

Abstract
Endochondral ossification, the process by which most of the skeleton is formed, is a powerful system for studying various aspects of the biological response to degraded extracellular matrix (ECM). In addition, the dependence of endochondral ossification upon neovascularization and continuous ECM remodeling provides a good model for studying the role of the matrix metalloproteases (MMPs) not only as simple effectors of ECM degradation but also as regulators of active signal-inducers for the initiation of endochondral ossification. The daunting task of elucidating their specific role during endochondral ossification has been facilitated by the development of mice deficient for various members of this family. Here, we discuss the ECM and its remodeling as one level of molecular regulation for the process of endochondral ossification, with special attention to the MMPs.
In the past decade, multiple discoveries have facilitated our understanding of skeletal development. Transcription factors and/or growth factors that are involved in the genetic and molecular control of OSTEOGENESIS (see glossary box), CHONDROGENESIS and joint formation have been identified and their pathways partially elucidated [1]. The attention now turns to a different level of molecular regulation – that provided by the extracellular matrix (ECM) of the developing bone and the proteases that remodel it. Recent studies attest to the importance of unique composition of distinct ECMs within the developing skeleton, as well as their influence on cell differentiation and function [2–4].
Bone develops in two different ways. Mesenchymal cells can directly differentiate into bone by the process of INTRAMEMBRANOUS OSSIFICATION, which is responsible for the formation of most of the craniofacial skeleton. Alternatively, these cells can differentiate into cartilage, which then provides a template for bone morphogenesis by the process of ENDOCHONDRAL OSSIFICATION; this process is responsible for the formation of most of the vertebrate appendicular and axial skeleton (Figure 1a). Endochondral ossification occurs at two distinct sites in the vertebrate long bone – the primary (diaphyseal) and the secondary (epiphyseal) sites of ossification. Bone development initiates at the primary site. The secondary (epiphyseal) site is under independent control and is ossified later (Figure 1b). During this process, a new structure, the GROWTH PLATE, is formed between the DIAPHYSIS and the EPIPHYSIS by the segregation of CHONDROCYTES at different stages of differentiation. Under the control of signaling through Indian hedgehog (Ihh), bone morphogenetic proteins (BMPs) and fibroblast growth factor 18, a region of resting chondrocytes feeds into a zone of proliferating chondrocytes that then undergo hypertrophy and subsequently apoptosis. The ECM produced by and surrounding the terminally differentiated hypertrophic chondrocytes is calcified, partially degraded by the hypertrophic chondrocytes themselves [5], as well as by CHONDROCLASTS and/or preosteoclasts, and becomes permissive of blood-vessel invasion. Following vascular invasion, osteogenic progenitors are recruited to this area, where they consequently deposit TRABECULAR BONE [1].
Figure 1.
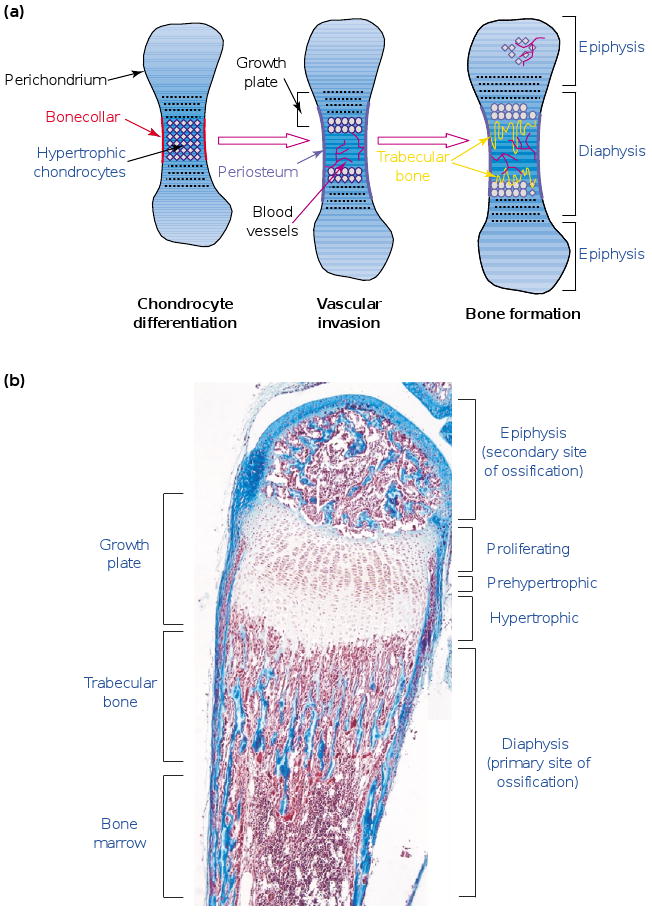
The process of endochondral ossification. (a) During endochondral ossification, mesenchymal cells differentiate into chondrocytes and lead to the formation of cartilage templates. Vascularization occurs around these templates, and osteoblasts differentiate around the central area in the bone collar. Chondrocytes in this central area differentiate into hypertrophic chondrocytes and allow vascular invasion. After complete differentiation, they die and extracellular matrix (ECM) remodeling is carried out by osteoclasts and chondroclasts recruited together with the blood vessels. This remodeling is necessary for trabecular-bone synthesis by osteoblasts precursors and for the formation of the bone-marrow cavity. (b) A section of a two-week-old mouse metatarsal stained with Masson trichrome. Chondrocytes proliferate and differentiate into prehypertrophic and hypertrophic chondrocytes. Endochondral ossification first takes place at the primary site at the center of the diaphysis, which allows formation of the two growth plates. The growth plates are ultimately responsible for the elongation of the long bones. Later, endochondral ossification occurs at a secondary site, in the epiphysis of the long bones. Scale bar represents 200 μm.
During endochondral ossification, an avascular tissue (cartilage) is gradually converted into one of the most highly vascularized tissues in the vertebrate body (bone). This conversion is dependent upon an angiogenic switch along the growth plate. A strong gradient of angiogenic factors, including vascular endothelial growth factor (VEGF), is established (Figure 2a); this is accompanied by changes in ECM synthesis and remodeling (Figure 2b) such as upregulation of collagen type X and matrix metalloprotease 13 (MMP 13) in hypertrophic chondrocytes. Thus, the process of ECM degradation is a key feature of both endochondral ossification and the subsequent bone remodeling.
Figure 2.

Endochondral ossification, extracellular matrix (ECM) remodeling and angiogenic switch. (a) The process of endochondral ossification is dependent upon neovascularization. During this process, cartilage is replaced by bone. The differentiation of chondrocytes into hypertrophic chondrocytes is accompanied by the expression of angiogenic growth factors, notably VEGF, allowing vascular invasion of the cartilage. In the diaphysis, this invasion is strictly limited to the front of ossification in the last row of hypertrophic chondrocytes in direct contact with blood vessels. (b) The process of endochondral ossification also depends upon a switch in the expression of ECM molecules. Proliferating chondrocytes express collagen type II, whereas hypertrophic chondrocytes express collagen type X, matrix metalloprotease 13 (MMP13) and high levels of angiogenic growth factors VEGF and CTGF. On the other side of the ossification front, osteoclasts express MMP9, whereas osteoblasts express MMP13 and collagen type I.
Among the key molecules acting in these processes, the MMPs have been implicated in the degradation of virtually all ECM molecules [6]. Recent analysis of transgenic mice deficient in MMP9 [7], MMP13 (Z. Werb; unpublished) and membrane-type 1 MMP (MT1-MMP) [8,9] has emphasized the importance of MMPs during particular stages of endochondral ossification. This review focuses on ECM remodeling by MMPs as a level of molecular regulation for the process of endochondral ossification. Special consideration is given to MMPs, in particular MMP9, MMP13 and MT1–MMP, as effectors and regulators of endochondral ossification.
Endochondral ossification and MMP expression
The MMP family of structurally related zinc-dependent proteases is able to cleave a variety of substrates including ECM proteins, extracellular non-ECM proteins and cell surface proteins [6]. MMPs are synthesized as secreted or transmembrane proenzymes and processed to an active form by the removal of an N-terminal propeptide. a covalent interaction between a cysteine residue in the N-terminal propeptide and the essential zinc ion bound to the catalytic domain of these proteases maintain them in the inactive conformations (Figure 3). Enzyme activation is generally accomplished by proteolytic cleavage of the propeptide [10]. For MT-MMPs, which contain a single-pass type I transmembrane domain, an extracellular N-terminal domain and a short cytoplasmic C-terminal domain, the activation might occur intracellularly in the secretory pathway through furin-like enzymes [11].
Figure 3.

Multiple roles of matrix metalloproteases (MMPs) in endochondral ossification. MMPs are directly involved in the remodeling of the extracellular matrix (ECM) through degradation of proteins such as collagens or proteoglycans. In addition, they have been implicated in more subtle functions such as apoptosis and the recruitment of cells, through the regulation of bioavailability and/or activation of factors sequestered into the ECM. For example, VEGF availability is regulated by this type of sequestration and release.
It has been known for decades that protease activity is present in the developing bone. Biochemical and immunohistochemical studies in the 1980s identified a collagenase and a collagenase inhibitor in the growth plate of embryonic chick and rat cells [5,12,13]. Subsequent immunolocalization studies in rabbits detected MMPs in distal femoral growth plate [14]. More recent developmental studies have described temporal and spatial expression patterns of specific MMPs and their inhibitors (Table 1). Several MMPs are expressed during endochondral ossification, including collagenases [MMP1 (human only) and MMP13], gelatinases (MMP2 and MMP9), stromelysins (MMP3 and MMP10) and a MT1-MMP; MMP9, MMP13 and, in particular, MT1-MMP are highly expressed during endochondral ossification. These MMPs can be regulated at multiple levels, including gene expression, spatial localization, zymogen activation and inhibition by naturally occurring inhibitors [e.g. tissue inhibitors of MMPs (TIMPs 1–4) which are also expressed during endochondral ossification] [15–20].
Table 1. MMP and TIMP expression during endochondral ossificationa.
| Cell type | MMP or TIMP expressed | Refs |
|---|---|---|
| Proliferating chondrocytes | MMP10 (Stromelysin 2) | [19,20,40,41] |
| TIMP1-4 | ||
| Hypertrophic chondrocytes | MMP10 (Stromelysin 2) | [19,20,24,25,40,41] |
| MMP13 (Collagenase 3) | ||
| TIMP1-4 | ||
| Osteoclasts and resorptive cells | MMP10 (Stromelysin 2) | [7,11,19,31,32] |
| MMP9 (Gelatinase B) | ||
| MMP14 (MT1-MMP) | ||
| Osteoblasts and bone-lining cells | MMP3 (Stromelysin 1) | [19,25–27] |
| MMP13 (Collagenase 3) | ||
| Osteocytes | MMP3 (Stromelysin 1) | [19] |
| Endothelial cells | MMP10 (Stromelysin 2) | [7,19] |
| MMP9 (Gelatinase B) | ||
| Monocytes | MMP10 (Stromelysin 2) | [7,21,31,32] |
| MMP9 (Gelatinase B) |
Abbreviations: MMP, matrix metalloproteases; MT1-MMP, membrane-type 1 MMP; TIMP, tissue inhibitor of matrix metalloproteases.
The MMPs expressed during endochondral ossification have been localized in a variety of temporospatial patterns. Pro-MMP3 (stromelysin-1) is present in the ECM along the PERIOSTEUM, whereas active MMP3 is present in OSTEOCYTES and OSTEOBLASTS. MMP10 (stromelysin-2) is expressed in resorptive cells at the chondro–osseous junction, OSTEOCLASTS, most mononuclear cells within the bone marrow, chondrocytes and blood vessels [21,19]. Although the expression patterns of these proteases suggest a potential role in endochondral ossification, no abnormal bone phenotype has been reported in mice deficient for either of these proteases.
During mouse embryogenesis, the expression of collagenolytic proteases is detected exclusively in the developing bone [22,23]. In these cells, the major collagenase expressed at the primary and secondary sites of ossification is MMP13. Detectable as early as 14.5 days of embryonic development in mice, MMP13 is highly expressed in the lower zone of hypertrophic cartilage, where its expression colocalizes with that of type X collagen [24]. At the chondro–osseous junction, MMP13 is expressed by the newly recruited osteoblasts adjacent to tartrate-resistant acid phosphatase (TRAP)-positive cells [25]. Particular intramembranous bones of the human fetus show the same expression pattern, with a strong expression first observed at 10 weeks of gestation [26] in osteoblasts lining the inner side of the calvaria [27]. At the secondary sites of ossification, MMP13 is expressed at the blind end of excavated canals formed by osteoclasts and along the walls of the forming-marrow space [28]. During bone formation, MMP13 transcription is controlled by Runx-2 (Cbfa-1) [29], a transcription factor of the runt gene family, by virtue of an osteoblast-specific element 2 (OSE2) site in the MMP13 promoter. In a model of fracture that recapitulates the steps of endochondral ossification, the observed expression pattern of MMP13 is similar to that detected during embryonic development [30].
The primary MMP involved in endochondral ossification is MMP9 (gelatinase B), which is highly expressed in monocytes, preosteoclasts and osteoclasts, as well as in chondroclastic cells of unknown origin, during mouse development [7,31,32]. Active MMP9 is concentrated at sites of cartilage resorption, proximal to the chondro–osseous junction, where vascular invasion occurs [7,33,34]. Analysis of MMP9−/− mice has pointed to a requirement for this protease during endochondral ossification, when it appears to coordinate ECM degradation, hypertrophic chondrocyte apoptosis, angiogenesis and osteoblastic differentiation [6]. By contrast, MMP2 (gelatinase A) is diffusely expressed in osteoblasts, bone marrow cells and periosteum. Surprisingly, humans in whom both MMP2 alleles are mutated display an osteolysis and arthritis syndrome typified by destruction and resorption of affected bones [35], similar to the phenotype of MT1-MMP−/− mice [8,9] described below. Although an abnormal bone phenotype has not been described in MMP2−/− mice [36], studies using different strains of mice are required as severity of bone phenotypes might be strain dependent.
The membrane localization of MT-MMPs makes them particularly suitable for pericellular proteolysis [10]. Among the six MT-MMPs identified, MT1-MMP is highly expressed in embryonic skeletal and periskeletal tissues [16,37]; similar to MMP9, its expression is particularly high in osteoclast precursors, osteoclasts and chondroclastic cells [38]. Analysis of MT1-MMP−/− mice indicates a role for this MMP in the degradation of noncalcified cartilage, a process distinct from that displayed by MMP9 [8,9]. Because MMPs contribute to ECM remodeling, it is reasonable to ask whether their inhibitors also play a role in this process. The major TIMP expressed during bone development is TIMP2 [16,39]; however, TIMP1, TIMP3 and TIMP4 are also expressed in skeletal elements during mouse and human development [15,20]. These inhibitors are mainly expressed by chondrocytes and bone-lining cells [20,40,41]. It is possible that the high concentration of these inhibitors contributes to the antiangiogenic properties of cartilage; however, no analysis of mice deficient for one or several TIMPs has reported abnormal bone phenotype.
MMPs and the growth of long bones
Endochondral ossification is a process that occurs from embryonic stages through adulthood and permits skeletal structures to be sustained during rapid bone growth. The crucial role of MMPs in this process can be observed by treating mice at 1–2 weeks of age (when the bones are growing very rapidly) with the broad-spectrum MMP inhibitor GM6001. This results in a dramatic expansion of hypertrophiccartilage in the growth plate at the primary site of ossification and an inhibition of the formation of secondary sites in the epiphysis (T.H. Vu, Z. Werb; unpublished). In addition, such experiments have demonstrated that MMPs are required for migration of preosteoclasts to the developing marrow cavity of primitive long bones [32]. Thus, the remodeling function of MMPs is crucial for the ability of endochondral ossification to perform more subtle functions such as directing the recruitment of different cell types, their communication and behavior. The development of mice deficient for one or several MMPs allows a better molecular understanding of protease function during bone formation.
Molecular insights from MMP-deficient mice
MMP9, MMP13 and MT1-MMP have been investigated for their involvement in endochondral ossification in developing bone using knockout mouse models [7,8,9]. There are many processes that these MMPs might mediate in this microenvironment including ECM degradation, vascular invasion, bioavailability of growth factors, chondrocyte apoptosis and recruitment of osteoprogenitor cells.
MMP9 is a key regulator of early growth plate angiogenesis and apoptosis of hypertrophic chondrocytes
MMP9−/− mice have a very pronounced phenotype at the primary site of ossification but little phenotype at the secondary site [7]. Compared with their wild-type litter-mates, they display substantially more hypertrophic cartilage, impaired endochondral ossification and a delay in the formation of the bone marrow cavity. In addition, these mice show a decrease in trabecular-bone formation, hypertrophic chondrocyte apoptosis and, in particular, early vascular invasion and osteoclast recruitment. This phenotype manifests during the developmental stage of rapid bone growth (from postnatal day one until four weeks of age); following this period, these mice fully recover. In older mice, the only persistent phenotype is the length of long bones, which is ∼10% shorter than that of wild-type mice. In addition to being expressed at the OSSIFICATION FRONT, MMP9 is expressed along the trabeculae and in osteoclasts. This expression pattern suggests a role for MMP9 not only in ECM degradation but also in invasion and migration of endothelial cells and osteoclasts. This is confirmed by studies that show a delay in the recruitment of osteoclasts and endothelial cells at the early stages of endochondral ossification in MMP9−/− mice. Moreover, this recruitment is under the influence of a major angiogenic factor, VEGF [42]. VEGF is made by the terminal hypertrophic chondrocytes under the control of Runx-2 [43] and is linked to the expression of another angiogenic factor, connective tissue growth factor (CTGF) [44]. The injection of a soluble VEGF receptor, the mFlt-1-Fc containing the terminal three Ig repeats of the receptor fused to the Fc portion of IgG, phenocopies the growth plate phenotype of MMP9−/− mice [45]. The same growth plate phenotype has been observed in mice expressing only the short isoform of VEGF [46,47]. In MMP9−/− mice, the amount of terminal hypertrophic chondrocytes increases and, according to in situ hybridization studies, a 10-fold increase in VEGF mRNA occurs. Moreover, in an in vitro angiogenesis assay, cartilage explants from MMP9−/− mice induce a delayed angiogenic response [7]. This implies a negative regulation of angiogenesis at high concentrations of VEGF. These observations led to the hypothesis that the protease regulates the availability of VEGF in hypertrophic chondrocytes, perhaps through its ECM remodeling function. Taken together, these observations point to a link between the functions of MMP9 and the VEGF system in coordinating processes involved in endochondral ossification. The precise mechanism for the involvement of MMP9 in controlling the bioavailability of VEGF during bone development is to be elucidated. However, MMP9 has many ECM substrates and the cleavage of one of these might activate VEGF function in situ. By contrast, CTGF−/− mice, which display a mild version of the MMP9−/− phenotype, show a decrease in VEGF expression in hypertrophic chondrocytes [48]. Thus, the molecular mechanisms coupling hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral ossification require further investigation.
After one week of postnatal development, an increase in osteoclast recruitment is observed in MMP9−/− mice. This correlates temporally with a decrease in trabecular-bone formation, suggesting that MMP9 might also play a role in the balance of osteoclast and/or osteoblast survival [6]. Another key feature of MMP9−/− mice is excessive hypertrophic cartilage, with a delay in the exit rate of chondrocytes from the late hypertrophic pool and an apparent defect in the apoptosis of hypertrophic chondrocytes [7]. Hypertrophic cartilages from wild-type and MMP9−/− mice show strikingly different protein expression patterns when compared by SDS–PAGE and silver-staining procedures. Indeed, many proteins accumulate in the absence of MMP9; these could be protein precursors or downstream signaling proteins that accumulate as a result of altered proteolysis. Among them, two substrates of MMP9, denatured collagen type II and galectin-3, might be key substrates controlling chondrocyte apoptosis (N. Ortega, Z. Werb; unpublished).
Experiments on fracture repair in the absence of MMP9 also suggest a role for this MMP in the regulation of chondrogenic and osteogenic cell differentiation during early stages of repair. Indeed, MMP9−/− mice generate a large cartilage callus even when fractured bones are stabilized, a situation usually repaired by intramembranous ossification without callus formation [49]. Intriguingly, stabilized fractures in MMP9−/− mice heal through endochondral rather than intramembranous ossification, suggesting that MMP9 might be involved in the mechanosensation that instructs the production of bone versus cartilage. Whether this indicates a switch in differentiation of mesenchymal progenitors or it is a manifestation of a defect in intramembranous ossification remains to be determined.
These results have implications for the role of MMP9 in the recruitment and mobilization of bone-marrow-derived stem, hematopoietic and endothelial precursors [50]. Indeed, while endochondral ossification is in progress, producing calcified bone from a cartilaginous template, hematopoietic cells also populate the bone marrow. In MMP9−/− mice, this process is delayed; however, at later stages, these mice display normal hematopoietic ability and have red- and white-blood-cell counts comparable to those of wild-type mice. The recruitment of hematopoietic stem cells, hematopoietic precursors and endothelial precursors from the bone marrow is a dynamic process that can be induced by various growth factors, including VEGF, in the presence of MMPs. In particular, VEGF cannot mobilize endothelial precursors into the blood circulation of wild-type mice treated with an MMP inhibitor or in MMP9−/− mice [50]. Activated MMP9 promotes the release of the stem-cell-active cytokine soluble Kit-ligand (sKitL) thereby driving stem, hematopoietic and endothelial progenitor cells from their quiescent niches to their proliferative niches within the bone marrow [50].
MMP9 and MMP13 act together for primary and secondary ossification
MMP9 and MMP13 are expressed in nonoverlapping cell types at the front of ossification [7,25]. MMP9 is expressed in osteoclasts, endothelial cells and other cell types, including bone marrow stromal cells, whereas MMP13 is expressed in the hypertrophic chondrocytes and osteoblasts. MMP9 is specifically required for the invasion of osteoclasts and endothelial cells into the discontinuously mineralized hypertrophic cartilage that fills the core of the diaphysis and is partially required for the passage of cells through unmineralized type I collagen of the nascent bone collar [42]. Early in postnatal development, MMP13−/− mice display a small increase in hypertrophic cartilage and trabecular bone (D. Stickens, Z. Werb; unpublished). Interestingly, mutant mice that have a collagenase-resistant collagen type I show no defect in the development of the appendicular skeleton during embryogenesis and the first week after birth, suggesting that the cleavage of collagen type I by a collagenolytic MMP might not be required for early endochondral ossification [51]. With age, these mice show increased endosteal bone deposition and osteocyte and osteoblast apoptosis. These observations confirm a role for collagenolytic MMPs in remodeling of mineralized ECM; however, the molecular mechanisms underlying these effects are not completely understood.
It has been proposed that MMP9 and MMP13 cooperate during the degradation of the unmineralized septa of hypertrophic chondrocytes [33]. Studies on MMP9−/− MMP13−/− mice support this hypothesis and show that these MMPs act synergistically in the initiation and development of primary and secondary sites of ossification. Indeed, these double-null mice, display delayed formation of bone marrow cavity, accumulation of hypertrophic cartilage and inhibition of trabecular-bone formation. The initiation of the secondary site of ossification is delayed until later than five weeks of postnatal development (Z. Werb; unpublished). Taken together, these data show that MMP9 and MMP13 are synergistic in their effects on bone development and remodeling.
MT1-MMP is essential for secondary ossification and chondrocyte proliferation
In contrast to MMP9−/− mice, MT1-MMP−/− mice have a very abnormal phenotype both at the site of intramembranous ossification and at the secondary site of ossification but little abnormal phenotype at the primary site of ossification. The particularly severe phenotype in these mice is increased neonatal mortality; those that survive develop skeletal dysplasia, arthritis and osteopenia [8]. Intramembranous ossification, as well as endochondral ossification, is disturbed with persistence of parietal cartilage and delayed initiation of endochondral ossification in the epiphyses; however, these mice do not display any obvious defects in the early process of endochondral ossification at the primary site. Only a slight delay with a very transient increase in hypertrophic zone was reported in the primary ossification sites of MT1-MMP−/− mice, whereas the vascularization of the growth plate at the chondro–osseous junction is normal [9]. Later in development and subsequent to the delay in secondary-site ossification, proliferation of chondrocytes in the growth plate at the primary site is impaired. The progressive osteopenia observed in these mice results from a decrease in bone deposition, as well as an increase in bone resorption, with a high number of osteoclasts at the chondro–osseous junction. Other studies have shown that osteoblast survival is actually regulated by MT1-MMP through activation of transforming growth factor-β (TGFβ) [52]. Interestingly, TGFβ has also been implicated in the process of differentiation of hypertrophic chondrocyte through the upregulation of parathyroid-hormone-related protein (PTHrP) [53]. PTHrP and another morphogenic factor, Ihh, regulate chondrocyte differentiation through the establishment of a negative feedback loop. Prehypertrophic chondrocytes differentiating into hypertrophic chondrocytes secrete Ihh, which induces PTHrP expression in the PERICHONDRIUM, which in turn inhibits hypertrophic differentiation [54,55]. Thus, MMPs might be upstream regulators of this important negative feedback loop, regulating TGFβ availability and, thereby adding another level of complexity to this system of molecular regulation.
Concluding remarks
The proteolytic activity of MMPs is essential for normal endochondral ossification. Indeed, MMPs act not only on matrix remodeling but also on more subtle functions such as directing the recruitment of different cell types, their communication and behavior (Figure 3). Many important regulatory factors are sequestered in the ECM and require MMP activity to be released and/or activated [56,57]. Therefore, the abnormal bone phenotypes observed in MMP-deficient mice and humans could be derived, at least in part, from the lack of activity of these sequestered factors. Even if MMP9 deficiency only partially phenocopies VEGF deprivation, it is clear that VEGF provides a molecular linkage between the degradation of the ECM, vascular invasion and hypertrophic chondrocyte apoptosis. In addition, because VEGF receptors are expressed on osteogenic precursors, osteoclasts and endothelial cells, it is possible that ECM remodeling is under strict regulation involving MMPs and VEGF release and/or activation. In addition to the regulation of growth factor bioavailability by MMPs, ECM remodeling also induces variations in calcium and phosphate ion concentrations. Interestingly, Adams and coworkers [58] demonstrated that, depending upon their extracellular concentrations, these ions could also regulate chondrocyte and osteoblast apoptosis. Thus, growth factors and ions might be involved in closely related molecular pathways that govern the equilibrium between survival and apoptosis of skeletal cells.
Determining the exact molecular targets of the MMPs during endochondral ossification will provide insights into this process and how it is regulated during normal bone development, repair and homeostasis. In addition to allowing for the elucidation of molecular mechanisms, these studies might also contribute to the understanding of the pathogenesis of diseases characterized by impaired skeletal growth particularly CHONDRODYSPLASIA, including RICKETS and OPSISMODYSPLASIA.
Acknowledgments
This work was supported by grants from the NIH(AG23218, AR46238 and DE13058). We apologize for our failure to cite many of the important and relevant studies in this field due to space limitations.
Glossary
- Chondroclast
Mononuclear cell implicated in the resorption of the transverse septa of hypertrophic cartilage; the precise origin of these cells is unknown.
- Chondrocyte
Unique cellular component of cartilage, which occupies a lacuna surrounded by a stiff flexible matrix formed from collagen fibers embedded in a cartilage proteoglycan gel.
- Chondrodysplasia
Abnormal cartilage development characteristic of many bone and cartilage disorders.
- Chondrogenesis
Cartilage development, especially chondrocyte differentiation.
- Diaphysis
Shaft of a long bone.
- Endochondral Ossification
Ossification over a cartilage template responsible for long-bone growth at the diaphyseal cartilage (center of long bone) and enlargement of epiphyses at the epiphyseal cartilage (ends of long bone).
- Epiphysis
End (head) of a long bone.
- Growth Plate
Cartilage zone between the diaphysis and the epiphysis, which allows for longitudinal bone growth. This structure can be temporary and disappear in adulthood, or can persist in some bones.
- Intramembranous Ossification
Ossification in the absence of a cartilage template, whereby bone develops directly from mesenchymal tissues.
- Opsismodysplasia
Pathology caused by chondrodysplasia and characterized by short hands and feet and thin lamellar vertebral bodies. In this disorder, cartilages show widening of the area containing hypertrophic chondrocytes.
- Osteoblast
Bone-forming cell, which secretes collagen type I and participates in the deposition of mineralized bone.
- Ossification Front
Area between hypertrophic chondrocytes and bone marrow, where bone trabeculae are initiated.
- Osteoclast
Multinucleated cell derived from monocytic lineage by fusion of precursor cells. They participate in the breakdown and resorption of bone and have been implicated in the resorption of the longitudinal septa of hypertrophic cartilages.
- Osteocyte
Mature bone cell, embedded within the calcified bone matrix, that participates in the maintenance of bone as a living tissue.
- Osteogenesis
The development of bone, with particular attention to the differentiation of and interactions/interplay between the cell types involved.
- Perichondrium
Membrane of fibrous connective tissue surrounding cartilage template.
- Periosteum
Dense fibrous membrane that covers the diaphysis and contains nerves and blood vessels necessary for the nutrition of the bone. This membrane also serves for attachment of muscles and tendons.
- Rickets
Childhood disease caused by deficiency of vitamin D and sunlight and associated with impaired metabolism of calcium and phosphorus. This disorder is characterized by soft and weakened bones showing a widened area of hypertrophic chondrocytes.
- Trabecular Bone
Mineralized connective tissue fibers forming bone spicules organized in a spongy latticework, particularly below hypertrophic cartilage.
References
- 1.Karsenty G, Wagner EF. Reaching a genetic and molecular understanding of skeletal development. Dev Cell. 2002;2:389–406. doi: 10.1016/s1534-5807(02)00157-0. [DOI] [PubMed] [Google Scholar]
- 2.Costell M, et al. Perlecan maintains the integrity of cartilage and some basement membranes. J Cell Biol. 1999;147:1109–1122. doi: 10.1083/jcb.147.5.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arikawa-Hirasawa E, et al. Perlecan is essential for cartilage and cephalic development. Nat Genet. 1999;23:354–358. doi: 10.1038/15537. [DOI] [PubMed] [Google Scholar]
- 4.Jacenko O, et al. A dominant interference collagen X mutation disrupts hypertrophic chondrocyte pericellular matrix and glycosaminoglycan and proteoglycan distribution in transgenic mice. Am J Pathol. 2001;159:2257–2269. doi: 10.1016/S0002-9440(10)63076-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blair HC, et al. Mechanisms balancing skeletal matrix synthesis and degradation. Biochem J. 2002;364:329–341. doi: 10.1042/BJ20020165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 7.Vu TH, et al. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell. 1998;93:411–422. doi: 10.1016/s0092-8674(00)81169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holmbeck K, et al. MT1-MMP-deficient mice develop dwarfism, osteopenia, arthritis, and connective tissue disease due to inadequate collagen turnover. Cell. 1999;99:81–92. doi: 10.1016/s0092-8674(00)80064-1. [DOI] [PubMed] [Google Scholar]
- 9.Zhou Z, et al. Impaired endochondral ossification and angiogenesis in mice deficient in membrane-type matrix metalloproteinase I. Proc Natl Acad Sci U S A. 2000;97:4052–4057. doi: 10.1073/pnas.060037197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van Wart HE, Birkedal-Hansen H. The cysteine switch: a principle of regulation of metalloproteinase activity with potential applicability to the entire matrix metalloproteinase gene family. Proc Natl Acad Sci U S A. 1990;87:5578–5582. doi: 10.1073/pnas.87.14.5578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sato H, et al. Activation of a recombinant membrane type 1-matrix metalloproteinase (MT1-MMP) by furin and its interaction with tissue inhibitor of metalloproteinases (TIMP)-2. FEBS Lett. 1996;393:101–104. doi: 10.1016/0014-5793(96)00861-7. [DOI] [PubMed] [Google Scholar]
- 12.Yasui N, et al. Production of collagenase inhibitor by the growth cartilage of embryonic chick bone: isolation and partial characterization. Coll Relat Res. 1981;1:59–72. doi: 10.1016/s0174-173x(80)80008-2. [DOI] [PubMed] [Google Scholar]
- 13.Dean DD, et al. Localization of collagenase in the growth plate of rachitic rats. J Clin Invest. 1985;76:716–722. doi: 10.1172/JCI112026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown CC, et al. Immunolocalization of metalloproteinases and their inhibitor in the rabbit growth plate. J Bone Joint Surg Am. 1989;71:580–593. [PubMed] [Google Scholar]
- 15.Flenniken AM, Williams BR. Developmental expression of the endogenous TIMP gene and a TIMP-lacZ fusion gene in transgenic mice. Genes Dev. 1990;4:1094–1106. doi: 10.1101/gad.4.7.1094. [DOI] [PubMed] [Google Scholar]
- 16.Apte SS, et al. The matrix metalloproteinase-14 (MMP-14) gene is structurally distinct from other MMP genes and is co-expressed with the TIMP-2 gene during mouse embryogenesis. J Biol Chem. 1997;272:25511–25517. doi: 10.1074/jbc.272.41.25511. [DOI] [PubMed] [Google Scholar]
- 17.Zeng Y, et al. Temporal and spatial regulation of gene expression mediated by the promoter for the human tissue inhibitor of metalloproteinases-3 (TIMP-3)-encoding gene. Dev Dyn. 1998;211:228–237. doi: 10.1002/(SICI)1097-0177(199803)211:3<228::AID-AJA4>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 18.Airola K, et al. Human TIMP-3 is expressed during fetal development, hair growth cycle, and cancer progression. J Histochem Cytochem. 1998;46:437–447. doi: 10.1177/002215549804600403. [DOI] [PubMed] [Google Scholar]
- 19.Bord S, et al. Tissue inhibitor of matrix metalloproteinase-1 (TIMP-1) distribution in normal and pathological human bone. Bone. 1999;24:229–235. doi: 10.1016/s8756-3282(98)00174-4. [DOI] [PubMed] [Google Scholar]
- 20.Huang W, et al. Tissue inhibitor of metalloproteinases-4 (TIMP-4) gene expression is increased in human osteoarthritic femoral head cartilage. J Cell Biochem. 2002;85:295–303. doi: 10.1002/jcb.10138. [DOI] [PubMed] [Google Scholar]
- 21.Bord S, et al. Stromelysin-1 (MMP-3) and stromelysin-2 (MMP-10) expression in developing human bone: potential roles in skeletal development. Bone. 1998;23:7–12. doi: 10.1016/s8756-3282(98)00064-7. [DOI] [PubMed] [Google Scholar]
- 22.Mattot V, et al. Expression of interstitial collagenase is restricted to skeletal tissue during mouse embryogenesis. J Cell Sci. 1995;108:529–535. doi: 10.1242/jcs.108.2.529. [DOI] [PubMed] [Google Scholar]
- 23.Gack S, et al. Expression of interstitial collagenase during skeletal development of the mouse is restricted to osteoblast-like cells and hypertrophic chondrocytes. Cell Growth Differ. 1995;6:759–767. [PubMed] [Google Scholar]
- 24.Tuckermann JP, et al. Collagenase-3 (MMP-13) and integral membrane protein 2a (Itm2a) are marker genes of chondrogenic/osteoblastic cells in bone formation: sequential temporal, and spatial expression of Itm2a, alkaline phosphatase, MMP-13, and osteocalcin in the mouse. J Bone Miner Res. 2000;15:1257–1265. doi: 10.1359/jbmr.2000.15.7.1257. [DOI] [PubMed] [Google Scholar]
- 25.Hayami T, et al. Spatiotemporal change of rat collagenase (MMP-13) mRNA expression in the development of the rat femoral neck. J Bone Miner Metab. 2000;18:185–193. doi: 10.1007/s007740070019. [DOI] [PubMed] [Google Scholar]
- 26.Stahle-Backdahl M, et al. Collagenase-3 (MMP-13) is expressed during human fetal ossification and re-expressed in postnatal bone remodeling and in rheumatoid arthritis. Lab Invest. 1997;76:717–728. [PubMed] [Google Scholar]
- 27.Johansson N, et al. Collagenase-3 (MMP-13) is expressed by hypertrophic chondrocytes, periosteal cells, and osteoblasts during human fetal bone development. Dev Dyn. 1997;208:387–397. doi: 10.1002/(SICI)1097-0177(199703)208:3<387::AID-AJA9>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 28.Davoli MA, et al. Enzymes active in the areas undergoing cartilage resorption during the development of the secondary ossification center in the tibiae of rats aged 0-21 days: II. Two proteinases, gelatinase B and collagenase-3, are implicated in the lysis of collagen fibrils. Dev Dyn. 2001;222:71–88. doi: 10.1002/dvdy.1160. [DOI] [PubMed] [Google Scholar]
- 29.Jimenez MJ, et al. Collagenase 3 is a target of Cbfa1, a transcription factor of the runt gene family involved in bone formation. Mol Cell Biol. 1999;19:4431–4442. doi: 10.1128/mcb.19.6.4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Uusitalo H, et al. Expression of cathepsins B, H, K, L, and S and matrix metalloproteinases 9 and 13 during chondrocyte hypertrophy and endochondral ossification in mouse fracture callus. Calcif Tissue Int. 2000;67:382–390. doi: 10.1007/s002230001152. [DOI] [PubMed] [Google Scholar]
- 31.Reponen P, et al. High expression of 92-kD type IV collagenase (gelatinase B) in the osteoclast lineage during mouse development. J Cell Biol. 1994;124:1091–1102. doi: 10.1083/jcb.124.6.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blavier L, Delaisse JM. Matrix metalloproteinases are obligatory for the migration of preosteoclasts to the developing marrow cavity of primitive long bones. J Cell Sci. 1995;108:3649–3659. doi: 10.1242/jcs.108.12.3649. [DOI] [PubMed] [Google Scholar]
- 33.Lee ER, et al. Active gelatinase B is identified by histozymography in the cartilage resorption sites of developing long bones. Dev Dyn. 1999;215:190–205. doi: 10.1002/(SICI)1097-0177(199907)215:3<190::AID-AJA2>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 34.Gustafsson E, et al. Role of collagen type II and perlecan in skeletal development. Ann N Y Acad Sci. 2003;995:140–150. doi: 10.1111/j.1749-6632.2003.tb03217.x. [DOI] [PubMed] [Google Scholar]
- 35.Martignetti JA, et al. Mutation of the matrix metalloproteinase 2 gene (MMP2) causes a multicentric osteolysis and arthritis syndrome. Nat Genet. 2001;28:261–265. doi: 10.1038/90100. [DOI] [PubMed] [Google Scholar]
- 36.Itoh T, et al. Reduced angiogenesis and tumor progression in gelatinase A-deficient mice. Cancer Res. 1998;58:1048–1051. [PubMed] [Google Scholar]
- 37.Kinoh H, et al. MT-MMP, the cell surface activator of proMMP-2 (pro-gelatinase A), is expressed with its substrate in mouse tissue during embryogenesis. J Cell Sci. 1996;109:953–959. doi: 10.1242/jcs.109.5.953. [DOI] [PubMed] [Google Scholar]
- 38.Sato T, et al. Identification of the membrane-type matrix metalloproteinase MT1-MMP in osteoclasts. J Cell Sci. 1997;110:589–596. doi: 10.1242/jcs.110.5.589. [DOI] [PubMed] [Google Scholar]
- 39.Blavier L, DeClerck YA. Tissue inhibitor of metalloproteinases-2 is expressed in the interstitial matrix in adult mouse organs and during embryonic development. Mol Biol Cell. 1997;8:1513–1527. doi: 10.1091/mbc.8.8.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Joronen K, et al. Temporospatial expression of tissue inhibitors of matrix metalloproteinases-1, -2 and -3 during development, growth and aging of the mouse skeleton. Histochem Cell Biol. 2000;114:157–165. doi: 10.1007/s004180000177. [DOI] [PubMed] [Google Scholar]
- 41.Dew G, et al. Localisation of matrix metalloproteinases and TIMP-2 in resorbing mouse bone. Cell Tissue Res. 2000;299:385–394. doi: 10.1007/s004419900166. [DOI] [PubMed] [Google Scholar]
- 42.Engsig MT, et al. Matrix metalloproteinase 9 and vascular endothelial growth factor are essential for osteoclast recruitment into developing long bones. J Cell Biol. 2000;151:879–889. doi: 10.1083/jcb.151.4.879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zelzer E, et al. Tissue specific regulation of VEGF expression during bone development requires Cbfa1/Runx2. Mech Dev. 2001;106:97–106. doi: 10.1016/s0925-4773(01)00428-2. [DOI] [PubMed] [Google Scholar]
- 44.Ivkovic S, et al. Connective tissue growth factor coordinates chondrogenesis and angiogenesis during skeletal development. Development. 2003;130:2779–2791. doi: 10.1242/dev.00505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gerber HP, et al. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat Med. 1999;5:623–628. doi: 10.1038/9467. [DOI] [PubMed] [Google Scholar]
- 46.Maes C, et al. Impaired angiogenesis and endochondral bone formation in mice lacking the vascular endothelial growth factor isoforms VEGF164 and VEGF188. Mech Dev. 2002;111:61–73. doi: 10.1016/s0925-4773(01)00601-3. [DOI] [PubMed] [Google Scholar]
- 47.Zelzer E, et al. Skeletal defects in VEGF(120/120) mice reveal multiple roles for VEGF in skeletogenesis. Development. 2002;129:1893–1904. doi: 10.1242/dev.129.8.1893. [DOI] [PubMed] [Google Scholar]
- 48.Ivkovic S, et al. Connective tissue growth factor coordinates chondrogenesis and angiogenesis during skeletal development. Development. 2003;130:2779–2791. doi: 10.1242/dev.00505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Colnot C, et al. Altered fracture repair in the absence of MMP9. Development. 2003;130:4123–4133. doi: 10.1242/dev.00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Heissig B, et al. Recruitment of stem and progenitor cells from the bone marrow niche requires MMP-9 mediated release of kit-ligand. Cell. 2002;109:625–637. doi: 10.1016/s0092-8674(02)00754-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhao W, et al. Osteocyte and osteoblast apoptosis and excessive bone deposition accompany failure of collagenase cleavage of collagen. J Clin Invest. 2000;106:941–949. doi: 10.1172/JCI10158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Karsdal MA, et al. Matrix metalloproteinase-dependent activation of latent transforming growth factor-beta controls the conversion of osteoblasts into osteocytes by blocking osteoblast apoptosis. J Biol Chem. 2002;277:44061–44067. doi: 10.1074/jbc.M207205200. [DOI] [PubMed] [Google Scholar]
- 53.Serra R, et al. Parathyroid hormone-related peptide (PTHrP)-dependent and -independent effects of transforming growth factor beta (TGF-beta) on endochondral bone formation. J Cell Biol. 1999;145:783–794. doi: 10.1083/jcb.145.4.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lanske B, et al. PTH/PTHrP receptor in early development and Indian hedgehog-regulated bone growth. Science. 1996;273:663–666. doi: 10.1126/science.273.5275.663. [DOI] [PubMed] [Google Scholar]
- 55.Vortkamp A, et al. Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science. 1996;273:613–622. doi: 10.1126/science.273.5275.613. [DOI] [PubMed] [Google Scholar]
- 56.Bergers G, et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol. 2000;2:737–744. doi: 10.1038/35036374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yu Q, et al. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000;14:163–176. [PMC free article] [PubMed] [Google Scholar]
- 58.Adams CS, et al. Matrix regulation of skeletal cell apoptosis. Role of calcium and phosphate ions. J Biol Chem. 2001;276:20316–20322. doi: 10.1074/jbc.M006492200. [DOI] [PubMed] [Google Scholar]