fit entirely into the F′ pocket, although α-GalCer can be presented without intracellular Ag processing (10). The acyl chain therefore may extend out of the groove, with its terminal portion free, or it may interact with some other part of mCD1.1. It remains to be determined whether compounds with even longer hydrophobic chains will need to be processed to be bound in the CD1 groove or whether there is some other mechanism for accommodating the long hydrophobic chains of lipid Ags.
Supplementary Material
Acknowledgments
We thank Drs. Kyoko Hayakawa, Olga Naidenko, Jennifer Matsuda, and Yasuhiko Koezuka for support and helpful discussions, and Dr. Chung-Ryu Wang for the gift of the 5C6 hybridoma. This work was supported by National Institutes of Health Grants RO1 CA52511 (M.K.) and RO1 CA58896 (I.A.W.) and by a grant from the Association pour la Recherche contre le Cancer (N.B.). This is manuscript no. 313 from the La Jolla Institute for Allergy and Immunology.
Abbreviations
- Ag
antigen
- α-GalCer
α-galactosylceramide
- APCs
antigen-presenting cells
- CDR
complementarity-determining region
- h
human
- m
mouse
- mfi
mean fluorescence intensity
- NK
natural killer
- TCR
T cell antigen receptor
References
- 1.Porcelli S A, Modlin R L. Annu Rev Immunol. 1999;17:297–329. doi: 10.1146/annurev.immunol.17.1.297. [DOI] [PubMed] [Google Scholar]
- 2.Burdin N, Kronenberg M. Curr Opin Immunol. 1999;11:326–331. doi: 10.1016/s0952-7915(99)80052-1. [DOI] [PubMed] [Google Scholar]
- 3.Zeng Z, Castaño A R, Segelke B W, Stura E A, Peterson P A, Wilson I A. Science. 1997;277:339–345. doi: 10.1126/science.277.5324.339. [DOI] [PubMed] [Google Scholar]
- 4.Hughes A L. Mol Biol Evol. 1991;8:185–201. doi: 10.1093/oxfordjournals.molbev.a040640. [DOI] [PubMed] [Google Scholar]
- 5.Bendelac A, Rivera M N, Park S H, Roark J H. Annu Rev Immunol. 1997;15:535–562. doi: 10.1146/annurev.immunol.15.1.535. [DOI] [PubMed] [Google Scholar]
- 6.Eberl G, Lees R, Smiley S T, Taniguchi M, Grusby M J, MacDonald H R. J Immunol. 1999;162:6410–6419. [PubMed] [Google Scholar]
- 7.Bendelac A, Lantz O, Quimby M E, Yewdell J W, Bennink J R, Brutkiewicz R R. Science. 1995;268:863–865. doi: 10.1126/science.7538697. [DOI] [PubMed] [Google Scholar]
- 8.Kawano T, Cui J, Koezuka Y, Toura I, Kaneko Y, Motoki K, Ueno H, Nakagawa R, Sato H, Kondo E, Koseki H, Taniguchi M. Science. 1997;278:1626–1629. doi: 10.1126/science.278.5343.1626. [DOI] [PubMed] [Google Scholar]
- 9.Burdin N, Brossay L, Koezuka Y, Smiley S T, Grusby M J, Gui M, Taniguchi M, Hayakawa K, Kronenberg M. J Immunol. 1998;161:3271–3281. [PubMed] [Google Scholar]
- 10.Naidenko O V, Maher J K, Ernst W A, Sakai T, Modlin R L, Kronenberg M. J Exp Med. 1999;190:1069–1080. doi: 10.1084/jem.190.8.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brossay L, Chioda M, Burdin N, Koezuka Y, Casorati G, Dellabona P, Kronenberg M. J Exp Med. 1998;188:1521–1528. doi: 10.1084/jem.188.8.1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spada F M, Koezuka Y, Porcelli S A. J Exp Med. 1998;188:1529–1534. doi: 10.1084/jem.188.8.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Couedel C, Peyrat M A, Brossay L, Koezuka Y, Porcelli S A, Davodeau F, Bonneville M. Eur J Immunol. 1998;28:4391–4397. doi: 10.1002/(SICI)1521-4141(199812)28:12<4391::AID-IMMU4391>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 14.Moody D B, Reinhold B B, Guy M R, Beckman E M, Frederique D E, Furlong S T, Ye S, Reinhold V N, Sieling P A, Modlin R L, et al. Science. 1997;278:283–286. doi: 10.1126/science.278.5336.283. [DOI] [PubMed] [Google Scholar]
- 15.Brossay L, Naidenko O, Burdin N, Matsuda J, Sakai T, Kronenberg M. J Immunol. 1998;161:5124–5128. [PubMed] [Google Scholar]
- 16.Grant E P, Degano M, Rosat J P, Stenger S, Modlin R L, Wilson I A, Porcelli S A, Brenner M B. J Exp Med. 1999;189:195–205. doi: 10.1084/jem.189.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sakai T, Naidenko O V, Iijima H, Kronenberg M, Koezuka Y. J Med Chem. 1999;42:1836–1841. doi: 10.1021/jm990054n. [DOI] [PubMed] [Google Scholar]
- 18.Morita M, Natori T, Akimoto K, Osawa T, Fukushima H, Koezuka Y. Bioorg Med Chem Lett. 1995;5:699–704. [Google Scholar]
- 19.Masuda K, Makino Y, Cui J, Ito T, Tokuhisa T, Takahama Y, Koseki H, Tsuchida K, Koike T, Moriya H, Amano M, Taniguchi M. J Immunol. 1997;158:2076–2082. [PubMed] [Google Scholar]
- 20.Brossay L, Tangri S, Bix M, Cardell S, Locksley R, Kronenberg M. J Immunol. 1998;160:3681–3688. [PubMed] [Google Scholar]
- 21.Lantz O, Bendelac A. J Exp Med. 1994;180:1097–1106. doi: 10.1084/jem.180.3.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kawano T, Tanaka Y, Shimizu E, Kaneko Y, Kamata N, Sato H, Osada H, Sekiya S, Nakayama T, Taniguchi M. Int Immunol. 1999;11:881–887. doi: 10.1093/intimm/11.6.881. [DOI] [PubMed] [Google Scholar]
- 23.Quiocho F A. Annu Rev Biochem. 1986;55:287–315. doi: 10.1146/annurev.bi.55.070186.001443. [DOI] [PubMed] [Google Scholar]
- 24.Park S H, Roark J H, Bendelac A. J Immunol. 1998;160:3128–3134. [PubMed] [Google Scholar]
- 25.Ding Y H, Smith K J, Garboczi D N, Utz U, Biddison W E, Wiley D C. Immunity. 1998;8:403–411. doi: 10.1016/s1074-7613(00)80546-4. [DOI] [PubMed] [Google Scholar]
- 26.Speir J A, Abdel-Motal U M, Jondal M, Wilson I A. Immunity. 1999;10:51–61. doi: 10.1016/s1074-7613(00)80006-0. [DOI] [PubMed] [Google Scholar]
- 27.Garcia K C, Degano M, Pease L R, Huang M, Peterson P A, Teyton L, Wilson I A. Science. 1998;279:1166–1172. doi: 10.1126/science.279.5354.1166. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}