

Abstract
Accessibility within chromatin is an important factor in the prompt removal of UV-induced DNA damage by nucleotide excision repair (NER). Chromatin remodeling by the SWI/SNF complex has been shown to play an important modulating role in NER in vitro and yeast in vivo. Nevertheless, the molecular basis of cross-talk between SWI/SNF and NER in mammalian cells is not fully understood. Here, we show that knockdown of Brg1, the ATPase subunit of SWI/SNF, negatively affects the elimination of cyclobutane pyrimidine dimers (CPD), but not of pyrimidine (6, 4)pyrimidone photoproducts (6-4PP) following UV irradiation of mammalian cells. Brg1-deficient cells exhibit a lower chromatin relaxation as well as impaired recruitment of downstream NER factors, XPG and PCNA, to UV lesions. However, the assembly of upstream NER factors, DDB2 and XPC, at the damage site was unaffected by Brg1 knockdown. Interestingly, Brg1 interacts with XPC within chromatin and is recruited to UV-damaged sites in a DDB2- and XPC-dependent manner. Also, postirradiation decrease of XPC levels occurred more rapidly in Brg1-deficient than normal cells. Conversely, XPC transcription remained unaltered upon Brg1 knockdown indicating that Brg1 affects the stability of XPC protein following irradiation. Thus, Brg1 facilitates different stages of NER by initially modulating UV-induced chromatin relaxation and stabilizing XPC at the damage sites, and subsequently stimulating the recruitment of XPG and PCNA to successfully culminate the repair.
In eukaryotic cells, DNA packaged into highly condensed chromatin presents a significant obstacle to DNA-based processes, e.g. DNA replication, transcription, and repair. Two major strategies are used by cells to alter chromatin structure and allow the necessary accessibility to regulatory proteins, i.e. the post-translational modifications of histones to mobilize histones along the DNA strand and the ATP-dependent chromatin remodeling to regulate histone-DNA interactions. These complexes utilize the energy derived from ATP hydrolysis to either replace histones or slide the nucleosomes along the DNA strand and consequently change the structure and accessibility of chromatin (1, 2). Besides their function in transcription regulation, ATP-dependent chromatin remodeling factors have been shown to play an important role in a number of DNA repair pathways including double strand break (DSB) repair, base excision repair (BER), as well as nucleotide excision repair (NER)3 (2).
NER is a versatile DNA repair pathway that can remove a broad range of structurally unrelated lesions including UV-induced bulky DNA adducts cyclobutane pyrimidine dimers (CPD) and pyrimidine(6-4)pyrimidone photoproducts (6-4PP). One subpathway of NER, global genome NER (GG-NER), removes damage from the entire genome, whereas DNA damage in the transcribed strand of active genes is preferentially eliminated by transcription-coupled NER (TC-NER) (3, 4). Repair of DNA damage by NER comprises four sequential steps: damage detection, excision of the damaged segment, repair synthesis, and ligation to restore the intact DNA (3, 5, 6). All of these steps require the access of DNA repair factors to the damaged DNA. Since the majority of the DNA damage exists in highly condensed nucleosomes, which restricts the accessibility of DNA and inhibits DNA repair (7), it is crucial to understand how the chromatin structure is modulated and affects the repair. NER occurs much more efficiently in naked DNA than in chromatin (8, 9). NER is also more efficient in the linker region of chromatin than in the nucleosome, indicating that binding of DNA repair factors to the DNA is inhibited by chromatin structure (10, 11). Two ATP-dependent chromatin remodeling complexes have been shown to stimulate NER in a number of in vitro studies. Chromatin assembly and modifying factor (ACF) is capable of moving nucleosomes along the DNA and its remodeling activity has been shown to facilitate NER dual incision in the linker DNA region. However, ACF does not seem to have an effect on NER in the nucleosome where most damage exists (12). SWI/SNF complex, on the other hand, enhances NER of DNA lesions located in the nucleosome core region in vitro and the remodeling activity of SWI/SNF depends on the presence of XPC, RPA, and XPA (13, 14). SWI/SNF has also been extensively studied in yeast and mammalian cells. A study in yeast by Gong et al. (15) demonstrated enhanced interaction between Rad4 (the yeast homologue of XPC) and two subunits of SWI/SNF complex, SNF5 and SNF6, after UV irradiation of yeast. In addition, SWI/SNF facilitates chromatin remodeling at the silent HML locus and expedites NER in response to UV. In mammalian cells, SWI/SNF protects cells against UV-induced DNA damage by modulating checkpoint activation and onset of apoptosis (16). However, it is still unclear if SWI/SNF directly affects NER in response to UV damage in mammalian cells.
In this study, we addressed the direct role of Brg1, the ATPase subunit of SWI/SNF, in the process of GG-NER in human cells. We have demonstrated, for the first time, that recruitment of Brg1 to UV-induced CPD depends on DDB2 and XPC, indicating that SWI/SNF functions downstream of damage recognition. In addition, XPC is shown to interact with Brg1 on the chromatin and this interaction is enhanced by UV irradiation. We speculate that the interaction of XPC with Brg1 helps recruit Brg1 to the UV-damage site. More importantly, upon arrival Brg1 enhanced the UV-induced chromatin relaxation to enable the recruitment of XPG and PCNA to the UV damage site for the repair of CPD. This is the first study demonstrating the Brg1 function specifically affecting the later stage of NER in mammalian cells.
EXPERIMENTAL PROCEDURES
Cells, Expression Constructs, and Treatments
Normal human fibroblast, OSU-2, were established in our laboratory as described previously (17). HeLa cells stably transfected with NH2-terminal FLAG-hemagglutinin (HA)-tagged DDB2 (HeLa-DDB2) were a gift from Dr. Yoshihiro Nakatani (Dana-Farber Cancer Institute, Boston, MA). HeLa cells were transfected with N-terminal GFP-HA-His-tagged XPC (kindly provided by Dr. Jan H. Hoeijamakers, Erasmus University, The Netherlands) and a stably transfected cell line (HeLa-XPC) was established in our laboratory. Li-Fraumeni syndrome fibroblast 041 cell line as described previously (18) was provided by Dr. Michael Tainsky (M.D. Anderson Cancer Center, Austin, TX). 041 cells stably transfected with V5-His-tagged DDB2 (N22 cells) were established in our laboratory. The NER-deficient cell line XP-C (GM15983) was obtained from NIGMS Human Genetic Cell Repository (Coriell Institute for Medical Research, Camden, NJ). These cell lines were cultured in either Dulbecco's modified Eagle's medium (DMEM) (OSU-2, XP-C, 041, and N22) or RPMI 1640 medium (HeLa-DDB2 and HeLa-XPC) supplemented with 10% fetal bovine serum at 37 °C in a humidified atmosphere with 5% CO2. The pcDNA3.1/XPC-V5-His expression constructs were generated in our laboratory as described previously (19) and transiently incorporated into XP-C cells using FuGENE 6 (Roche Diagnostics Corp., Indianapolis, IN). To study the function of Brg1, cells were transfected with hBrg1 siRNA (Dharmacon, Lafayette, CO) using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) 48 h before UV treatments.
UV Irradiation
Cells were washed in PBS once and exposed for desired times to 254 nm UV light from a germicidal lamp. For localized micropore UV irradiation, OSU-2 cells growing on glass coverslips were washed with PBS and UV irradiated through a 5 μm isopore polycarbonate filter (Millipore, Bedford, MA) as described previously (20).
Western Blotting
Western blot analysis was carried out as previously described (21). Brg1 protein levels in cells were determined by Western blotting using a rabbit polyclonal antibody purchased from Santa Cruz Biotechnology (Santa Cruz, CA). XPC protein levels were detected using a rabbit polyclonal XPC antibody generated in our laboratory (19). To serve as a loading control, total Lamin B levels in the cell lysates were detected using anti-Lamin B antibody (Santa Cruz Biotechnology).
Quantification of CPD and 6-4PP by Immuno-slot Blot (ISB) Analysis
A highly sensitive ISB assay was used to quantify the existence of UV-induced CPD or 6-4PP as described previously (22). In short, after UV irradiation, cells were lysed immediately or further incubated for the desired times. DNA was then isolated by phenol/chloroform. Same amount of DNA was loaded onto nitrocellulose membranes, and the amounts of CPD or 6-4PP were determined using monoclonal anti-CPD (TDM-2) or anti-6-4PP (64 M-2) antibody (MBL International Corporation, Woburn, MA).
Isolation of Chromatin-bound Proteins and HA or Flag Tag Pull-down
HeLa-XPC cells were treated with UV and allowed to repair for 30 min. Cells were then lysed in 1 ml of cytoplasmic lysis buffer (10 mm Tris-HCl, pH 7.9; 0.34 m sucrose, 3 mm CaCl2, 2 mm MgOAc, 0.1 mm EDTA, 1 mm dithiothreitol, 0.5% Nonidet P-40, and protease inhibitors) for 10 min on ice and the nuclei were collected by centrifugation at 3500 × g for 15 min. Nuclei were further lysed with 1 ml of nuclear lysis buffer (20 mm HEPES, pH 7.9, 3 mm EDTA, 10% glycerol, 150 mm KOAc, 1.5 mm MgCl2, 1 mm dithiothreitol, 0.1% Nonidet P-40, and protease inhibitors) for 10 min on ice and centrifuge at 10,000 × g for 30 min. The chromatin-enriched pellet was resuspended in nuclease incubation buffer (150 mm HEPES, pH 7.9, 150 mm KOAc, and protease inhibitors) and digested overnight at 4 °C with 250 units of Benzonase (Novagen, Gibbstown, NJ). The chromatin-bound proteins were collected by centrifugation at 18,000 × g for 30 min. Immunoprecipitation with anti-HA affinity matrix (Roche Applied Science) was conducted overnight at 4 °C to pull-down XPC, and the immunoprecipitates were subjected to Western blot analysis with anti-Brg1 antibody. HeLa-DDB2 cells were treated and processed the same way as HeLa-XPC cells except anti-FLAG M2 affinity gel (Sigma) was used to pull-down DDB2.
Chromatin Immunoprecipitation (ChIP) Assay
After desired UV treatments, cells were resuspended in 1% formaldehyde in PBS and incubated for 10 min at room temperature. Cross-linking reactions were quenched by the addition of glycine to a final concentration of 125 mm. Cells were then washed in PBS three times and resuspended in radioimmune precipitation assay buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 5 mm EDTA, 0.5% sodium deoxycholate, 0.1% SDS, 1% Nonidet P-40, and protease inhibitors) and kept on ice for 20 min. The cell lysates were sonicated to shear the chromatin and centrifuged at 18,000 × g for 10 min. The supernatants with 1 mg of protein were incubated with protein A/G-agarose for 2 h at 4 °C to preclear the lysate. After removal of protein A/G-agarose, the supernatant was incubated with 5 μg of anti-Brg1 or anti-acetyl-histone H3 (K9,14) antibody (Upstate Biotechnology, Billerica, MA) and protein A/G-agarose at 4 °C overnight. For control, the cell lysate was incubated with protein A/G-agarose and normal rabbit IgG. After extensive washing, the agarose-bound proteins/DNA were recovered by incubating in elution buffer (1% SDS and 0.1 m NaHCO3) at room temperature followed by incubating in 0.2 m NaCl for 5 h at 65 °C to reverse formaldehyde cross-linking. The samples were treated with RNase at 37 °C for 1 h followed by proteinase K at 37 °C overnight. DNA was isolated with phenol/chloroform and subjected to ISB analysis with anti-CPD antibody as described earlier.
Reverse Transcriptase PCR (RT-PCR)
OSU-2 cells were irradiated with UV at 20 J/m2 and allowed to repair for desired times. Total RNA was isolated with RNeasy Mini Kit (Qiagen, Valencia, CA) following the manufacturer's procedures. First strand cDNA was synthesized with 300 ng of total RNA using Superscript III reverse transcriptase (Invitrogen). Expression level of XPC and GAPDH were detected with touchdown PCR as follows: 95 °C for 5 min, 1 cycle; 94 °C for 30 s, 65 °C for 1 min, 72 °C for 1 min. The annealing temperature was decreased incrementally from 65 to 50 °C over ten cycles and then held at 50 °C for an additional 15 PCR cycles. The protocol was completed with one cycle of 72 °C for 10 min. The primers used for assessing XPC or GAPDH expression were 5′-CCA TGA ATG AAG ACA GCA ATG-3′ and 5′-ATT CCT CAT CAT CTC GAG CA-3′; 5′-GAA GGT GAA GGT CGG AGT-3′, and 5′-GAA GAT GGT GAT GGG ATT TC-3′, respectively.
Micrococcal Nuclease (MNase) Digestion Assay
MNase digestion assay, as described previously (21), was used to study UV-induced chromatin relaxation. Briefly, OSU-2 cells with or without hBrg1 siRNA transfection were UV irradiated at 200 J/m2 and lysed in hypotonic cell lysis buffer (10 mm Tris-HCl, pH 8.0, 10 mm MgCl2, 1 mm dithiothreitol, 25% glycerol, 0.2% Nonidet P-40, 0.5 μm spermidine, 0.15 μm spermine, and protease inhibitor mixture) for 10 min. Nuclei were pelleted by centrifugation and resuspended in 200 μl MNase buffer (10 mm Tris-HCl, pH 8.0, 50 mm NaCl, 300 mm sucrose, 3 mm MgCl2, and 1 mm CaCl2) and digested with MNase (Sigma) at room temperature for 5 min. The reaction was then stopped by adding 5× stop solution (0.1 m EDTA, 0.01 m EGTA, pH 8.0). DNA was isolated by phenol/chloroform and separated on a 1.2% agarose gel.
Cellular Protein Fractionation
OSU-2 cells with or without Brg1 siRNA transfection were UV irradiated at 20 J/m2 and incubated in the medium for 30 min. Cellular protein fractionation procedure, described earlier (23), was used to study the distribution of Brg1 or DDB2 in cells. In brief, the same number of cells in different treatments were lysed in hypotonic buffer (10 mm HEPES, pH 7.9, 10 mm KCl, 1.5 mm MgCl2, and protease inhibitor mixture) and treated with 0.1% Triton X-100. After centrifugation, the supernatant was saved as cytoplasmic proteins (S). The pellet containing nuclear protein was treated with higher concentrations of Triton X-100 (1%) in low salt (LS) buffer (10 mm Tris-HCl, pH 7.4, 0.2 mm MgCl2, and protease inhibitor) to recover the fraction of nucleoplasmic soluble proteins (TW). The proteins within the pellet were further separated with increasing concentrations (0.3, 0.5, and 2.0 m) of NaCl in LS buffer, and the fractions were designated as 0.3, 0.5, and 2.0, respectively. The protein fractions were analyzed by Western blotting with anti-Brg1 or anti-DDB2 antibodies.
Immunofluorescent Staining
After the desired treatment, OSU-2 cells were fixed with 2% paraformaldehyde in 0.5% Triton X-100 as previously described (20), and double stained with rabbit anti-XPC and mouse anti-CPD antibodies to study the recruitment of XPC to the UV damage site. The cells were also stained with rabbit anti-XPB (Santa Cruz Biotechnology) and mouse anti-CPD antibodies to study the recruitment of XPB to UV lesion. Mouse anti-XPG (Santa Cruz Biotechnology) or mouse-anti-PCNA (Santa Cruz Biotechnology) antibodies were used to localize XPG or PCNA to the damage sites. Anti-rabbit and anti-mouse IgG conjugated with Texas Red or FITC, respectively, were simultaneously used for the detection of primary antibody binding. Fluorescence images were obtained with a Nikon fluorescence microscope E80i (Nikon, Tokyo, Japan) fitted with appropriate filters for FITC and Texas Red. The digital images were then captured with a cooled CCD camera and processed with the help of its SPOT software (Diagnostic Instruments, Sterling Heights, MI).
RESULTS
Brg1 Is Required for Efficient Removal of UV-induced DNA Damage CPD but Not 6-4PP
A few existing studies in vitro as well as in vivo imply that SWI/SNF chromatin remodeling complex is an indispensable factor in NER occurring within the context of chromatin (24, 25). Furthermore, SWI/SNF seems involved in the cellular response to UV damage through checkpoint modulation (16). To test if Brg1, the ATPase subunit of SWI/SNF, affects the removal of UV-induced DNA damage in human normal cells, we knocked down the expression of Brg1 by transfecting OSU-2 cells with specific hBrg1 siRNA. After 24 or 48 h of transfection, cells were harvested, and cell lysates were subjected to Western blotting using anti-Brg1 antibodies. As shown in Fig. 1A, in comparison with control siRNA-transfected cells, Brg1 level was dramatically reduced at 24 h after Brg1 siRNA transfection and maintained at very low expression even at 48-h post-transfection. Next, we compared the efficiency of the removal of UV-induced CPD and 6-4PP between Brg1-proficient and Brg1 knockdown cells. OSU-2 cells with or without Brg1 siRNA transfection were UV irradiated at a dose of 10 J/m2 and allowed to repair for varying time periods. The removal of UV-induced CPD or 6-4PP was analyzed using ISB assay with anti-CPD or anti-6-4PP antibody. Upon Brg1 knockdown the efficiency of NER was markedly compromised as reflected by a decreased removal rate of UV-induced CPD. As shown in Fig. 1B, the difference in the rate of repair between Brg1-proficient and -deficient OSU-2 cells can be seen as early as 4 h following UV irradiation and is maximal at 24 h as CPD remaining was 42 and 62%, respectively. However, the repair of another UV-induced DNA lesion, 6-4PP, was not affected by Brg1 knockdown (Fig. 1C). These results with normal human fibroblasts are consistent with previously published data in human adrenal carcinoma cell line SW13 cells, which are deficient in Brg1 (16). All these findings indicate an important role of Brg1 in the efficient removal of CPD. However, it is still unclear whether Brg1 directly affects NER via regulating the expression, the recruitment of NER core factors, or is a consequence of its potential role in checkpoint regulation in response to UV damage (16).
FIGURE 1.

Brg1 is required for efficient removal of CPD but not 6-4PP. A, knockdown of Brg1 level by hBrg1 siRNA. OSU-2 cells were transfected with Brg1 siRNA (20 nm) using Lipofectamine 2000. Cell lysate was collected 24 or 48 h after transfection and subjected to Western blot analysis with anti-Brg1 antibody. Lamin B was detected to serve as an internal control. B and C, effect of Brg1 on the efficiency of CPD (B) or 6-4PP (C) removal. After 24 h of transfection with Brg1 siRNA, OSU-2 cells were cultured in serum-free medium for an additional 24 h. Cells were UV irradiated at 10 J/m2 and allowed to repair for indicated times. The same amount of genomic DNA was subjected to ISB analysis and the amount of CPD (B) or 6-4PP (C) was detected with anti-CPD or anti-6-4PP antibody, respectively, at each time point. Data presented in the graph on the right represent the relative CPD or 6-4PP remaining in each sample. Percentage CPD or 6-4PP was calculated from the intensity relative to initial irradiated sample. The data points represent an average of three independent measurements, with error bars representing standard deviation.
Brg1 Depletion Affects UV-induced Chromatin Remodeling but Has No Effect on the Recruitment of DDB2, XPC, and XPB
SWI/SNF, as a chromatin remodeling complex, is involved in DNA damage-induced chromatin decondensation, which enhances the access of DNA damage response proteins to the damage sites and facilitates repair (24). Therefore, we speculated that Brg1 may affect CPD removal by increasing UV-induced chromatin relaxation, and thus enhancing the recognition of DNA damage by NER factors. To affirm the role of Brg1 in UV-induced chromatin remodeling, OSU-2 cells, with or without Brg1 siRNA transfection, were grown in serum-free medium for 48 h before UV treatment to arrest cells in G0/G1 phase. The G0/G1 phase OSU-2 cells were then UV irradiated at 200 J/m2 and allowed to repair for additional 30 min. Cell nuclei were isolated and digested with different concentrations of MNase to determine the sensitivity of chromatin to nuclease. We used three concentrations of MNase and found that MNase at 0.5 unit/ml gave distinct digestion patterns between cells that were unirradiated versus UV irradiated as well as in the absence and presence of Brg1. Chromatin decondensation indicated by a more thorough digestion with MNase was observed at 30 min after UV irradiation in Brg1-proficient cells. However, in cells lacking Brg1, UV-induced chromatin relaxation was dramatically compromised (Fig. 2A). This result indicates that UV-induced chromatin decondensation heavily depends on functional Brg1.
FIGURE 2.

Brg1 is indispensable for UV-induced chromatin relaxation but has no effect on DDB2, XPC, and XPB recruitment to damage sites. A, knockdown of Brg1 affects UV-induced chromatin relaxation. OSU-2 cells were transfected with control siRNA or Brg1 siRNA for 24 h and serum starved for an additional 24 h and then UV irradiated at 200 J/m2. Cells were cultured in the same medium for 30 min, and nuclei were isolated and subjected to MNase digestion assay to determine the extent of chromatin relaxation. B, binding of Brg1 or DDB2 to UV-damaged chromatin. OSU-2 cells were transfected with control siRNA or Brg1 siRNA for 24 h and UV irradiated at 20 J/m2. Cells were incubated in the same medium for another 30 min and subjected to cellular protein fractionation. Five fractions were obtained according to the resistance to detergent and salt, e.g. S, cytoplasmic soluble protein; TW, nucleoplasmic soluble proteins; 0.3, proteins binding to chromatin loosely; 0.5, proteins binding to chromatin with intermediate affinity; 2.0, proteins binding to chromatin tightly. Each protein fraction, corresponding to an equivalent cell number, was analyzed by Western blotting with anti-Brg1 or anti-DDB2 antibody. C, recruitment of XPC or XPB to UV-damage. OSU-2 cells with or without Brg1 siRNA transfection were grown on coverslips and UV irradiated (40 J/m2) through a 5-μm isopore polycarbonate filter. Cells were incubated in medium for 30 min and then fixed with 2% paraformaldehyde. Cells were double stained with mouse anti-CPD antibody and rabbit anti-XPC antibody or rabbit anti-XPB antibody.
UV-DDB2 has emerged to be a critical factor for UV lesion detection and repair especially in the context of chromatin (26, 27). To study the recruitment of DDB2 to UV-damaged chromatin, we analyzed the binding of DDB2 to chromatin from differences of resistance in loosely or tightly bound proteins to extraction in different concentrations of detergent and salt. OSU-2 cells with or without Brg1 knockdown were UV irradiated at 20 J/m2 and further incubated for 30 min to allow the repair. The same number of cells were lysed and separated into five fractions as indicated in Fig. 2B, and proteins in each fraction were subjected to Western blot analysis using anti-Brg1 or anti-DDB2 antibody. As expected, Brg1 was tightly bound to the chromatin and mainly present in the 0.5 m and 2 m salt fractions. UV irradiation did not affect the distribution of Brg1 within cells. While OSU-2 cells transfected with Brg1 siRNA showed a dramatic reduction of Brg1 expression in each fraction, the distribution of Brg1 also did not exhibit any distinguishable change. In contrast, DDB2 demonstrated a clear translocation from low salt fraction in unirradiated cells (upper panel) to high salt fraction in UV-irradiated cells (lower panel) indicating a tighter association of DDB2 with UV-damaged chromatin. Once again, knockdown of Brg1 did not change the pattern of DDB2 distribution in different fractions of both unirradiated and irradiated cells (Fig. 2B). These data indicated that Brg1 does not influence the recruitment of DDB2 to UV-damaged chromatin.
XPC is another critical early damage recognition factor in NER (28, 29). To study the recruitment of XPC to damaged DNA, cells were treated via local micropore UV irradiation (40 J/m2) and allowed to repair for 30 min. Cells were then fixed and subjected to double staining with anti-XPC and anti-CPD antibodies. Recruitment of XPC to the UV-induced damage sites was distinctly observed after UV irradiation in both Brg1-proficient and Brg1-deficient cells (Fig. 2C). In fact, in both sets, more than 95% of CPD foci were co-stained with XPC, indicating the recruitment of XPC to the UV-damaged DNA sites was comparable between Brg1-proficient and -deficient cells. We also tested the co-localization of XPB, a component of the transcription factor TFIIH, which is essential for NER (6), to the damage sites and found that cells had normal recruitment of XPB to the UV damage sites despite Brg1 deficiency. These results indicate that Brg1 does not affect the incoming upstream damage recognition factors DDB2 and XPC as well as THIIH to UV-damaged DNA sites. Therefore, it prompted us to investigate whether Brg1 acts downstream of these early NER steps and possibly affects the later stage of NER.
Brg1 Interacts with XPC and Its Recruitment to CPD Depends on DDB2 and XPC
We have already shown that Brg1 does not affect DDB2 and XPC recruitment, indicating Brg1 may come to damage sites at a later stage. In fact, an in vitro study seems to indicate that function of Brg1 depends on XPC, which might come to the damage site without Brg1 and actually help recruit Brg1 itself to the damaged chromatin (14). To check this possibility in cells, we investigated the recruitment of Brg1 to damaged DNA in normal cells as well as in cells deficient in DDB2 or XPC. First, we determined if Brg1 is recruited to the UV damage site with a ChIP-ISB assay. OSU-2 cells were UV irradiated at 20 J/m2 and allowed to repair for 30 min. Cells were cross-linked with formaldehyde and sonicated to breakdown the chromatin. The supernatants with 1 mg total protein were incubated with anti-Brg1 antibody overnight and the DNA was recovered from the protein/DNA complex and subjected to ISB analysis with anti-CPD antibody. As shown in Fig. 3A, CPD level in the Brg1-bound chromatin is markedly higher compared with IgG control indicating the recruitment of Brg1 to CPD. However, as for 6-4PP, there was no obvious binding of Brg1 to this lesion indicating that Brg1 is recruited only to CPD but not to 6-4PP sites.
FIGURE 3.

Brg1 interacts with XPC, and its recruitment to CPD depends on both DDB2 and XPC. A, recruitment of Brg1 to UV-damage sites. OSU-2 cells were UV irradiated at 20 J/m2 and repaired for 30 min. Cells were fixed with 1% formaldehyde and chromatin was isolated and subjected to immunoprecipitation with anti-Brg1 or anti-AcH3 (Lys-9, 14) antibodies or normal rabbit IgG. An input sample represents 2% of the total chromatin subjected to ChIP. ISB was carried out with anti-CPD or anti-6-4PP antibody to determine the level of damage (CPD or 6-4PP) in the isolated chromatin. B, Brg1 recruitment to CPD depends on DDB2. 041 or N22 cells were treated with UV and processed as in A. C, Brg1 recruitment to CPD depends on XPC. XP-C or XP-C cells transiently transfected with XPC plasmid were treated and processed as in A. The IgG-nonspecific background was deducted from each specific band, and the relative amount of CPD or 6-4PP bound to Brg1 or AcH3 was then calculated relative to the respective input levels set as 100%. D, Brg1 interacts with XPC on the chromatin. HeLa-XPC cells were treated with UV at 20 J/m2 and allowed to repair for 30 min. Cell nuclei were isolated and digested with Benzonase nuclease to release chromatin-associated proteins and immunoprecipitated with anti-HA affinity gel. IP mixture was subjected to Western blot analysis with anti-Brg1 antibody. E, Brg1 does not interact with DDB2 on the chromatin. HeLa-DDB2 cells were treated with UV at 20 J/m2 and allowed to repair for 30 min. Cell nuclei were isolated and digested with Benzonase nuclease to release chromatin-associated proteins and immunoprecipitated with anti-FLAG M2 affinity gel. IP mixture was subjected to Western blot analysis with anti-Brg1 antibody.
To determine if Brg1 recruitment requires DDB2, we treated 041 cells (DDB2-deficient) and N22 cells (which stably express DDB2) with UV and the ChIP-ISB assay was performed to test the recruitment of Brg1 to CPD in the presence or absence of DDB2. As shown in Fig. 3B, cells lacking DDB2 (041 cells) do not have Brg1 recruitment to CPD whereas in DDB2-proficient cells there is a marked increase of CPD level in Brg1-bound chromatin, indicating that the recruitment of Brg1 to CPD happens only in the presence of DDB2. Similarly, using XPC-deficient XP-C cells and XP-C cells transiently transfected with XPC plasmid, we showed that Brg1 recruitment to CPD also requires XPC (Fig. 3C). These results indicate that Brg1 is recruited to UV-induced CPD, and this recruitment depends on both DDB2 and XPC.
Evidence from yeast has shown that SNF5 and SNF6, two subunits of SWI/SNF, interact with XPC on the chromatin and are involved in NER (15). To determine if Brg1 directly interacts with the core NER factor XPC in mammalian cells, HeLa-XPC cells were UV irradiated at 20 J/m2, and the soluble chromatin was immunoprecipitated with anti-HA affinity gel. IP product was subjected to Western blotting using anti-XPC antibody. As shown in Fig. 3D, the amount of Brg1 associated with XPC-HA dramatically increased after UV treatment, indicating a clear interaction of Brg1 with XPC on the chromatin in response to UV irradiation. We further detected the interaction between DDB2 and Brg1 in HeLa-DDB2 cells expressing FLAG-tagged DDB2 by using the same assay with anti-FLAG M2 affinity gel. As shown in Fig. 3E, Brg1 was not pulled down by DDB2 irrespective of cells being UV irradiated or not. This suggests that DDB2 may indirectly influence the recruitment of Brg1 to CPD through XPC since DDB2 has been shown to be required for the recruitment of XPC to UV-damaged sites (30, 31). Taken together, these results indicate that Brg1 recruitment to UV-damaged chromatin depends on DDB2 and XPC and is initiated via interaction with XPC on the chromatin.
Brg1 Protects XPC from UV-induced Degradation
It is not clear whether Brg1 affects the removal of UV lesions indirectly through checkpoint pathway modulation or directly, by affecting NER factors in mammalian cells. Here we provide evidence that Brg1 is involved in NER by affecting XPC protein stability in normal cells. Human fibroblasts with or without Brg1 siRNA transfection were exposed to UV at 20 J/m2 and allowed to repair for various time periods. In both cells, XPC levels rapidly decreased after UV irradiation. However, UV-induced XPC degradation was dramatically faster in cells lacking Brg1 (Fig. 4A). To rule out the possibility that Brg1 could be affecting XPC expression at the transcription level, OSU-2 cells were treated with UV and allowed to repair up to a 24-h period. RNA was isolated and subjected to RT-PCR. As shown in Fig. 4B, XPC mRNA level, normalized to GAPDH, was not significantly different between Brg1-proficient and Brg1-deficient cells either before or after UV treatment. Therefore, we conclude the observed difference in XPC protein level was not due to reduced transcription, but from postirradiation protein degradation.
FIGURE 4.

Brg1 protects XPC from degradation after UV. A, Brg1 affects XPC protein levels in response to UV. OSU-2 cells were transfected with control siRNA or Brg1 siRNA for 48 h and treated with UV at 20 J/m2 and allowed to repair for indicated time periods. Cell lysates were subjected to Western blotting with anti-XPC antibody. Brg1 level was detected to check the efficiency of siRNA knockdown and Lamin B was used as an internal control. B, Brg1 did not affect transcription level of XPC before or after UV. OSU-2 cells were transfected with either control siRNA or Brg1 siRNA for 48 h and treated with UV at 20 J/m2 and further cultured for the indicated time periods. RT-PCR was used to analyze mRNA levels of XPC. GAPDH mRNA was used as an internal control for normalization between samples. Values are expressed as fold increase relative to the XPC mRNA levels without UV irradiation. Each point represents the mean of three determinations with error bars representing standard deviations.
Brg1 Is Indispensable for XPG and PCNA Recruitment to UV Damage Site
We have shown that Brg1 was required for UV-induced chromatin relaxation and making DNA damage accessible to repair proteins. Nevertheless, the factors involved in the early NER, e.g. DDB2, XPC, and TFIIH were seen to recruit normally to the DNA damage site in the absence of Brg1. Furthermore, Brg1 also played a definitive role in maintaining the requisite level of XPC protein after UV irradiation. Thus, we surmised that Brg1, by affecting UV-induced chromatin decondensation and XPC stability, could be instrumental in repair process through its influence in the recruitment of factors functioning further downstream during late NER. To test this hypothesis, OSU-2 cells with or without Brg1 siRNA transfection were UV irradiated through a 5-μm pore filter and allowed to repair the damage for 30 min. Because we had already seen XPC co-recruitment in more than 95% of the CPD foci in both Brg1-proficient and -deficient cells, we used the XPC foci as an indicator of UV damage to study the recruitment of other downstream NER factors. As shown by double staining with rabbit anti-XPC and mouse anti-XPG antibodies in Fig. 5A, about 99% of XPG foci co-recruited to the XPC-marked UV damage sites in normal OSU-2 cells. However, in the cells transfected with Brg1 siRNA, the recruitment of XPG to UV-damaged DNA sites was drastically diminished. In fact, only 53% of XPG co-localized with XPC foci in Brg1-deficient cells. Because XPG is required for PCNA recruitment (32), we also tested the co-localization of PCNA to UV lesions. As shown in Fig. 5B, co-localization of PCNA with XPC foci was reduced by a half in Brg1-deficient cells. These results indicate that Brg1 facilitates NER through modulating the recruitment of downstream NER factors like XPG and PCNA to the damage sites.
FIGURE 5.
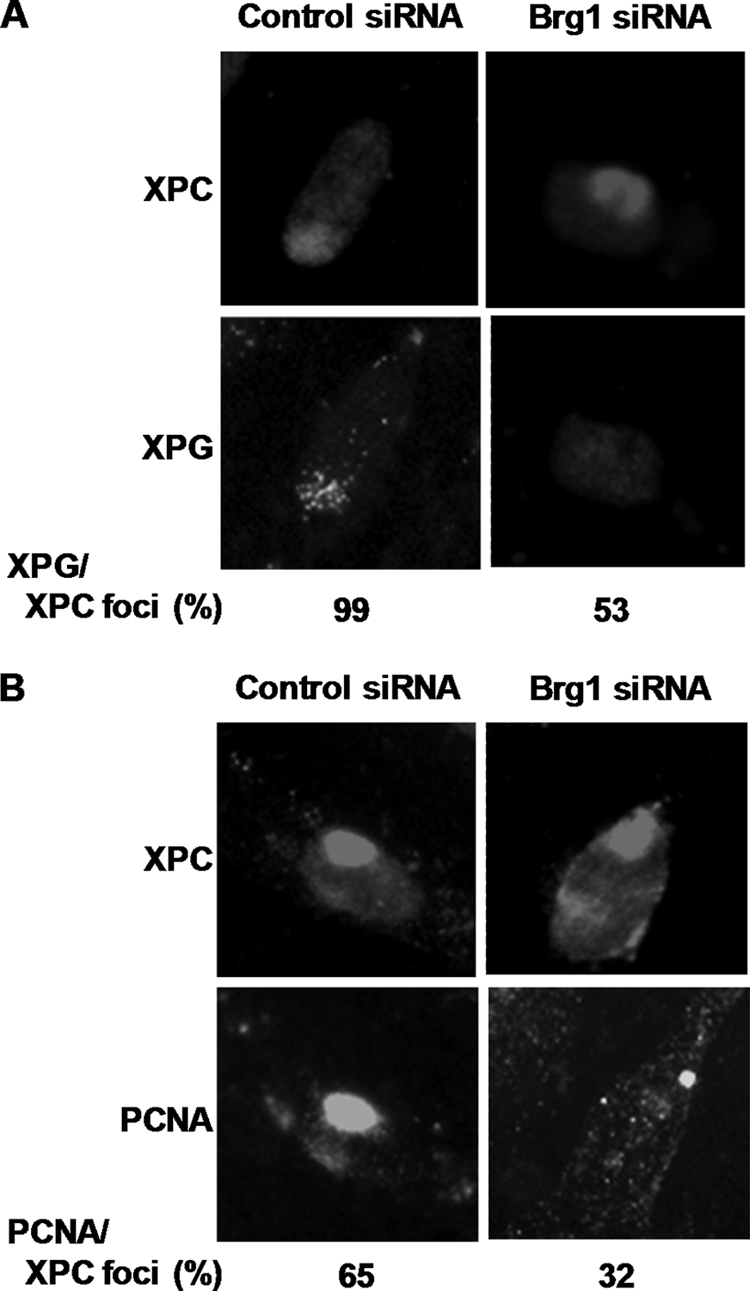
Brg1 affects the recruitment of XPG and PCNA to UV damage sites. A, recruitment of XPG to DNA damage sites. OSU-2 cells were UV treated at 40 J/m2 through a filter containing pores of 5 μm in diameter and double stained with rabbit anti-XPC antibody and mouse anti-XPG antibody. B, OSU-2 cells were treated and processed as in A and double stained with rabbit anti-XPC antibody and mouse anti-PCNA antibody. The percentage of foci-containing cells were obtained from scoring >300 cells within a uniformly defined field.
DISCUSSION
SWI/SNF ATP-dependent chromatin remodeling complex is an important player in DNA repair pathways including DSB repair, BER, and NER (2). Although SWI/SNF has been implicated in regulating the chromatin remodeling process in NER by a number of in vitro excision assays (13, 14), its exact role in NER has not been investigated in mammalian cells. In this report, we provide the first time evidence that Brg1 directly interacts with NER factor XPC, affecting NER protein stability (XPC) and/or factor recruitment (XPG and PCNA) leading to efficient repair in human cells.
Brg1 Modulation against Different Types of UV Lesions
We found that Brg1 is required for the removal of UV-induced DNA lesion CPD. This is expected since in vitro studies have suggested that SWI/SNF has an important role in chromatin remodeling and efficient NER of UV lesions (13, 14). Studies in yeast as well as in mammalian cells also indicated that SWI/SNF is indispensable for NER (15, 16). However, we found that Brg1 does not bind to another UV lesion 6-4PP and does not affect its repair. Although Hara et al. (13) also show an effect of SWI/SNF on NER they found that SWI/SNF is required for the repair of 6-4PP but not CPD. Because their repair assay was carried out in vitro with reconstituted NER factors and yeast SWI/SNF, whereas our study determined the repair efficiency in human cells, these disparate observations could be accounted by the differences of in vitro and in vivo system as well as human and yeast SWI/SNF activities. It should be noted that the repair of CPD within the nucleosomes is more severely inhibited than the repair of 6-4PP within the nucleosomes, indicating that the repair of CPD needs greater chromatin remodeling than 6-4PP (13). Thus, the access and repair of 6-4PP does not seem to strictly depend on chromatin remodeling as CPD. Besides, in the in vitro assay system, addition of SWI/SNF alone while enough for the repair of 6-4PP is insufficient for CPD repair. Moreover, there are additional factors in cells, besides SWI/SNF, to remodel chromatin. Although knockdown of one of them, e.g. SWI/SNF, would have some effect on chromatin decondensation, the chromatin can be partially opened to allow the repair of 6-4PP but not CPD. In fact, the study in yeast also shows that inactivation of SWI/SNF inhibits the removal of CPD from the silent HML locus (15).
Brg1 and NER
SWI/SNF plays an important role in chromatin remodeling by utilizing the energy from ATP hydrolysis. Thus, the ATPase function is crucial for its chromatin remodeling activity. SWI/SNF has two ATPase subunits Brg1 and Brm. Each of them form a discrete complex by interacting with other Brg1/Brm-associated factors (BAFs) and may have distinct roles in cellular processes (24). Here we show that Brg1 is indispensable for chromatin relaxation caused by UV in normal human fibroblasts. However, the accessibility of damaged DNA to NER factors DDB2, XPC, and XPB was not affected by Brg1, indicating that the recruitment of early repair initiation factors, DDB2, XPC and THIIH, to the damage site does not need the function of Brg1. Interestingly, on the other hand, binding of Brg1 to the UV lesion indeed depends on DDB2 and XPC. Furthermore, while DDB2 is indispensable for Brg1 recruitment, DDB2 did not show any interaction with Brg1 before or after UV. In contrast, the interaction between XPC and Brg1 clearly existed in non-irradiated cells and was further enhanced by UV treatment. Taken together, we propose a model for the sequence of these early events. After UV irradiation, DDB2 is recruited to the damage site that then helps recruit XPC to the UV lesion. XPC binds avidly to Brg1 and assists in loading Brg1 to the UV-damage site. Brg1 then modulates the chromatin structure which affects the assembly of downstream NER factors in a later stage of repair and consequently the repair efficiency. In fact, the sequence of these early events has been implied in a previous study conducted in vitro. This study has demonstrated that XPC, RPA, and XPA stimulate the remodeling activity of SWI/SNF and in turn stimulate the excision of the AAF-G adduct from the nucleosome core by NER (14). Both, this in vitro study and our in vivo study in mammalian cells, suggest that Brg1 does not affect the early stage (damage recognition) of NER and may be required for the late stage of the damage processing.
In support of the aforementioned model, we found the recruitment of incision factor XPG was inhibited in Brg1-deficient cells. Because PCNA recruitment to a UV lesion has been shown to require the presence of XPG at the damage site (33, 34), we checked the recruitment of PCNA and found that in Brg-1-deficient cells when XPG recruitment was impaired the PCNA recruitment was also hindered. Since both XPG and PCNA are crucial for the repair of UV lesions, this impaired recruitment of XPG and PCNA could be envisaged in the NER inhibition by Brg1 down-regulation. Since chromatin remodeling may be operational at multiple levels in the repair process, it is important to ascertain the steps of repair that are crucially affected by the chromatin remodeling. While a number of studies suggest that chromatin remodeling factors mostly impact the later stage of repair (14, 35–38), several other studies have also presented evidence for the involvement of chromatin remodeling in the early stage of repair (39–41). Our study has provided strongly supporting data for the role of human SWI/SNF chromatin remodeling factor in the later steps of NER.
Although we found that damage recognition by DDB2 and XPC does not require Brg1 activity, it does not necessarily preclude the possibility that DDB2 and XPC initial binding to DNA damage does need chromatin relaxation to access the damaged DNA. Histone modifications especially acetylation have been found to be stimulated by UV and enhance the accessibility of chromatin and recruitment of repair factors (42–44). Therefore, histone acetylation happening immediately after UV irradiation could initiate chromatin decondensation to make damaged DNA more accessible to damage recognition factors, DDB2 and XPC. Binding of XPC together with XPA, RPA, and TFIIH unfold and stabilize the disrupted nucleosome for recruiting of chromatin remodeling factor SWI/SNF and subsequent NER proteins (45).
Brg1 and XPC Degradation
An intriguing finding in this study is that Brg1 deficiency stimulates the UV-induced XPC protein degradation. XPC degradation, occurring within 30 min of UV exposure of cells, is a critical processing event for the recruitment of XPG and efficiency of NER (46). However, we found that in Brg1-deficient cells, where XPC degraded faster and at greater level than in normal cells, XPG recruitment and NER efficiency were both compromised. It seems that despite the need for XPC degradation to free up the damage site and render sufficient space for XPG recruitment, a basal level of XPC is still needed to assemble functional NER machinery. This would include the recruitment of factors like SWI/SNF and their functional participation to help recruit downstream factors for damage processing. Future studies are needed to clarify the mechanism of how Brg1 regulates XPC degradation and its role in NER. One possibility is that Brg1 protects XPC from degradation by promoting XPC modification. In support of this Wang et al. has reported that XPC degradation is ubiquitylation-independent. In fact, XPC modifications, including ubiquitylation and sumoylation, protect XPC from degradation (19, 46). Here too, we have found that in Brg1-deficient cells XPC has significantly fewer modifications compared with Brg1-proficient cells explaining the more dramatic degradation of XPC in Brg1-deficient cells. Therefore, it is easy to speculate that Brg1 is recruited by XPC, which in turn stabilizes it by regulating XPC modifications.
In summary, we provide evidence that Brg1 interacts with XPC and is recruited to a UV lesion in a DDB2- and XPC-dependent manner. Brg1, in turn, modulates UV-induced chromatin remodeling and XPC stability, and consequently promotes damage excision and repair synthesis by facilitating the recruitment of XPG and PCNA to the damage site. Collectively, our findings demonstrate the essential role of Brg1 in prompt elimination of UV-induced DNA damage by NER in mammalian cells.
Acknowledgments
We thank Drs. Yoshihiro Nakatani and Michael Tainsky for providing cell lines and Dr. Jan H. Hoeijmakers for GFPHA-His-tagged XPC construct.
This work was supported, in whole or in part, by Public Health Service National Institutes of Health Grants ES2388, ES12991, and CA93413 (to A. A. W.).
- NER
- nucleotide excision repair
- CPD
- cyclobutane pyrimidine dimer
- 6-4PP
- pyrimidine(6-4)pyrimidone photoproduct
- DDB
- damaged DNA-binding protein
- GG-NER
- global genomic NER
- SWI/SNF
- SWI/SNF chromatin remodeling complex
- ChIP
- chromatin immunoprecipitation assay
- PBS
- phosphate-buffered saline
- MNase
- micrococcal nuclease
- HA
- hemagglutinin
- ISB
- Immuno-slot Blot.
REFERENCES
- 1.Misteli T., Soutoglou E. (2009) Nat. Rev. Mol. Cell Biol. 10, 243–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dinant C., Houtsmuller A. B., Vermeulen W. (2008) Epigenetics. Chromatin. 1, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Laat W. L., Jaspers N. G., Hoeijmakers J. H. (1999) Genes Dev. 13, 768–785 [DOI] [PubMed] [Google Scholar]
- 4.Hanawalt P. C. (2002) Oncogene 21, 8949–8956 [DOI] [PubMed] [Google Scholar]
- 5.Sancar A. (1996) Annu. Rev. Biochem. 65, 43–81 [DOI] [PubMed] [Google Scholar]
- 6.Sugasawa K. (2006) J. Mol. Histol. 37, 189–202 [DOI] [PubMed] [Google Scholar]
- 7.Green C. M., Almouzni G. (2002) EMBO Rep. 3, 28–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Z. G., Wu X. H., Friedberg E. C. (1991) J. Biol. Chem. 266, 22472–22478 [PubMed] [Google Scholar]
- 9.Hara R., Mo J., Sancar A. (2000) Mol. Cell. Biol. 20, 9173–9181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smerdon M. J., Thoma F. (1990) Cell 61, 675–684 [DOI] [PubMed] [Google Scholar]
- 11.Wellinger R. E., Thoma F. (1997) EMBO J. 16, 5046–5056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ura K., Araki M., Saeki H., Masutani C., Ito T., Iwai S., Mizukoshi T., Kaneda Y., Hanaoka F. (2001) EMBO J. 20, 2004–2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hara R., Sancar A. (2003) Mol. Cell. Biol. 23, 4121–4125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hara R., Sancar A. (2002) Mol. Cell. Biol. 22, 6779–6787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gong F., Fahy D., Smerdon M. J. (2006) Nat. Struct. Mol. Biol. 13, 902–907 [DOI] [PubMed] [Google Scholar]
- 16.Gong F., Fahy D., Liu H., Wang W., Smerdon M. J. (2008) Cell Cycle 7, 1067–1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Venkatachalam S., Denissenko M. F., Wani A. A. (1995) Carcinogenesis 16, 2029–2036 [DOI] [PubMed] [Google Scholar]
- 18.Li J., Wang Q. E., Zhu Q., El-Mahdy M. A., Wani G., Praetorius-Ibba M., Wani A. A. (2006) Cancer Res. 66, 8590–8597 [DOI] [PubMed] [Google Scholar]
- 19.Wang Q. E., Zhu Q., Wani G., El-Mahdy M. A., Li J., Wani A. A. (2005) Nucleic Acids Res. 33, 4023–4034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Q. E., Zhu Q., Wani M. A., Wani G., Chen J., Wani A. A. (2003) DNA Repair 2, 483–499 [DOI] [PubMed] [Google Scholar]
- 21.Zhao Q., Barakat B. M., Qin S., Ray A., El-Mahdy M. A., Wani G., Arafa el-S., Mir S. N., Wang Q. E., Wani A. A. (2008) J. Biol. Chem. 283, 32553–32561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wani A. A., D'Ambrosio S. M., Alvi N. K. (1987) Photochem. Photobiol. 46, 477–482 [DOI] [PubMed] [Google Scholar]
- 23.Wang Q. E., Zhu Q., Wani G., Chen J., Wani A. A. (2004) Carcinogenesis 25, 1033–1043 [DOI] [PubMed] [Google Scholar]
- 24.Reisman D., Glaros S., Thompson E. A. (2009) Oncogene 28, 1653–1668 [DOI] [PubMed] [Google Scholar]
- 25.Zhang L., Jones K., Gong F. (2009) Biochem. Cell Biol. 87, 265–272 [DOI] [PubMed] [Google Scholar]
- 26.Hwang B. J., Toering S., Francke U., Chu G. (1998) Mol. Cell. Biol. 18, 4391–4399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tang J. Y., Hwang B. J., Ford J. M., Hanawalt P. C., Chu G. (2000) Mol. Cell 5, 737–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sugasawa K., Ng J. M., Masutani C., Iwai S., Van der Spek P. J., Eker A. P., Hanoaka F., Bootsma D., Hoeijmakers J. H. (1998) Mol. Cell 2, 223–232 [DOI] [PubMed] [Google Scholar]
- 29.Riedl T., Hanaoka F., Egly J. M. (2003) EMBO J. 22, 5293–5303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fitch M. E., Nakajima S., Yasui A., Ford J. M. (2003) J. Biol. Chem. 278, 46906–46910 [DOI] [PubMed] [Google Scholar]
- 31.Moser J., Volker M., Kool H., Alekseev S., Vrieling H., Yasui A., Van Zeeland A. A., Mullenders L. H. (2005) DNA Repair 4, 571–582 [DOI] [PubMed] [Google Scholar]
- 32.Mocquet V., Lainé J. P., Riedl T., Yajin Z., Lee M. Y., Egly J. M. (2008) EMBO J. 27, 155–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Staresincic L., Fagbemi A. F., Enzlin J. H., Gourdin A. M., Wijgers N., Dunand-Sauthier I., Giglia-Mari G., Clarkson S. G., Vermeulen W., Schärer O. D. (2009) EMBO J. 28, 1111–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aboussekhra A., Wood R. D. (1995) Exp. Cell Res. 221, 326–332 [DOI] [PubMed] [Google Scholar]
- 35.Downs J. A., Allard S., Jobin-Robitaille O., Javaheri A., Auger A., Bouchard N., Kron S. J., Jackson S. P., Côté J. (2004) Mol. Cell 16, 979–990 [DOI] [PubMed] [Google Scholar]
- 36.Morrison A. J., Highland J., Krogan N. J., Arbel-Eden A., Greenblatt J. F., Haber J. E., Shen X. (2004) Cell 119, 767–775 [DOI] [PubMed] [Google Scholar]
- 37.van Attikum H., Fritsch O., Hohn B., Gasser S. M. (2004) Cell 119, 777–788 [DOI] [PubMed] [Google Scholar]
- 38.Nakanishi S., Prasad R., Wilson S. H., Smerdon M. (2007) Nucleic Acids Res. 35, 4313–4321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bao Y., Shen X. (2007) Curr. Opin. Genet. Dev. 17, 126–131 [DOI] [PubMed] [Google Scholar]
- 40.Chai B., Huang J., Cairns B. R., Laurent B. C. (2005) Genes Dev. 19, 1656–1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Park J. H., Park E. J., Lee H. S., Kim S. J., Hur S. K., Imbalzano A. N., Kwon J. (2006) EMBO J. 25, 3986–3997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ramanathan B., Smerdon M. J. (1986) Carcinogenesis 7, 1087–1094 [DOI] [PubMed] [Google Scholar]
- 43.Smerdon M. J., Lan S. Y., Calza R. E., Reeves R. (1982) J. Biol. Chem. 257, 13441–13447 [PubMed] [Google Scholar]
- 44.Ramanathan B., Smerdon M. J. (1989) J. Biol. Chem. 264, 11026–11034 [PubMed] [Google Scholar]
- 45.Baxter B. K., Smerdon M. J. (1998) J. Biol. Chem. 273, 17517–17524 [DOI] [PubMed] [Google Scholar]
- 46.Wang Q. E., Praetorius-Ibba M., Zhu Q., El-Mahdy M. A., Wani G., Zhao Q., Qin S., Patnaik S., Wani A. A. (2007) Nucleic Acids Res. 35, 5338–5350 [DOI] [PMC free article] [PubMed] [Google Scholar]