

Abstract
The mechanisms of free fatty acid-induced lipoapoptosis are incompletely understood. Here we demonstrate that Mcl-1, an anti-apoptotic member of the Bcl-2 family, was rapidly degraded in hepatocytes in response to palmitate and stearate by a proteasome-dependent pathway. Overexpression of a ubiquitin-resistant Mcl-1 mutant in Huh-7 cells attenuated palmitate-mediated Mcl-1 loss and lipoapoptosis; conversely, short hairpin RNA-targeted knockdown of Mcl-1 sensitized these cells to lipoapoptosis. Palmitate-induced Mcl-1 degradation was attenuated by the novel protein kinase C (PKC) inhibitor rottlerin. Of the two human novel PKC isozymes, PKCδ and PKCθ, only activation of PKCθ was observed by phospho-immunoblot analysis. As compared with Jurkat cells, a smaller PKCθ polypeptide and mRNA were expressed in hepatocytes consistent with an alternative splice variant. Short hairpin RNA-mediated knockdown of PKCθ reduced Mcl-1 degradation and lipoapoptosis. Likewise, genetic deletion of Pkcθ also attenuated Mcl-1 degradation and cytotoxicity by palmitate in primary hepatocytes. During treatment with palmitate, rottlerin inhibited phosphorylation of Mcl-1 at Ser159, a phosphorylation site previously implicated in Mcl-1 turnover. Consistent with these results, an Mcl-1 S159A mutant was resistant to degradation and improved cell survival during palmitate treatment. Collectively, these results implicate PKCθ-dependent destabilization of Mcl-1 as a mechanism contributing to hepatocyte lipoapoptosis.
Current evidence suggests that hepatic steatosis is present in up to 30% of the American population (1). A subset of these individuals develop severe hepatic lipotoxicity, a syndrome referred to as NASH2 (2), which can progress to cirrhosis and its chronic sequela (3, 4). A major risk factor for hepatic lipotoxicity is insulin resistance (5–7), resulting in excessive lipolysis within peripheral adipose tissue with release of high levels of free fatty acids (FFA) to the circulation. Circulating FFA are taken up by the liver via fatty acid transporter 5 and CD36 (8–10), and the bulk of hepatic neutral fat is derived from re-esterification of circulating FFA (8). Current concepts indicate that FFA, and not their esterified product (triglyceride), mediate hepatic lipotoxicity (11, 12). Elevated serum FFA correlate with liver disease severity (13–15), and therapies that enhance insulin sensitivity ameliorate hepatic lipotoxicity, in part, by decreasing plasma FFA (16). Hepatic FFA also accumulate in experimental steatohepatitis, further supporting a role for these nutrients in hepatic lipotoxicity (17). Saturated FFA are more strongly implicated in hepatic lipotoxicity than unsaturated FFA (18, 19). Saturated FFA induce hepatocyte apoptosis (20, 21), a cardinal feature of nonalcoholic fatty liver disease (22), and serum biomarkers of apoptosis are useful for identifying hepatic lipotoxicity (23). Thus, FFA-mediated lipotoxicity occurs, in part, by apoptosis.
Apoptosis is regulated by members of the Bcl-2 protein family (24). These proteins can be categorized into three subsets as follows: the guardians or anti-apoptotic members of this family, which include Bcl-2, A1, Mcl-1, Bcl-xL, and Bcl-w; the multidomain executioners or proapoptotic members of this family, which include Bax and Bak; and the messengers or biosensors of cell death, which share only the third Bcl-2 homology domain and are referred to as BH3-only proteins. This last group of proteins includes Bid, Bim, Bmf, Puma, Noxa, Hrk, Bad, and Bik. We have previously reported that cytotoxic FFA induce Bim expression by a FoxO3a-dependent mechanism that contributes, in part, to lipoapoptosis by activating Bax (20, 21). However, Bax activation can be held in check by anti-apoptotic members of the Bcl-2 family suggesting their function may also be dysregulated during FFA-mediated cytotoxicity.
Bcl-2 is not expressed in hepatocytes at the protein level (25), whereas Bcl-w and Bfl-1/A1 knock-out mice have no liver phenotype (26–28). However, both potent anti-apoptotic proteins Bcl-xL and Mcl-1 are expressed by hepatocytes and exhibit a liver phenotype in knock-out mice (29, 30), whereas up-regulation of Mcl-1 renders hepatocytes resistant to apoptosis (31–33). It has also been posited that cellular elimination of Mcl-1 is a critical step in certain proapoptotic cascades (34, 35). Mcl-1 is unique among Bcl-2 proteins in that it has a short half-life, 30–120 min in most cell types, due to the presence of two sequences rich in proline, glutamic acid, serine, and threonine, which target the protein for rapid degradation by the proteasome (36). Proteasomal degradation of Mcl-1 is promoted by ubiquitination, which in turn is regulated by various kinase cascades (36). Despite its potential importance, a role for Mcl-1 in regulating hepatocyte FFA-mediated lipoapoptosis remains unexplored.
Given that FFA induce insulin resistance (37), the kinases potentially regulating lipoapoptosis are likely those also identified in insulin resistance syndromes, especially the novel PKC isoforms PKCδ and PKCθ (38). The novel PKC isoforms are activated by diacylglycerol, which rises in the presence of FFA (39–41), and diacylglycerol levels are significantly increased in NASH (42). A role for PKCδ in apoptosis has not been described. PKCθ has recently been shown to be activated by endoplasmic reticulum stress in liver cells (43) and lipids in vivo (44, 45). Furthermore, PKCθ has also been implicated in apoptosis of Jurkat cells, neuroblastoma cells, and myeloid leukemia cells (46, 47). However, neither its role in mediating lipoapoptosis nor modulating levels/activity of Bcl-2 proteins has been examined.
This study addresses the role of Mcl-1 and PKCθ in FFA-induced lipoapoptosis. We identify a pathway that involves PKCθ-dependent proteasomal degradation of Mcl-1. Using inhibitors of various steps along this pathway, along with Mcl-1 mutants that are resistant to proteasomal degradation or Ser159 phosphorylation, our studies implicate Mcl-1 degradation via a PKCθ-dependent process as a critical step in lipoapoptosis.
EXPERIMENTAL PROCEDURES
Materials
Isopropyl alcohol and bovine serum albumin were from Sigma. Rottlerin (a novel PKC inhibitor) and glycogen synthase kinase-3β (GSK-3β) inhibitor IX were from Calbiochem. Z-VAD-fmk was from Enzyme Systems (Livermore, CA).
Cell Culture
Huh-7 cells, a human hepatoma cell line, and primary mouse hepatocytes were cultured in Dulbecco's modified Eagle's medium containing high glucose (25 mm), 100,000 units/liter penicillin, 100 mg/liter streptomycin, and 10% (v/v) heat-inactivated fetal bovine serum. Mouse hepatocytes were isolated from C57BL/6 wild type (The Jackson Laboratory, Bar Harbor, ME), human MCL-1 transgenic (Ruth W. Craig, Dartmouth University, Hanover, NH), and Pkcθ−/− mice (The Jackson Laboratories) by collagenase perfusion, purified by Percoll gradient centrifugation, and plated as primary cultures as we described previously (48).
Fatty Acid Treatment
Palmitate, stearate, and oleate (Sigma) were individually dissolved in isopropyl alcohol at a concentration of 80 mm. The concentration of the vehicle, isopropyl alcohol, was 1% in the final incubations. For all studies employing FFA, the media contained 1% (w/v) bovine serum albumin to ensure a ratio between bound and unbound FFA analogous to the ratio present in human plasma (49). The final concentration of fatty acids used ranged from 0 to 800 μm based on concentrations found in human NASH (13, 16, 50). Although these concentrations are similar to total fasting FFA plasma concentrations found in the pathophysiologic condition of NASH, the concentrations are elevated compared with each species of FFA present in human plasma.
Immunoblot Analysis
Cells were incubated for 20 min on ice with lysis buffer (50 mm Tris-HCl, pH 7.4; 1% (w/v) Nonidet P-40; 0.25% (w/v) sodium deoxycholate; 150 mm NaCl; 1 mm EDTA; 1 mm phenylmethylsulfonyl fluoride; 1 μg/ml aprotinin, leupeptin, and pepstatin; 1 mm Na3VO4; 1 mm NaF) and centrifuged at 14,000 × g for 15 min at 4 °C. Protein concentration was determined by the Bradford assay (Sigma), and aliquots containing 80 μg of protein were electrophoretically resolved by SDS-PAGE on a pre-cast 12% acrylamide gel (Bio-Rad) and immobilized on nitrocellulose membrane. Nonspecific binding sites were blocked with 5% (w/v) nonfat dry milk in Tris-buffered saline (20 mm Tris, 150 mm NaCl, pH 7.4) containing 0.1% (w/v) Tween 20. Blots were incubated with primary antibodies at room temperature for 2 h or at 4 °C overnight and then, after washing, with secondary antibodies at room temperature for 1 h.
The following primary antibodies were employed: rabbit polyclonal antibody, which is strongly reactive to human but not murine Mcl-1 at 1:1000 (sc-819, Santa Cruz Biotechnology, Santa Cruz, CA); rabbit anti-mouse Mcl-1 (600-401-394, Rockland, Gilbertsville, PA); monoclonal mouse anti-human PKCθ (clone 27, Pharmingen); polyclonal rabbit anti-human PKCθ (2059, Cell Signaling Technology, Danvers, MA; data not shown); polyclonal rabbit anti-human phospho-Thr538 PKCθ (9377, Cell Signaling Technology); polyclonal rabbit anti-human PKCδ (2058, Cell Signaling Technology); polyclonal rabbit anti-mouse phospho-Thr505 PKCδ (9374, Cell Signaling Technology); polyclonal goat anti-β-actin antibody at 1:2000 (C11, Santa Cruz Biotechnology); and mouse monoclonal anti-γ-tubulin (T6557, Sigma). Immune complexes were visualized using the Odyssey infrared imaging system (LI-COR Biosciences, Lincoln, NE) or a chemiluminescent substrate (ECL, Amersham Biosciences) and Kodak X-Omat film (Eastman Kodak Co.). Quantitation of immune complexes was performed using Odyssey 3.0 software per the manufacturer's protocol.
RNA Isolation, Reverse Transcription, and Quantitative Real Time PCR
Total RNA was isolated from Huh-7 cells using TRIzol (Invitrogen) per the manufacturer's protocol. RNA concentration was estimated by measuring absorbance at 260 nm using a NanoDrop spectrophotometer (Thermo Fisher Scientific, Wilmington, DE), and 1 μg of total RNA was reverse-transcribed using the SuperScript® III first-strand synthesis system (Invitrogen) and random hexamer primers per the manufacturer's protocol. A 1.5-μl aliquot of a 20-μl reverse transcription reaction was subjected to real time PCR analysis. PCR primers used for human MCL-1 were as follows: forward 5′-AAGCCAATGGGCAGGTCT-3′ and reverse 5′-TGTCCAGTTTCCGAAGCAT-3′, which generated an amplicon of 121 bp from the cDNA. The forward primer is located in exon 1 and the reverse primer in exon 2, and no detectable genomic product was generated under the PCR conditions employed. Universal ProbeLibrary probe 4 (Roche Applied Science) bound in the amplicon, and cleavage of the 5′-fluorescein during amplification generated the fluorescent signal. Commercially available primers for ribosomal 18 S RNA (Ambion, Austin, TX) were used for copy controls. The Lightcycler TaqMan master kit (Roche Applied Science) was employed for all reactions. Quantitative real time PCR using a capillary thermocycler (LightCycler, Roche Applied Science) was performed as we described previously in detail (21).
Generation of Mcl-1 Mutants and Their Transfection
Sequential mutagenesis of the established ubiquitination sites in human Mcl-1 (amino acids 5, 40, 136, 194, and 197) from lysine to arginine (KR Mcl-1) and mutation of serine at amino acid 159 to alanine (S159A Mcl-1) were performed using the QuikChange II site-directed mutagenesis kit (Stratagene, Cedar Creek, TX) per the manufacturer's protocol. S peptide-tagged human Mcl-1 cDNA subcloned into the pcDNA3 vector (Invitrogen), which has been previously described (51), was used as the template. The entire Mcl-1 insert was sequenced by automated sequencing to verify that the correct mutation was present and the construct remained in-frame. The preparation of the S159A mutant has been described previously (51). These plasmids were prepared for transfection using a plasmid miniprep kit (Bio-Rad). Transfection was performed with Lipofectamine (Invitrogen). Huh-7 cells were either transfected with an enhanced green fluorescent protein (GFP) expressing construct alone (pEGFP-N, Clontech) or with GFP plus a pcDNA3 vector expressing the mutated human Mcl-1 at a 1:2 ratio. Transfected cells were identified by expression of GFP.
Quantitation of Apoptosis
Characteristic morphological changes of apoptosis were assessed by staining nuclei with 4′,6-diamidino-2-phenylindole dihydrochloride (Sigma) followed by fluorescence microscopy using excitation and emission wavelengths of 380 and 430 nm, respectively (48, 52). Caspase-3/7 activity was quantified by measuring rhodamine release from the caspase-3/7 substrate rhodamine 110, bis(N-CBZ-l-aspartyl-l-glutamyl-l-valyl-l-aspartic acid amide), using the Apo-ONE™ homogeneous caspase-3/7 kit (Promega Corp., Madison, WI) following the supplier's instructions.
shRNA-targeted Knockdown of Mcl-1 and PKCθ
A previously described construct expressing an shRNA-targeting Mcl-1 (53) or a human PKCθ shRNA construct purchased from Sigma were employed to knock down their respective expression. The constructs were transfected using a standard lipofection method. Stably transfected Huh-7 clones were selected in medium containing 1200 μg/ml G418. Individual colonies were subcloned and screened for Mcl-1 or PKCθ expression by immunoblot analysis.
Northern Blot Analysis
RNA was extracted from Jurkat and Huh-7 cells using the FastTrack MAG mRNA isolation kit (Invitrogen) and used for Northern blot analysis. Digoxigenin-labeled probe and Northern blot examination were performed using the digoxigenin Northern starter kit (Roche Applied Science) following the manufacturer's instructions. Digoxigenin-labeled probe was created from a PCR product corresponding to exons 12–14 of the human PKCθ gene.
Mcl-1 Ubiquitination
Huh-7 cells were stably transfected with S peptide-tagged human Mcl-1 as we have described previously for other cell types (51). HA-tagged ubiquitin in a pcDNA plasmid was obtained from Dr. Mark A. McNiven (Mayo Clinic, Rochester, MN) and transiently transfected into the S peptide Mcl-1-expressing Huh-7 cells. Cells were treated with palmitate (800 μm) plus MG-132 (10 mm) for the desired time intervals. S peptide Mcl-1 was affinity-purified from whole cell lysates using S protein coupled to agarose beads as described previously (51). The affinity-purified protein complexes were resolved by SDS-PAGE and immobilized on nitrocellulose. Immunoblot analysis was performed for S peptide and HA.
Phosphorylation Site Analysis
Huh-7 cells stably transfected with S peptide-tagged Mcl-1 were cultured in 10-cm dishes. After the cells were solubilized in lysis buffer, S peptide-tagged Mcl-1 was recovered on S protein-agarose as described previously (51). The tagged polypeptide was released into SDS sample buffer, resolved by SDS-PAGE, and stained with Coomassie Brilliant Blue (Bio-Rad) in 20% methanol and 7.5% acetic acid. The Mcl-1 band was excised and subjected to tryptic digestion followed by quadruple time-of-flight tandem mass spectrometry analysis at the Taplin Biological Mass Spectrometry Facility, Harvard Medical School.
Statistical Analysis
All data represent at least three independent experiments expressed as the mean ± S.E. Differences between groups were compared using the two-tailed Student's t test. Analysis of variance with a Bonferroni test to correct for multiple comparisons was employed when appropriate.
RESULTS
Cellular Mcl-1 Is Preferentially Degraded by the Proteasome in Response to Saturated FFA
We initially assessed that if saturated, cytotoxic FFA altered the cellular fate of Mcl-1 in Huh-7 cells. Palmitate (16:0 saturated FFA) induced a rapid decrease of Mcl-1 protein that was apparent within 1 h and maximal at 4 h (Fig. 1A). Levels of the anti-apoptotic protein Bcl-xL, however, were unaltered. Treatment with the proapoptotic FFA stearate (18:0 saturated FFA) also resulted in a rapid loss of cellular Mcl-1 (Fig. 1B). In contrast, the nontoxic FFA oleate (18:1 mono-unsaturated FFA) did not significantly reduce Mcl-1 cellular protein levels (Fig. 1C).
FIGURE 1.
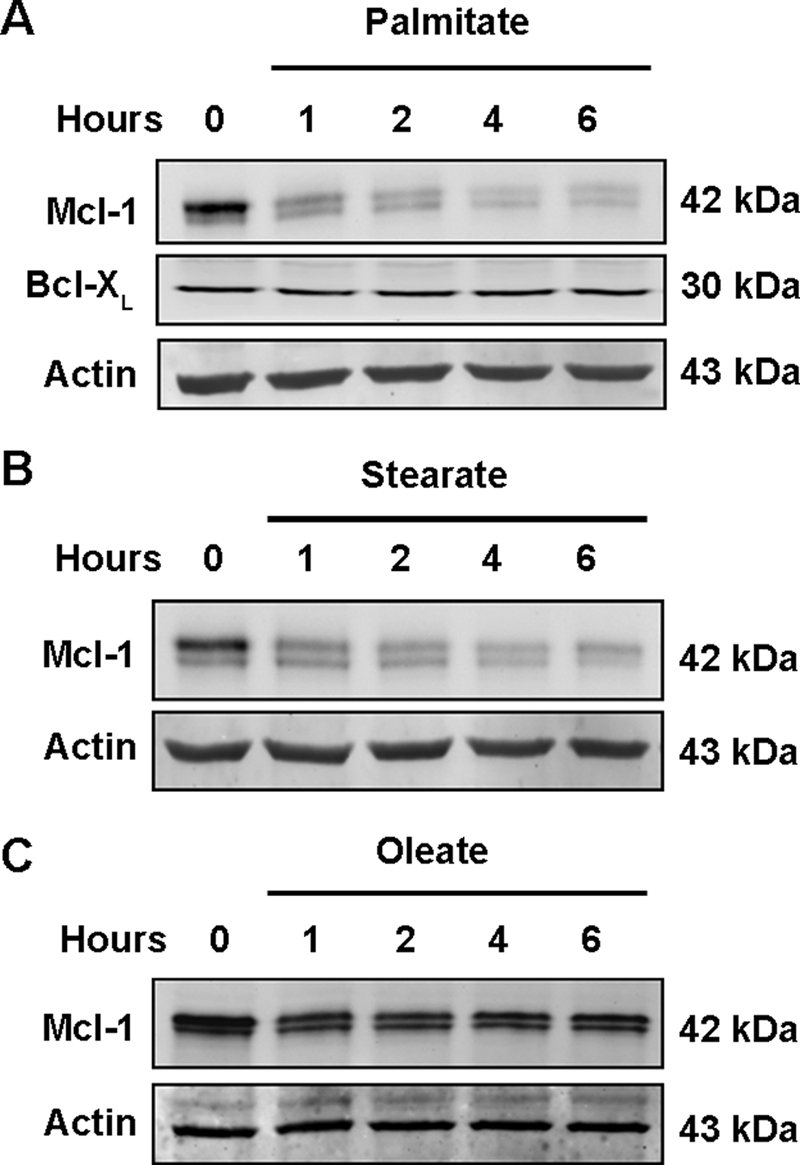
Mcl-1 protein levels decrease in Huh-7 cells in response to treatment with saturated free fatty acids. Immunoblot of whole cell extracts was obtained from Huh-7 cells treated with 800 μm palmitate (A), 600 μm stearate (B), or 800 μm oleate (C) for the indicated times. Endogenous human Mcl-1 in Huh-7 cells consistently migrated as a doublet of 42 kDa. Immunoblot analysis was performed for the proteins of interest, Mcl-1 (sc-819), Bcl-xL, and β-actin, a control for protein loading.
Additional experiments were performed to assess the cause of the Mcl-1 down-regulation. Treatment with palmitate did not alter steady state levels of MCL-1 mRNA, indicating that FFA are unlikely to affect either MCL-1 mRNA generation or stability (Fig. 2A). Loss of Mcl-1 was also not simply a consequence of lipoapoptosis, as the pan-caspase inhibitor Z-VAD-fmk, which blocks FFA-mediated apoptosis (52), did not prevent loss of cellular Mcl-1 (Fig. 2B, upper and lower panels). Analysis of replicate immunoblots for palmitate-induced Mcl-1 degradation in the presence or absence of Z-VAD-fmk revealed no significant difference (p > 0.25) in Mcl-1 levels at any of the analyzed time points (Fig. 2B, lower panel).
FIGURE 2.

Palmitate destabilizes Mcl-1 protein and not mRNA. A, palmitate does not alter steady state Mcl-1 mRNA levels. Mcl-1 mRNA levels were measured by quantitative real time PCR performed on cDNA from Huh-7 cells treated with 800 μm palmitate for the time intervals indicated. Ribosomal 18 S RNA was used as a copy control. Each point is the absolute number of copies of Mcl-1 mRNA divided by the number of copies of 18 S RNA determined by comparison with known standards. B, caspase inhibition does not prevent palmitate-induced loss of Mcl-1 protein level. Huh-7 cells were incubated in the presence of 800 μm palmitate plus 50 μm Z-VAD-fmk for the time intervals indicated. Whole cell extracts were obtained, and proteins were resolved by SDS-PAGE. Immunoblot analysis was performed for Mcl-1 and, as a control for protein loading, β-actin. C, MG-132, a proteasome inhibitor, blocks palmitate-induced loss of cellular Mcl-1 protein. Huh-7 cells were incubated in the presence of 800 μm palmitate plus 10 μm MG-132. At the time intervals indicated, total protein was isolated for immunoblot analysis. D, proteasome-resistant Mcl-1 mutant is resistant to cellular degradation. Huh-7 cells were transiently transfected with a construct expressing a human Mcl-1 with mutations (lysine to arginine) of five established ubiquitination sites (KR Mcl-1). Twenty four h after transfection, cells were treated with 800 μm palmitate. Immunoblot analysis for Mcl-1 was performed on whole cell extracts obtained at the time intervals indicated. In contrast to endogenous Mcl-1, KR Mcl-1 migrated as a single 42-kDa band.
Mcl-1 protein is known to be rapidly turned over in many cell types by a ubiquitin-dependent proteasome-mediated pathway. Consistent with this concept, the proteasome inhibitor MG-132 blocked loss of cellular Mcl-1 during palmitate treatment (Fig. 2C). To further confirm this mechanism for Mcl-1 cellular elimination, Huh-7 cells were transiently transfected with a ubiquitin-resistant Mcl-1 mutant in which lysines 5, 40, 136, 194, and 197 were mutated to arginine (termed KR Mcl-1), thereby reducing Mcl-1 turnover by the ubiquitin proteasomal pathway (54). Palmitate-mediated cellular Mcl-1 elimination was attenuated in cells transfected with this ubiquitin-resistant mutant (Fig. 2D). Collectively, these data suggest that saturated FFA result in Mcl-1 protein elimination by a ubiquitin-dependent proteasome degradation pathway.
Mcl-1 Degradation Contributes to Lipoapoptosis
If Mcl-1 degradation contributes to lipoapoptosis, then transfection with the ubiquitin-resistant mutant should reduce palmitate lipotoxicity. Consistent with this hypothesis, transfection of Huh-7 cells with KR Mcl-1 not only preserved cellular Mcl-1 levels (Fig. 2D) but also attenuated palmitate lipotoxicity (Fig. 3, A and B). We next tested the converse concept, i.e. the ability of Mcl-1 depletion to sensitize cells to lipotoxicity by an otherwise nontoxic palmitate concentration. Consistent with a pivotal role for Mcl-1 in lipoapoptosis, shRNA-targeted knockdown of Mcl-1 sensitized cells to otherwise nontoxic concentrations of palmitate (Fig. 3, C and D).
FIGURE 3.

Overexpression of a ubiquitin-resistant Mcl-1 mutant (KR Mcl-1) protects Huh-7 cells from lipoapoptosis, and targeted Mcl-1 knockdown sensitizes cells to palmitate-induced lipoapoptosis. A and B, Huh-7 cells were transiently transfected with a plasmid expressing KR Mcl-1 or GFP as control. Twenty four h following transfection, the cells were treated with 800 μm palmitate for 16 h at 37 °C. Apoptosis was quantified morphologically (A) as described under “Experimental Procedures” or biochemically (B) by measuring caspase-3/7 activity. *, statistically significant difference (p < 0.01) compared with GFP vehicle cells; **, statistically significant difference (p < 0.01) compared with GFP palmitate cells. C, Mcl-1 knockdown was performed by stable transfection of Huh-7 cells with a plasmid encoding shRNA directed to Mcl-1. Efficiency of the targeted knockdown of Mcl-1 was established by immunoblot analysis of whole cell extracts. D, knockdown of Mcl-1 increases sensitivity of Huh-7 cells to lipoapoptosis by palmitate. Huh-7 cells and Huh-7 cells stably transfected with shRNA targeted against Mcl-1 (shMcl-1) were incubated with 400 μm palmitic acid for 16 h. Apoptosis was quantified morphologically (C) and biochemically (D) by measuring caspase-3/7 activity. **, statistically significant difference (p < 0.01) for palmitate-treated shMcl-1 cells versus the palmitate-treated parental cell line.
In additional experiments, we confirmed these observations in primary mouse hepatocytes. In these cells, palmitate again induced loss of cellular Mcl-1 (Fig. 4A). Hepatocytes isolated from mice expressing a human MCL-1 transgene had sustained Mcl-1 protein levels after palmitate treatment (Fig. 4B). As the primary sequence of human and mouse Mcl-1 proteins is not identical (76% identity and 82% similarity), the preservation of human Mcl-1 suggests that one or more components of the degradation pathway (potentially the ubiquitin ligase or proteosome itself) exhibits species-specific preferences. Consistent with the preservation of the hMcl-1 protein levels, these murine hepatocytes also displayed reduced lipoapoptosis as compared with cells from wild type animals (Fig. 4C). Taken together, these observations implicate a role for Mcl-1 in regulating lipoapoptosis.
FIGURE 4.

Primary hepatocytes isolated from mice overexpressing human Mcl-1 are resistant to palmitate-induced loss of Mcl-1 and lipoapoptosis. A, primary murine hepatocytes were isolated from wild type C57BL/6 mice and cultured overnight. Cells were treated with 400 μm palmitate for the time intervals indicated, and whole cell extracts were procured. Immunoblot analysis was performed for murine Mcl-1 (Rockland) and β-actin. Murine Mcl-1 migrated as a doublet with a dominant band of ∼37 kDa. B, primary murine hepatocytes were obtained from C57BL/6 mice transgenic for human MCL-1. Studies were performed as described in A with Mcl-1 immunoblot using sc-819 anti-human Mcl-1. C, primary mouse hepatocytes from C57BL/6, wild type (WT), and transgenic mice for human MCL-1 (Mcl-1 Tg) were treated with 400 μm palmitate for 8 h. Apoptosis was quantified based on nuclear morphology. *, statistically significant difference (p < 0.01) compared with vehicle-treated wild type cells; **, statistically significant difference (p < 0.001) compared with palmitate-treated wild type cells.
Degradation of Mcl-1 Is Mediated by PKCθ
Further experiments were designed to identify the pathway involved in FFA-induced apoptosis. Phosphorylation of Mcl-1 Ser159 by GSK-3β has been shown previously to accelerate Mcl-1 degradation during growth factor withdrawal via enhanced ubiquitination (55, 56). However, GSK-3β inhibition did not prevent palmitate-induced degradation of Mcl-1 (Fig. 5A). In contrast, rottlerin, a cell-permeable pharmacological inhibitor of novel PKC isozymes, diminished palmitate-induced Mcl-1 degradation (Fig. 5B), raising the possibility that a novel PKC isoform might contribute to Mcl-1 down-regulation. PKC-dependent ubiquitination of Mcl-1 was investigated in Huh-7 cells expressing S peptide-tagged Mcl-1 and HA-tagged ubiquitin. To facilitate the detection of ubiquitinated Mcl-1, the proteasome inhibitor MG-132 was employed. Ubiquitinated Mcl-1 was detected in Huh-7 cells treated with palmitate, although cells co-treated with rottlerin displayed decreased ubiquitinated Mcl-1 signal, consistent with PKC-dependent ubiquitination (Fig. 5C).
FIGURE 5.
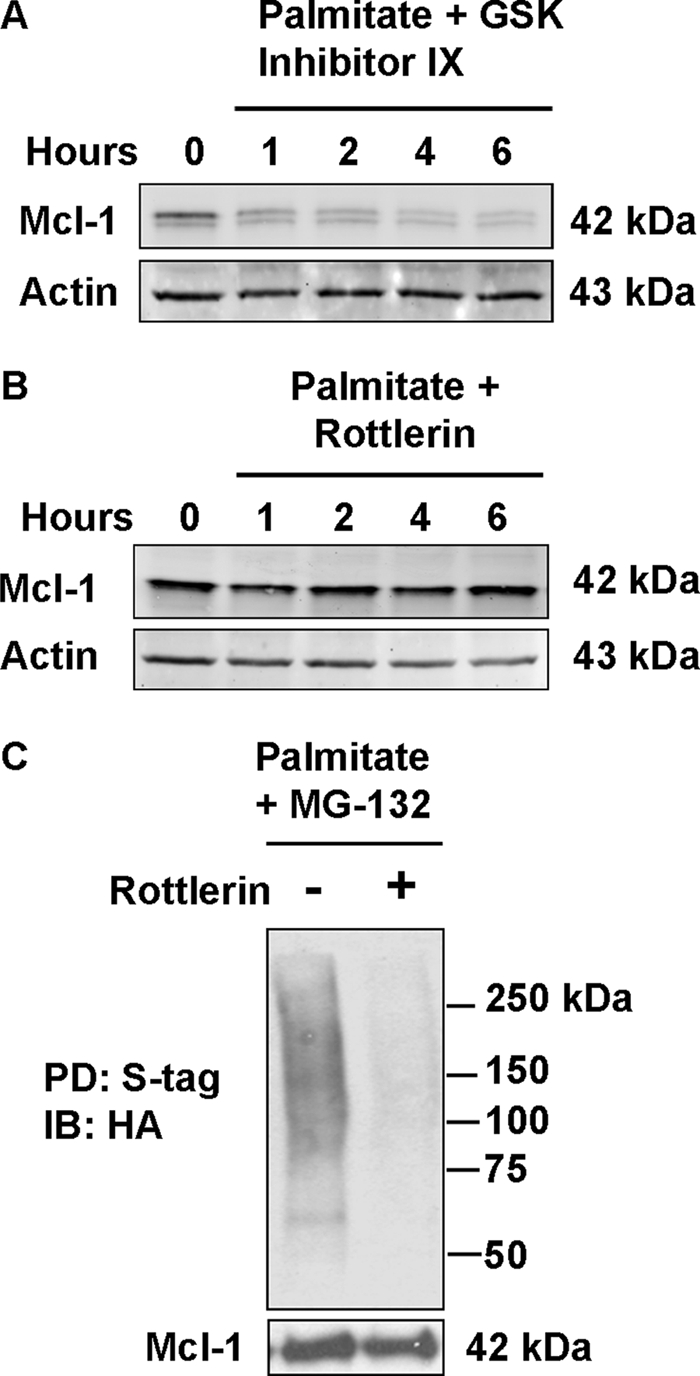
Inhibition of PKCθ, a novel PKC, blocks Mcl-1 degradation by palmitate. Huh-7 cells were incubated in the presence of 800 μm palmitate plus 3 μm GSK-3β inhibitor IX (A) or 10 μm of the novel PKC inhibitor rottlerin (B). At the time intervals indicated, whole cell extracts were prepared for immunoblot analysis. C, ubiquitination of Mcl-1 is dependent on novel PKC activity. Whole cell extracts were prepared from Huh-7 cells stably expressing HA-tagged ubiquitin that were treated with 800 μm palmitate and the proteasome inhibitor MG-132 for 8 h either with or without the PKCθ inhibitor rottlerin. Affinity pulldown (PD) was performed for S-tagged (S-tag) Mcl-1 using protein S-agarose followed by immunoblot (IB) analysis of ubiquitin with anti-HA peptide tag antibody.
Members of the novel PKC isozymes contain autoinhibitory domains that fold into the substrate-binding site within the catalytic domain to prevent substrate recognition. Phosphorylation of the autoinhibitory domain results in a conformational change, permitting recognition of substrates by this enzyme (57). Palmitate induces rapid Thr538 phosphorylation within this autoinhibitory domain, consistent with PCKθ activation (Fig. 6A). In contrast, PKCδ was not strongly phosphorylated at Thr505 within its autoinhibitory domain following exposure to palmitate (Fig. 6B). Of note, blotting with either the Thr538 phospho-specific PKCθ or total PKCθ antiserum displayed a single polypeptide of 55 kDa (also observed using an unrelated polyclonal anti-PKCθ antibody, data not shown), although the expected molecular mass of PKCθ has been reported to be 79 kDa, which we observed with whole cell extracts from Jurkat cells (supplemental Fig.). Based on this observation, we posited that hepatocytes might express a unique PKCθ isoform. Consistent with this possibility, Northern blot analysis demonstrated a 1.9-kb band in Huh-7 cells compared with the 2.8-kb band observed in Jurkat cells, suggesting that an alternative splice variant is the predominant PKCθ isoform in hepatocytes (Fig. 6C). Exploration of the mRNA by reverse transcription followed by long distance PCR demonstrated exons 2–15 in both Jurkat and Huh-7 cells, although a product was identified using primers in exons 2–18 only in Jurkat but not Huh-7 cells (supplemental Fig.). Likely the mRNA is alternatively spliced at or near the 3′-terminal exon.
FIGURE 6.

Palmitate induces phosphorylation of PKCθ but not PKCδ. A, Huh-7 cells were incubated in the presence of 800 μm palmitate over the time intervals indicated. Whole cell lysates were subjected to immunoblot analysis for phospho-Thr538 PKCθ and total PKCθ or (B) phospho-Thr505 PKCδ and total PKCδ. Immunoblots were also probed for α-tubulin or β-actin as loading controls. C, Northern blot analysis demonstrates expression of a PKCθ mRNA in Huh-7 cells that is smaller than the transcript in Jurkat cells.
To explore the function of this PKCθ isoform in lipoapoptosis, we knocked down its expression using shRNA that targeted nucleotides within the catalytic domain of the enzyme. This shRNA construct successfully knocked down expression of the 55-kDa PKCθ isoform (Fig. 7A), further confirming the immunoreactive band at 55 kDa represents PKCθ. shRNA-targeted knockdown of PCKθ prevented palmitate-induced degradation of Mcl-1 and actually resulted in an increased level of this anti-apoptotic protein (Fig. 7B). Retention of Mcl-1 during palmitate treatment was also observed in primary mouse hepatocytes from Pkcθ−/− animals (Fig. 7C). Taken together, these data implicated PKCθ in the degradation of Mcl-1 by saturated FFA.
FIGURE 7.

shRNA-targeted knockdown of PKCθ prevents palmitate-induced loss of Mcl-1. A, immunoblot analysis for PKCθ using whole cell extracts from Huh-7 cells stably transfected with empty vector expression construct as a control and cells stably transfected with the PKCθ-targeted shRNA. B, stable cell lines (as described in A) were incubated with 800 μm palmitate over 6 h. Immunoblot analysis was performed on whole cell extracts for Mcl-1 and β-actin as a control for protein loading. C, hepatocytes from Pkcθ−/− mice maintain Mcl-1 protein levels following palmitate treatment. Primary murine hepatocytes from Pkcθ knock-out mice were treated with palmitate 400 μm. At desired time intervals, whole cell lysates were prepared and blotted for Mcl-1 and actin (cf. Fig. 4A). In primary hepatocytes from Pkcθ−/− mice, actin was detected as a doublet, possibly due to detection of both β and γ isoforms.
Inhibition of PKCθ Activity or Expression Attenuates Palmitate-induced Lipoapoptosis
If PKCθ inhibition attenuates Mcl-1 loss, then PKCθ inhibition should also ameliorate lipoapoptosis. Consistent with this prediction, inhibition of PKCθ activity with rottlerin or reduction of PKCθ by shRNA reduced lipotoxicity by palmitate in Huh-7 cells (Fig. 8, A and B). Moreover, palmitate-induced apoptosis was significantly reduced in primary hepatocytes from Pkcθ−/− versus wild type mice (Fig. 8C). Thus, inhibition of PKCθ activity or expression not only maintains cellular Mcl-1 levels upon palmitate challenge, but also mitigates lipoapoptosis.
FIGURE 8.

Inhibition, shRNA-mediated knockdown, and genetic deletion of Pkcθ reduce lipoapoptosis. A, Huh-7 cells were treated with the novel PKC inhibitor rottlerin (10 μm) beginning 30 min prior to the addition of 800 μm palmitate. After 16 h of incubation, apoptosis was quantified by characteristic nuclear morphologic changes. B, Huh-7 cells were stably transfected with an shRNA targeting PKCθ or the empty expression construct (Huh-7; see Fig. 7). After 16 h of incubation with 800 μm palmitate, apoptosis was quantified by nuclear morphology. C, apoptosis was also quantified in primary murine hepatocytes from wild type and Pkcθ−/− mice at 8 h following addition of 400 μm palmitate. *, statistically significant difference (p < 0.01) compared with vehicle-treated cells; **, statistically significant difference (p < 0.01) compared with palmitate-treated cells in the absence of rottlerin.
Ser159 Phosphorylation Contributes to Mcl-1 Degradation and Lipoapoptosis by Palmitate
To further explore the role of PKCθ in Mcl-1 degradation, we next determined whether Mcl-1 phosphorylation required PKCθ catalytic activity. For these studies we employed Huh-7 cells stably transfected with an S peptide-tagged Mcl-1 to facilitate affinity purification of Mcl-1. Huh-7 cells stably expressing S peptide-tagged Mcl-1 were treated with palmitate with or without rottlerin, and Mcl-1 was affinity-purified and analyzed by mass spectrometry. Ser159, a previously described phosphorylation site of Mcl-1, was found to be phosphorylated in palmitate-treated cells but not in cells treated with palmitate plus rottlerin, suggesting PKCθ-dependent Mcl-1 Ser159 phosphorylation.
The potential Mcl-1 Ser159 phosphorylation following PKCθ activation was further investigated by antiserum specific for Mcl-1 phosphorylated at both Ser159 and Thr163. To confirm the specificity of this antiserum, we expressed wild type, S159A, and T163A mutant Mcl-1 in Huh-7 cells followed by S protein-based affinity purification and immunoblot analysis. Phosphorylated wild type Mcl-1 was readily identified, whereas substitution of either Ser159 or Thr163 with alanine prevented recognition by this phospho-specific antiserum (Fig. 9A). There was a slight increase in phosphorylated Mcl-1 from cells treated with palmitate (Fig. 9A, 4th lane) compared with vehicle (1st lane). To determine whether phosphorylation was dependent upon PKCθ activity, we incubated cells in the presence or absence of rottlerin, with or without palmitate. Inhibition of novel PKC activity decreased the phospho-Mcl-1 signal (Fig. 9B).
FIGURE 9.

Mcl-1 Ser159 is phosphorylated in a PKC-dependent manner, and S159A mutation decreases palmitate-induced Mcl-1 degradation and apoptosis. A, affinity-purified S peptide-tagged Mcl-1 from Huh-7 cells treated with vehicle or 800 μm palmitate was blotted using antiserum specific for phosphorylation at Ser159/Thr163. Mutation of either site to Ala prevents detection with this antibody. PD, pulldown; IB, immunoblot; WT, wild type. B, whole cell lysates of Huh-7 cells treated with MG-132 as well as rottlerin (Rott) (10 μm) and palmitate (PA) (800 μm) as indicated were blotted with anti-phospho-Mcl-1 or total Mcl-1. Veh, vehicle. The proteasome inhibitor MG-132 was included to prevent degradation and facilitate detection of phosphorylated Mcl-1. C, Huh-7 cells were stably transfected with a construct expressing either S peptide-tagged wild type Mcl-1 (left) or S159A Mcl-1 (right). The stable transfectants were treated with palmitate (800 μm) for the indicated time and then subjected to immunoblot analysis with anti-S peptide antibody. The ratio of intensities of S peptide-tagged Mcl-1 to β-actin is shown. S peptide-tagged human Mcl-1 migrates as a single band of 42 kDa. D, stable transfectants were treated with 800 μm palmitate for 16 h, and apoptosis was quantitated by characteristic morphologic features. *, statistically significant difference (p < 0.01) compared with vehicle-treated cells transfected with wild type Mcl-1;**, statistically significant difference (p < 0.001) compared with palmitate-treated cells transfected with wild type Mcl-1.
We reasoned that blocking phosphorylation of Mcl-1 at Ser159 would prevent the degradation of Mcl-1 and promote survival upon palmitate treatment. Mutation of Ser159 to alanine results in decreased palmitate-induced Mcl-1 degradation (Fig. 9C) and lipoapoptosis (Fig. 9D). Thus, whether PKCθ directly or indirectly phosphorylates Mcl-1 at Ser159, phosphorylation at this site regulates Mcl-1 turnover and cell survival during exposure to palmitate.
DISCUSSION
Results of this study provide new insight into the mechanism of FFA-induced lipoapoptosis by showing in Huh-7 cells and primary murine hepatocytes the following: (i) the saturated FFA palmitate and stearate, but not the unsaturated FFA oleate, induce loss of cellular Mcl-1 protein by a proteasome-dependent pathway; (ii) inhibition of Mcl-1 degradation attenuates apoptosis by saturated FFA; and (iii) Mcl-1 degradation and apoptosis by saturated FFA appear to be mediated, in part, by PKCθ. Each of these results is discussed in greater detail below.
In the multiple model systems described here, treatment of liver-derived cells with saturated FFA results in rapid loss of cellular Mcl-1 protein. The observations that saturated FFA such as palmitate and stearate, but not the unsaturated FFA oleate, result in Mcl-1 loss are consistent with prior observations regarding the specificity of these FFA in mediating lipoapoptosis (52, 58). Although Mcl-1 regulation is multifaceted (reviewed in Ref. 36), our data suggest that saturated FFA induce Mcl-1 degradation by a ubiquitin-dependent proteasome degradation pathway. For example, loss of Mcl-1 was attenuated by the proteasome inhibitor MG-132 and, more importantly, by mutation of the five key lysine residues within its proline-, glutamic acid-, serine-, and threonine-rich sequence that are necessary for ubiquitination. In addition ubiquitinated Mcl-1 was detected in palmitate-treated cells. Two ubiquitin-protein isopeptide ligases, β-TrCP and Mule, have been reported to ubiquitinate this polypeptide promoting its recognition and rapid degradation by the proteasome (54, 59). Although we cannot rule out the possibility that saturated FFA regulate the activity of these ubiquitin-protein isopeptide ligase, our data suggest that the major contributor to FFA-induced Mcl-1 down-regulation is a direct post-translational modification of Mcl-1 by a PKCθ-mediated pathway, thereby rendering it susceptible to ubiquitin-protein isopeptide ligase ubiquitination (see below).
Several observations suggest loss of cellular Mcl-1 is not simply a consequence of cell death but rather contributes to FFA cytotoxicity. First, a pan-caspase inhibitor that prevents cell death (52) did not avert loss of cellular Mcl-1. We also did not observe Mcl-1 degradation products by immunoblot analysis as has been identified in processes associated with caspase-mediated Mcl-1 cleavage (60). Second, expression of a ubiquitin-resistant mutant, which undergoes limited degradation in response to saturated FFA, attenuates cell death. Finally, shRNA-induced reduction in Mcl-1 levels further sensitizes the cells to lipotoxicity. These observations are consistent with published data indicating that cellular elimination of Mcl-1 is a pivotal step in several apoptotic pathways (34, 35). Prior studies on the regulation of hepatocyte lipoapoptosis by Bcl-2 proteins have focused on the role of proapoptotic members of this family, especially Bim-mediated activation of Bax (52). This study extends these observations by demonstrating a key role for the anti-apoptotic protein Mcl-1 in hepatocyte lipoapoptosis. Mcl-1 may attenuate apoptosis by binding to and sequestering Bim, thereby preventing its activation of Bax (24, 61, 62). Thus, a broader picture of lipoapoptosis by saturated FFA is emerging where loss of Mcl-1 likely facilitates Bim activation of Bax, which in turn triggers the mitochondrial cell death pathway.
Our observations suggest that the novel PKC isoform PKCθ promotes Mcl-1 degradation by cytotoxic FFA in hepatocytes. Pharmacologic inhibition of PKCθ with rottlerin, inhibition of PKCθ protein expression by targeted shRNA, and genetic deletion of Pkcθ all reduced loss of Mcl-1 and lipoapoptosis by palmitate. Interestingly, the PKCθ expressed in hepatocytes migrates significantly faster on gels than that in Jurkat cells. Evidence that the 55-kDa band in hepatocytes represents PKCθ includes recognition of a band of the same molecular weight by an unrelated PKCθ antiserum (phospho-PKCθ epitope), as well as modulation of expression by an shRNA targeting PKCθ. Northern blot and PCR analyses suggest that a smaller alternative splice form is responsible for the polypeptide identified on SDS-PAGE. The faster migrating PKCθ protein and reduced size of mRNA have not previously been described and may well be liver-specific splice variants, raising the possibility of unique regulation of this kinase in liver.
Although PKCθ has been implicated in apoptosis of Jurkat, neuroblastoma, and myeloid leukemia cells (46, 47), its effects on cellular Mcl-1 protein levels have not been reported previously. The present data suggest that PKCθ activity contributes to Mcl-1 Ser159 phosphorylation. Mcl-1 phosphorylation at this residue has been shown previously to prime Mcl-1 for ubiquitin-protein isopeptide ligase-directed ubiquitination and subsequent proteasome degradation (55). Although GSK-3β can phosphorylate Mcl-1 at Ser159, a GSK-3β inhibitor did not prevent Mcl-1 degradation by palmitate treatment. More importantly, as identified by mass spectrometry, the novel PKC inhibitor rottlerin blocked Mcl-1 Ser159 phosphorylation and ubiquitination during exposure to palmitate. These composite observations suggest that PKCθ may directly phosphorylate Mcl-1 at Ser159, an interpretation supported by the in silico observation that this site is a PKC consensus phosphorylation site (Netphos 2.0).
In summary, regulation of the mitochondrial death pathway by Bcl-2 proteins is a complex and hierarchal process that displays cell type- and stimulus-specific pathways. Lipoapoptosis appears to involve all three subsets of Bcl-2 proteins. Expression of the BH3-only protein Bim, which promotes Bax activation, is increased by the transcription factor FoxO3a (21, 52). This work extends these observations by also implicating PKCθ-driven Mcl-1 elimination as a critical event in this cytotoxic process. Consistent with activation of a complex cytotoxic network, we note that inhibition of Bim expression or PKCθ activation individually only partially ameliorates hepatocyte lipotoxicity. Thus, inhibition of any single component is not sufficient to totally abrogate lipoapoptosis. Other overlapping signaling processes may also contribute to saturated FFA-mediated hepatocyte cytotoxicity. The optimal therapeutic strategy to minimize hepatic lipotoxicity will require knowledge of the composite cytotoxic network induced by FFA. Once these networks are elucidated, therapeutic strategies can be developed to minimize liver damage in human fatty liver diseases.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grants R01 DK41876 (to G. J. G.), T32 07198 (to H. C. M.), K01 DK79875 (to J. L. M.), R01 CA69008 (to S. H. K.), and the optical microscopy core of P309K 84567. This work was also supported by the Mayo Foundation.

The on-line version of this article (available at http://www.jbc.org) contains a supplemental figure.
- NASH
- nonalcoholic steatohepatitis
- FFA
- free fatty acid
- GFP
- green fluorescent protein
- GSK-3β
- glycogen synthase kinase-3β
- KR Mcl-1
- lysine to arginine ubiquitin-resistant Mcl-1 mutant
- PEST
- sequences rich in proline, glutamic acid, serine, threonine
- PKC
- protein kinase C
- shRNA
- short hairpin RNA
- Z-
- benzyloxycarbonyl-
- fmk
- fluoromethyl ketone
- HA
- hemagglutinin.
REFERENCES
- 1.Browning J. D., Szczepaniak L. S., Dobbins R., Nuremberg P., Horton J. D., Cohen J. C., Grundy S. M., Hobbs H. H. (2004) Hepatology 40, 1387–1395 [DOI] [PubMed] [Google Scholar]
- 2.Adams L. A., Lymp J. F., St Sauver J., Sanderson S. O., Lindor K. D., Feldstein A., Angulo P. (2005) Gastroenterology 129, 113–121 [DOI] [PubMed] [Google Scholar]
- 3.Ekstedt M., Franzén L. E., Mathiesen U. L., Thorelius L., Holmqvist M., Bodemar G., Kechagias S. (2006) Hepatology 44, 865–873 [DOI] [PubMed] [Google Scholar]
- 4.Ratziu V., Poynard T. (2006) Hepatology 44, 802–805 [DOI] [PubMed] [Google Scholar]
- 5.Chavez-Tapia N. C., Mendez-Sanchez N., Uribe M. (2006) Ann. Intern. Med. 144, 379–380 [DOI] [PubMed] [Google Scholar]
- 6.Angulo P. (2002) N. Engl. J. Med. 346, 1221–1231 [DOI] [PubMed] [Google Scholar]
- 7.Parekh S., Anania F. A. (2007) Gastroenterology 132, 2191–2207 [DOI] [PubMed] [Google Scholar]
- 8.Donnelly K. L., Smith C. I., Schwarzenberg S. J., Jessurun J., Boldt M. D., Parks E. J. (2005) J. Clin. Invest. 115, 1343–1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doege H., Grimm D., Falcon A., Tsang B., Storm T. A., Xu H., Ortegon A. M., Kazantzis M., Kay M. A., Stahl A. (2008) J. Biol. Chem. 283, 22186–22192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou J., Febbraio M., Wada T., Zhai Y., Kuruba R., He J., Lee J. H., Khadem S., Ren S., Li S., Silverstein R. L., Xie W. (2008) Gastroenterology 134, 556–567 [DOI] [PubMed] [Google Scholar]
- 11.Listenberger L. L., Han X., Lewis S. E., Cases S., Farese R. V., Jr., Ory D. S., Schaffer J. E. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 3077–3082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamaguchi K., Yang L., McCall S., Huang J., Yu X. X., Pandey S. K., Bhanot S., Monia B. P., Li Y. X., Diehl A. M. (2007) Hepatology 45, 1366–1374 [DOI] [PubMed] [Google Scholar]
- 13.Nehra V., Angulo P., Buchman A. L., Lindor K. D. (2001) Dig. Dis. Sci. 46, 2347–2352 [DOI] [PubMed] [Google Scholar]
- 14.de Almeida I. T., Cortez-Pinto H., Fidalgo G., Rodrigues D., Camilo M. E. (2002) Clin. Nutr. 21, 219–223 [DOI] [PubMed] [Google Scholar]
- 15.Araya J., Rodrigo R., Videla L. A., Thielemann L., Orellana M., Pettinelli P., Poniachik J. (2004) Clin. Sci. 106, 635–643 [DOI] [PubMed] [Google Scholar]
- 16.Belfort R., Harrison S. A., Brown K., Darland C., Finch J., Hardies J., Balas B., Gastaldelli A., Tio F., Pulcini J., Berria R., Ma J. Z., Dwivedi S., Havranek R., Fincke C., DeFronzo R., Bannayan G. A., Schenker S., Cusi K. (2006) N. Engl. J. Med. 355, 2297–2307 [DOI] [PubMed] [Google Scholar]
- 17.Larter C. Z., Yeh M. M., Haigh W. G., Williams J., Brown S., Bell-Anderson K. S., Lee S. P., Farrell G. C. (2008) J. Hepatol. 48, 638–647 [DOI] [PubMed] [Google Scholar]
- 18.Gentile C. L., Pagliassotti M. J. (2008) J. Nutr. Biochem. 19, 567–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang D., Wei Y., Pagliassotti M. J. (2006) Endocrinology 147, 943–951 [DOI] [PubMed] [Google Scholar]
- 20.Malhi H., Barreyro F. J., Isomoto H., Bronk S. F., Gores G. J. (2007) Gut 56, 1124–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barreyro F. J., Kobayashi S., Bronk S. F., Werneburg N. W., Malhi H., Gores G. J. (2007) J. Biol. Chem. 282, 27141–27154 [DOI] [PubMed] [Google Scholar]
- 22.Feldstein A. E., Canbay A., Angulo P., Taniai M., Burgart L. J., Lindor K. D., Gores G. J. (2003) Gastroenterology 125, 437–443 [DOI] [PubMed] [Google Scholar]
- 23.Wieckowska A., Zein N. N., Yerian L. M., Lopez A. R., McCullough A. J., Feldstein A. E. (2006) Hepatology 44, 27–33 [DOI] [PubMed] [Google Scholar]
- 24.Youle R. J., Strasser A. (2008) Nat. Rev. Mol. Cell Biol. 9, 47–59 [DOI] [PubMed] [Google Scholar]
- 25.Tzung S. P., Fausto N., Hockenbery D. M. (1997) Am. J. Pathol. 150, 1985–1995 [PMC free article] [PubMed] [Google Scholar]
- 26.Print C. G., Loveland K. L., Gibson L., Meehan T., Stylianou A., Wreford N., de Kretser D., Metcalf D., Köntgen F., Adams J. M., Cory S. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 12424–12431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ross A. J., Waymire K. G., Moss J. E., Parlow A. F., Skinner M. K., Russell L. D., MacGregor G. R. (1998) Nat. Genet. 18, 251–256 [DOI] [PubMed] [Google Scholar]
- 28.Hamasaki A., Sendo F., Nakayama K., Ishida N., Negishi I., Nakayama K., Hatakeyama S. (1998) J. Exp. Med. 188, 1985–1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takehara T., Tatsumi T., Suzuki T., Rucker E. B., 3rd, Hennighausen L., Jinushi M., Miyagi T., Kanazawa Y., Hayashi N. (2004) Gastroenterology 127, 1189–1197 [DOI] [PubMed] [Google Scholar]
- 30.Vick B., Weber A., Urbanik T., Maass T., Teufel A., Krammer P. H., Opferman J. T., Schuchmann M., Galle P. R., Schulze-Bergkamen H. (2009) Hepatology 49, 627–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schulze-Bergkamen H., Brenner D., Krueger A., Suess D., Fas S. C., Frey C. R., Dax A., Zink D., Büchler P., Müller M., Krammer P. H. (2004) Hepatology 39, 645–654 [DOI] [PubMed] [Google Scholar]
- 32.Kahraman A., Mott J. L., Bronk S. F., Werneburg N. W., Barreyro F. J., Guicciardi M. E., Akazawa Y., Braley K., Craig R. W., Gores G. J. (2009) Dig. Dis. Sci. 54, 1908–1917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kodama Y., Taura K., Miura K., Schnabl B., Osawa Y., Brenner D. A. (2009) Gastroenterology 136, 1423–1434 [DOI] [PubMed] [Google Scholar]
- 34.Nijhawan D., Fang M., Traer E., Zhong Q., Gao W., Du F., Wang X. (2003) Genes Dev. 17, 1475–1486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cuconati A., Mukherjee C., Perez D., White E. (2003) Genes Dev. 17, 2922–2932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Warr M. R., Shore G. C. (2008) Curr. Mol. Med. 8, 138–147 [DOI] [PubMed] [Google Scholar]
- 37.Solinas G., Naugler W., Galimi F., Lee M. S., Karin M. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 16454–16459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schenk S., Saberi M., Olefsky J. M. (2008) J. Clin. Invest. 118, 2992–3002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Itani S. I., Ruderman N. B., Schmieder F., Boden G. (2002) Diabetes 51, 2005–2011 [DOI] [PubMed] [Google Scholar]
- 40.Timmers S., Schrauwen P., de Vogel J. (2008) Physiol. Behav. 94, 242–251 [DOI] [PubMed] [Google Scholar]
- 41.Yu C., Chen Y., Cline G. W., Zhang D., Zong H., Wang Y., Bergeron R., Kim J. K., Cushman S. W., Cooney G. J., Atcheson B., White M. F., Kraegen E. W., Shulman G. I. (2002) J. Biol. Chem. 277, 50230–50236 [DOI] [PubMed] [Google Scholar]
- 42.Puri P., Baillie R. A., Wiest M. M., Mirshahi F., Choudhury J., Cheung O., Sargeant C., Contos M. J., Sanyal A. J. (2007) Hepatology 46, 1081–1090 [DOI] [PubMed] [Google Scholar]
- 43.Sakaki K., Wu J., Kaufman R. J. (2008) J. Biol. Chem. 283, 15370–15380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim J. K., Fillmore J. J., Sunshine M. J., Albrecht B., Higashimori T., Kim D. W., Liu Z. X., Soos T. J., Cline G. W., O'Brien W. R., Littman D. R., Shulman G. I. (2004) J. Clin. Invest. 114, 823–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Savage D. B., Petersen K. F., Shulman G. I. (2007) Physiol. Rev. 87, 507–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schultz A., Jönsson J. I., Larsson C. (2003) Cell Death Differ. 10, 662–675 [DOI] [PubMed] [Google Scholar]
- 47.Datta R., Kojima H., Yoshida K., Kufe D. (1997) J. Biol. Chem. 272, 20317–20320 [DOI] [PubMed] [Google Scholar]
- 48.Faubion W. A., Guicciardi M. E., Miyoshi H., Bronk S. F., Roberts P. J., Svingen P. A., Kaufmann S. H., Gores G. J. (1999) J. Clin. Invest. 103, 137–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Richieri G. V., Kleinfeld A. M. (1995) J. Lipid Res. 36, 229–240 [PubMed] [Google Scholar]
- 50.Sanyal A. J., Campbell-Sargent C., Mirshahi F., Rizzo W. B., Contos M. J., Sterling R. K., Luketic V. A., Shiffman M. L., Clore J. N. (2001) Gastroenterology 120, 1183–1192 [DOI] [PubMed] [Google Scholar]
- 51.Kobayashi S., Lee S. H., Meng X. W., Mott J. L., Bronk S. F., Werneburg N. W., Craig R. W., Kaufmann S. H., Gores G. J. (2007) J. Biol. Chem. 282, 18407–18417 [DOI] [PubMed] [Google Scholar]
- 52.Malhi H., Bronk S. F., Werneburg N. W., Gores G. J. (2006) J. Biol. Chem. 281, 12093–12101 [DOI] [PubMed] [Google Scholar]
- 53.Taniai M., Grambihler A., Higuchi H., Werneburg N., Bronk S. F., Farrugia D. J., Kaufmann S. H., Gores G. J. (2004) Cancer Res. 64, 3517–3524 [DOI] [PubMed] [Google Scholar]
- 54.Zhong Q., Gao W., Du F., Wang X. (2005) Cell 121, 1085–1095 [DOI] [PubMed] [Google Scholar]
- 55.Maurer U., Charvet C., Wagman A. S., Dejardin E., Green D. R. (2006) Mol. Cell 21, 749–760 [DOI] [PubMed] [Google Scholar]
- 56.Zhao Y., Altman B. J., Coloff J. L., Herman C. E., Jacobs S. R., Wieman H. L., Wofford J. A., Dimascio L. N., Ilkayeva O., Kelekar A., Reya T., Rathmell J. C. (2007) Mol. Cell. Biol. 27, 4328–4339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu Y., Graham C., Li A., Fisher R. J., Shaw S. (2002) Biochem. J. 361, 255–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wei Y., Wang D., Topczewski F., Pagliassotti M. J. (2006) Am. J. Physiol. Endocrinol. Metab. 291, E275–E281 [DOI] [PubMed] [Google Scholar]
- 59.Ding Q., He X., Hsu J. M., Xia W., Chen C. T., Li L. Y., Lee D. F., Liu J. C., Zhong Q., Wang X., Hung M. C. (2007) Mol. Cell. Biol. 27, 4006–4017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Han J., Goldstein L. A., Gastman B. R., Froelich C. J., Yin X. M., Rabinowich H. (2004) J. Biol. Chem. 279, 22020–22029 [DOI] [PubMed] [Google Scholar]
- 61.Meng X. W., Lee S. H., Dai H., Loegering D., Yu C., Flatten K., Schneider P., Dai N. T., Kumar S. K., Smith B. D., Karp J. E., Adjei A. A., Kaufmann S. H. (2007) J. Biol. Chem. 282, 29831–29846 [DOI] [PubMed] [Google Scholar]
- 62.Gavathiotis E., Suzuki M., Davis M. L., Pitter K., Bird G. H., Katz S. G., Tu H. C., Kim H., Cheng E. H., Tjandra N., Walensky L. D. (2008) Nature 455, 1076–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.