

Abstract
One key mechanism for endothelial dysfunction is eNOS uncoupling, whereby eNOS generates O2•− rather than NO, due to deficient eNOS cofactor tetrahydrobiopterin (BH4). This study was designed to examine the effect of BH4 deficiency on cardiac morphology and function as well as the impact of metallothionein (MT) on BH4 deficiency-induced abnormalities, if any. FVB and cardiac-specific MT transgenic mice were exposed to 2,4-diamino-6-hydroxy-pyrimidine (DAHP, 10 mmol/l, 3 wks), an inhibitor of the BH4 synthetic enzyme GTP cyclohydrolase I. DAHP reduced plasma BH4 levels by 85% and elevated blood pressure in both FVB and MT mice. Echocardiography found decreased fractional shortening and increased end systolic diameter in DAHP-treated FVB mice. Cardiomyocytes from DAHP-treated FVB mice displayed enhanced O2•− production, contractile and intracellular Ca2+ defects including depressed peak shortening and maximal velocity of shortening/relengthening, prolonged duration of relengthening, reduced intracellular Ca2+ rise and clearance. DAHP triggered mitochondrial swelling/myocardial filament aberrations and mitochondrial O2•− accumulation, assessed by TEM and MitoSOX Red fluorescence, respectively. DAHP also promoted the L-NAME inhibitable O2•− production and eNOS phosphorylation at Thr497. Although MT had little effect on cardiac mechanics and ultrastructure, it attenuated DAHP-induced defects in cardiac function, morphology, O2•− production and eNOS phosphorylation (Thr497). The DAHP-induced cardiomyocyte mechanical responses were alleviated by in vitro BH4 treatment. DAHP inhibited mitochondrial biogenesis, mitochondrial uncoupling protein 2 (UCP2) and chaperone HSP90, all but UCP2 was rescued by MT. Our data suggest a role of BH4 deficiency in cardiac dysfunction and therapeutic potential of antioxidants against eNOS uncoupling in the hearts.
Keywords: BH4, eNOS uncoupling, cardiomyocyte mechanics, metallothionein, superoxide
INTRODUCTION
Nitric oxide (NO) is an essential regulator of cardiac contraction although a consensus as to the main cardiac physiological function of NO has only recently emerged by means of genetic deletion or overexpression of all 3 NOS isoforms in cardiomyocytes 1, 2. Despite their apparent promiscuity, each NOS isozyme appears to correlate with specific signaling events, partially due to their subcellular compartmentation with colocalized effectors and limited NO diffusibility. eNOS is believed to sustain normal cardiac excitation-contraction coupling 3, which is consistent with our data showing that eNOS gene transfer improves cardiomyocyte contractile and intracellular Ca2+ properties via a PI-3 kinase-mediated mechanism 4. An ample amount of experimental evidence has indicated that eNOS can be uncoupled due to enhanced oxidative stress under certain disease states such as hypertension, atherosclerosis and diabetes 5–7. Functional eNOS oxidizes L-arginine to L-citrulline and NO in the presence of the essential NOS cofactor tetrahydrobiopterin (BH4) 8, 9. In disease state, BH4 is oxidized by high peroxynitrite (ONOO-) formed by the NO - superoxide (O2•−) reaction, rendering eNOS to produce O2•− rather than NO in the vasculature (a phenomena known as “eNOS uncoupling”) 9, 10. Although eNOS uncoupling plays an important role for the pathogenesis of vascular diseases such as endothelial dysfunction and hypertension 10, 11, its impact on the pathophysiology of heart diseases remains poorly understood. We hypothesized that eNOS uncoupling may contribute to the pathogenesis of cardiac dysfunction whereas “eNOS recoupling” through the preserved redox balance is permissive to the maintenance of physiological cardiac function. To this end, the present study was designed to examine the effect of eNOS uncoupling through inhibition of the rate-limiting BH4 synthetic enzyme GTP cyclohydrolase I on murine myocardial contractile function and ultrastructure. Given the prominent role of free radical accumulation and antioxidant reserve in the development of eNOS uncoupling 7, 10, the impact of GTP cyclohydrolase I inhibition was also examined in transgenic mice with overexpression of the thiol-rich metallothionein (MT), a heavy metal scavenger known to preserve the hearts against environmental toxic insults and oxidative damage 12, 13. 2,4-diamino-6-hydroxy-pyrimidine (DAHP), a selective inhibitor of GTP cyclohydrolase I, was used to block BH4 synthesis 14. In an effort to elucidate the cellular mechanisms involved in DAHP and MT-induced myocardial alterations, if any, special attention was drawn towards O2•− production and the peroxisome proliferator-activated receptor γ (PPARγ) coactivator 1α (PGC-1α)-regulated mitochondrial biogenesis. Mitochondrial uncoupling protein 2 (UCP2), a novel member of the mitochondrial anion carrier family, and heat shock protein 90 (HSP90), an essential mitochondrial chaperone, are both involved in the control of mitochondrial function and reactive oxygen species (ROS) generation 15, 16. Expression of both proteins in association with mitochondrial O2•− accumulation were evaluated in myocardium from DAHP-treated wild-type Friend Virus B (FVB) and MT transgenic mice.
MATERIALS AND METHODS
See the Methods section in the online supplement for additional details (http://hyper.ahajournals.org).
Experimental animals and DAHP treatment
The experimental protocols have been approved by the Animal Use and Care Committee at the University of Wyoming. Adult male wild-type FVB and cardiac-specific MT overexpression transgenic mice (5 months of age) were fed 2,4-diamino-6-hydroxy-pyrimidine (DAHP, 10 mmol/l) in drinking water for 3 weeks 14. Since DAHP affects neurotransmitter release such as serotonin and catecholamine which may alter glucose metabolism 17, serum glucose levels were monitored.
Measurement of total biopterin and BH4 levels
Plasma BH4 and total biopterin (BH4, dihydropterin [BH2] and oxidized biopterin [B]) levels were measured by HPLC with florescence detection as described 18.
Echocardiographic assessment
Cardiac geometry and function were evaluated using a 2-D guided M-mode echocardiography equipped with a 15-6 MHz linear transducer. Fractional shortening was calculated from end-diastolic diameter (EDD) and end-systolic diameter (ESD) using the equation of (EDD-ESD)/EDD 19.
Intracellular fluorescence measurement of O2•−
Myocytes loaded with dihydroethidium (DHE, 5 μmol/l) were sampled using an Olympus BX-51 microscope equipped with an Olympus MagnaFire™ SP digital camera and an ImagePro image analysis software 20.
HPLC fluorescence detection of O2•−
2-hydroxyethidium (2-OH-E+), a product formed from the reaction of O2•− and hydroethidine (HE), is considered a more suitable probe for intracellular O2•− 21. HE, ethidium (E+) and 2-OH-E+ were separated by an HPLC system equipped with a fluorescence detector 21.
MitoSOX Red fluorescence measurement of mitochondrial O2•−
Cells loaded with MitoSOX Red (2 μmol/l) were rinsed and MitoSOX Red fluorescence intensity was captured at 510/580 nm using an Olympus BX51 microscope equipped with a digital cooled charged-coupled device camera 22.
Cardiomyocyte isolation and mechanics
Mouse cardiomyocytes were isolated as described 12. Mechanical properties of myocytes were assessed using an IonOptix™ soft-edge system 12. Cell shortening and relengthening were assessed using the following indices: peak shortening (PS), time-to-PS (TPS), time-to-90% relengthening (TR90) and maximal velocities of shortening/relengthening (± dL/dt).
Intracellular Ca2+ transients and sacroplasmic reticulum (SR) Ca2+ release
Myocytes loaded with fura-2/AM (0.5 μmol/l) were exposed to lights emitted through either a 360 or a 380 nm filter. Fluorescence emissions were detected between 480–520 nm 12. SR Ca2+ release was assessed by rapid puff of caffeine (10 mmol/l)-induced intracellular Ca2+ transients in fura-2-loaded myocytes 23.
Electron microscopy
Following perfusion fixation, left ventricular and interventricular septal tissues were minced to 1 mm3 followed by fixation and postfixation. Tissue blocks were embedded in Epon/Araldite and cured 48 hr at 60°C. Thin sections were collected on naked copper (300-mesh) grids, stained with lead citrate and uranyl acetate, and imaged with a Hitachi 7500 transmission electron microscope 19.
Western blot analysis
Membrane proteins were separated on SDS-polyacrylamide gels and were transferred to polyvinylidene difluoride membranes. The membranes were blocked with 5% milk and then incubated overnight with specific antibodies. The antigens were detected by the luminescence method 19.
Total RNA extraction, cDNA synthesis, reverse transcription and real-time PCR
Total RNA extraction, cDNA synthesis and real-time PCR were carried out as described 12.
Data analysis
Mean ± SEM. Statistical significance (p < 0.05) was estimated by ANOVA followed by a Tukey's test for post hoc analysis where appropriate.
RESULTS
General morphometric and echocardiographic characteristics
As shown in Table 1, DAHP and MT transgene failed to elicit any effects on body, heart, liver or kidney weights, organ size (normalized to body weight), heart rate, EDD, LV mass (absolute and normalized) and blood glucose. DAHP moderately but significantly elevated both systolic and diastolic blood pressure, the effect of which was unaffected by MT. DAHP treatment significantly increased ESD but reduced wall thickness and fractional shortening, which were significantly attenuated by MT transgene. MT itself did not affect myocardial geometry and fractional shortening. As expected, blockade of GTP cyclohydrolase I with DAHP significantly reduced total biopterin (by 64%) and BH4 (by 85%) levels regardless of MT overexpression or not. Measurement of O2•− levels using DHE fluorescence revealed enhanced O2•− levels in cardiomyocytes from DAHP-treated FVB but not MT mice. DAHP-elicited increased of O2•− production was significantly attenuated by the NOS inhibitor L-NAME (100 μmol/l), indicating the presence of eNOS uncoupling (Fig. 1A–1C). Furthermore, DAHP promoted eNOS phosphorylation at the Thr497 residue without any effect on total eNOS expression, which is another hallmark of eNOS uncoupling 9. Consistent with its effect on the DAHP-induced O2•− production, MT nullified the DAHP-elicited phosphorylation of eNOS at Thr497 (Fig. 1D). Given that oxidation of HE to 2-OH-E+ was used as a more suitable measurement to quantify the production of O2*− 21, HPLC fluorescence detection was performed for 2-OH-E+ levels Our data revealed that DAHP stimulated the SOD-inhibitable O2•− production in FVB but not MT mice. Our result also indicated that MT transgene did not have any effect on 2-OH-E+ production (Fig. 2).
Table 1.
General feature of FVB and MT mice with or without DAHP treatment for 3 weeks.
| Parameter | FVB (34) | MT (34) | FVB-DAHP (32) | MT-DAHP (31) |
|---|---|---|---|---|
| Body Weight (g) | 26.44 ± 0.42 | 27.48 ± 0.67 | 26.68 ± 0.45 | 27.56 ± 0.65 |
| Heart Weight (HW, mg) | 153 ± 7 | 156 ± 8 | 156 ± 8 | 148 ± 7 |
| HW/Body Weight (mg/g) | 5.77 ± 0.23 | 5.61 ± 0.20 | 5.89 ± 0.30 | 5.41 ± 0.21 |
| Liver Weight (LW, g) | 1.42 ± 0.05 | 1.40 ± 0.07 | 1.41 ± 0.05 | 1.38 ± 0.05 |
| LW/Body Weight (mg/g) | 53.7 ± 1.7 | 50.4 ± 1.6 | 53.1 ± 1.4 | 50.3 ± 1.4 |
| Kidney Weight (KW, g) | 0.341 ± 0.011 | 0.346 ± 0.014 | 0.360 ± 0.013 | 0.369 ± 0.014 |
| KW/Body Weight (mg/g) | 12.89 ± 0.30 | 12.45 ± 0.27 | 13.60 ± 0.41 | 13.34 ± 0.31 |
| Systolic BP (mmHg) | 103.3 ± 3.1 | 105.5 ± 3.2 | 145.8 ± 10.7* | 140.5 ± 6.3* |
| Diastolic BP (mmHg) | 72.5 ± 10.0 | 75.5 ± 5.5 | 117.0 ± 12.7* | 101.8 ± 8.6* |
| Blood Glucose (mM) | 5.10 ± 0.26 | 5.34 ± 0.27 | 5.41 ± 0.25 | 5.22 ± 0.30 |
| Heart Rate (bpm) | 545 ± 15 | 541 ± 20 | 557 ± 24 | 544 ± 19 |
| Wall Thickness (mm) | 0.871 ± 0.049 | 0.927 ± 0.063 | 0.712 ± 0.043* | 0.868 ± 0.019† |
| EDD (mm) | 2.32 ± 0.16 | 2.29 ± 0.21 | 2.04 ± 0.14 | 2.24 ± 0.16 |
| ESD (mm) | 1.15 ± 0.11 | 1.11 ± 0.12 | 1.42 ± 0.09* | 1.23 ± 0.10† |
| LV Mass (mg) | 66.9 ± 6.2 | 62.4 ± 9.2 | 53.6 ± 9.6 | 66.9 ± 8.5 |
| Normalized LV Mass (mg/g) | 2.35 ± 0.20 | 2.27 ± 0.30 | 1.92 ± 0.34 | 2.42 ± 0.28 |
| Fractional Shortening (%) | 50.6 ± 2.6 | 50.9 ± 4.1 | 36.7 ± 1.9* | 45.3 ± 2.8† |
Mean ± SEM,
p < 0.05 vs. FVB group,
p < 0.05 vs. FVB-DAHP group,
number of mice per group is given in parentheses.
Fig. 1.

Effect of DAHP on content of total biopterin (panel A), BH4 (panel B), O2*_ levels in the absence or presence of the NOS inhibitor L-NAME (100 μmol/l) (panel C) and p-eNOS (Thr497) (panel D) in FVB and MT mice. Inset: Representative gel blots of p-eNOS and total eNOS. Mean ± SEM, n = 5–7, * p<0.05 vs. FVB, # p<0.05 vs. FVB-DAHP group.
Fig. 2.

HPLC identification of 2-OH-E+ from the O2•_/HE reaction in cardiomyocytes. Panels A – D display the HPLC chromatograms from FVB (A), FVB-DAHP (B), MT (C) and MT-DAHP (D) groups. Panel E: Standard HPLC peak intensity of authentic 2-OH-E+. Panel F: Levels of 2-OH-E+ in FVB, FVB-DAHP, MT and MT-DAHP groups with or without copper zinc SOD (100 units/ml). 2-OH-E+: 2-Hydroxyethidium (labeled peak 1); E+: Ethidium (labeled peak 2). Mean ± SEM, n = 100–120 cells per group, * p<0.05 vs. FVB, # p<0.05 vs. respective FVB-DAHP group, ** p<0.05 vs. FVB-DAHP group in the absence of SOD.
Effect of DAHP and BH4 on cardiomyocyte mechanical and fluorescent properties
DAHP treatment had no overt effect on cell phenotype (data not shown). The resting cell length (CL) of cardiomyocytes was comparable between FVB and MT groups, regardless of DAHP treatment. Cardiomyocytes from the FVB-DAHP group exhibited significantly depressed PS, reduced ± dL/dt and prolonged TR90 associated with comparable TPS. Interestingly, cardiac overexpression of MT abrogated the DAHP-induced changes in PS, ± dL/dt and TR90 without eliciting any significant effect by itself. To further examine the role of BH4 deficiency in these mechanical aberrations, cardiomyocytes from DAHP-treated FVB mice were incubated in vitro with BH4 (10 μmol/l) for 4 hrs prior to mechanical assessment. Our results depicted that BH4 significantly alleviated the DAHP-induced cardiomyocyte mechanical dysfunction in a manner reminiscent of MT transgene (Fig. 3). To explore the mechanism of action behind DAHP, MT and BH4-induced mechanical responsiveness, intracellular Ca2+ transients were recorded using fura-2 fluorescence in cardiomyocytes from FVB and MT mice with or without DAHP treatment. Fig. 4 displays that DAHP depressed the electrically-stimulated rise in fura-2 fluorescent intensity (ΔFFI), SR Ca2+ release and prolonged intracellular Ca2+ decay without affecting baseline intracellular Ca2+ FFI. While MT itself did not affect any of these intracellular Ca2+ properties, it prevented the DAHP-induced depression in ΔFFI, SR Ca2+ release and intracellular Ca2+ clearance. Consistent with the mechanical data, in vitro BH4 incubation significantly attenuated the DAHP-induced dysregulation in intracellular Ca2+ homeostasis.
Fig. 3.

Effect of DAHP-induced response on cardiomyocyte contractile function in FVB and MT mice. A cohort of cardiomyocytes from FVB and FVB-DAHP groups was incubated with BH4 (10 μmol/l) for 4 hrs prior to mechanical assessment. (A): Resting cell length; (B): Peak cell shortening; (C): + dL/dt; (D): − dL/dt; (E): TPS; and (F): TR90. Mean ± SEM, n = 132–133 cells from 6 mice per group, * p<0.05 vs. FVB, # p<0.05 vs. FVB-DAHP.
Fig. 4.

Effect of DAHP on intracellular Ca2+ transients and SR Ca2+ release in cardiomyocytes from FVB and MT mice. A cohort of cardiomyocytes from FVB and FVB-DAHP groups was incubated with BH4 (10 μmol/l) for 4 hrs prior to intracellular Ca2+ assessment. (A): Baseline intracellular Ca2+ fura-2 fluorescent intensity (FFI); (B): Change of fura-2 fluorescence intensity in response to electrical stimulation (ΔFFI); (C): Intracellular Ca2+ fluorescence decay rate; and (D): SR Ca2+ release. Mean ± SEM, n = 68–70 cells (7 cells for SR Ca2+ release) per group, * p<0.05 vs. FVB; # p<0.05 vs. FVB-DAHP.
Electron microscopy in myocardium from FVB and MT mice following DAHP treatment
Without DAHP treatment, little ultrastructural difference was noticeable in myocardial samples between FVB and MT groups. DAHP treatment triggered extensive focal damage in left ventricular and intraventricular septal (data not shown) myocardial tissue sections in FVB group. This was evidenced by cytoarchitectural damage including mitochondrial swelling and overtly disrupted sarcomeres and myofilament array. Consistent with the mechanical observations, MT alleviated the DAHP-induced cardiac unltrastructural damage (Fig. 5). Myocardial tissues from MT-DAHP mice were ultrastructurally indistinguishable from control samples.
Fig. 5.
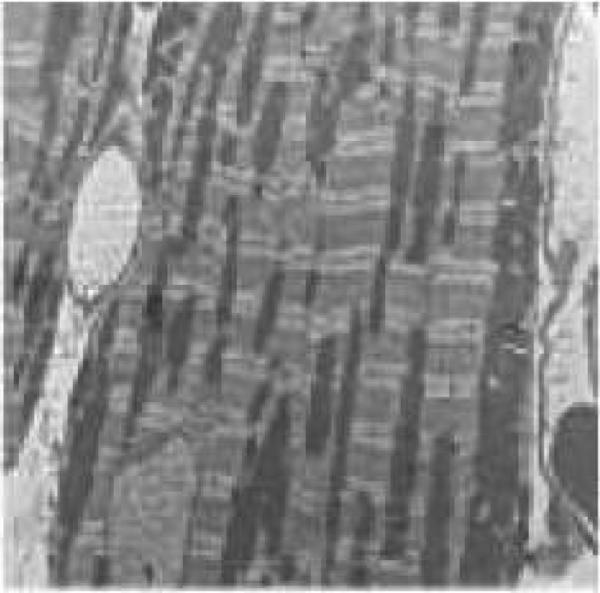
Transmission electron microscopic micrographs of left ventricular heart tissues from FVB and MT mice with or without DAHP treatment. (A): FVB; (B): FVB-DAHP; (C): MT; and (D): MT-DAHP. Myocardial tissues in A, C and D appear normal with myofibrils composed of regular and uninterrupted sarcomeres separated by continuous rows of normal mitochondria. In the myocardium from FVB-DAHP mice, irregularly shaped mitochondria and myelin figures are randomly distributed between highly disrupted myofibrils.
Mitochondrial biogenesis: PGC-1α, its downstream factors and mtDNA copy number
Given the above ultrastructural finding of mitochondrial anomalies in response to DAHP treatment and the proven association between eNOS uncoupling and mitochondrial damage in endothelial cells 24, the impact of DAHP and MT on the expression of the mitochondrial biogenesis regulator PGC-1α and its downstream factors including nuclear respiratory factor 1, 2 (NRF1, NRF2), mitochondrial transcription factor A (mtTFA) as well as mtDNA copy number 25 was examined. Our data revealed that DAHP significantly down-regulated PGC-1α by ~20% associated with marked reductions in mRNA levels of NRF1, NRF2 and mtTFA. MT effectively alleviated the DAHP-induced depression of PGC-1α and its downstream nuclear factors with little effect by itself. The mtDNA copy number, a key index for oxidative mitochondrial damage and mitochondrial biogenesis, was also downregulated following DAHP treatment, the effect of which was reversed by MT (Fig. 6, S1).
Fig. 6.

RT-PCR measurement of (A) nuclear respiratory factor 1 (NRF1), (B) nuclear respiratory factor 2 (NRF2), (C) mitochondrial transcription factor A (mtTFA) and (D) mtDNA copy number in left ventricles from FVB and MT mice with or without DAHP treatment. Mean ± SEM, n = 5–6, * p<0.05 vs. FVB, # p<0.05 vs. FVB-DAHP.
Western blot analysis for UCP2, HSP90 and mitochondrial O2•− production
Immunoblot analysis exhibited that DAHP treatment significantly reduced expression of mitochondrial uncoupling protein UCP2 and mitochondrial chaperone HSP90, both of which are closely associated with mitochondrial dysfunction, oxidative stress and heart failure 15, 16. Although MT transgene itself did not alter protein expression of UCP2 and HSP90, it attenuated the DAHP-induced decline in protein expression of HSP90 but not UCP2 (see online supplement Fig. S1). To further evaluate mitochondrial damage and its potential contribution to the enhanced intracellular O2•− generation, mitochondrial O2•− production was detected using the MitoSOX Red probe. DAHP significantly promoted mitochondrial O2•− production in cardiomyocytes from FVB but not MT mice. MT itself did not elicit any significant effect on mitochondrial O2•− production (see online supplement Fig. S2).
DISCUSSION
The major findings of our study are that BH4 deficiency with DAHP treatment directly leads to moderate elevation in blood pressure, cardiac contractile dysfunction, intracellular Ca2+ dysregulation, deranged myocardial ultrastructure, loss of mitochondrial biogenesis, reduced expression of UCP2 and HSP90, as well as enhanced intracellular/mitochondrial O2•− levels and theronine 497 phosphorylation of eNOS, all of which with the exception of elevated blood pressure and downregulated UCP2 were alleviated by the free radical scavenger MT. Our study demonstrated for the first time myocardial contractile and intracellular Ca2+ dysfunctions associated with morphological aberration associated with BH4 deficiency and eNOS uncoupling. The deranged cardiac ultrastructural integrity including mitochondrial swelling and anomalies in contractile filament further confirmed that the myopathy is focal to cardiomyocytes and mitochondria. Our observation that MT abrogates the DAHP-induced changes in cardiac mechanical function and ultrastructural integrity indicates the therapeutic potential of antioxidants in eNOS uncoupling-induced cardiac injury.
Inhibition of GTP cyclohydrolase I is considered a rather selective tool to assess the role of de novo synthesis of BH4 in a given biological system 14, 17. To this end, DAHP has been widely used as the prototypical GTP cyclohydrolase I inhibitor 14, 17, 26. Our present study revealed that specific GTP cyclohydrolase I inhibition leads to elevated blood pressure which is not affected by cardiac overexpression of MT, thus not favoring a role of blood pressure regulation in the DAHP and MT-induced responses on myocardial function and ultrastructure. DAHP markedly reduced levels of both total biopterin and BH4, depressed peak shortening, depressed maximal velocities of shortening/relengthening, prolonged relengthening duration and intracellular Ca2+ clearance as well as reduced intracellular Ca2+ recruitment and SR Ca2+ release capacity. These observations indicated the presence of compromised cardiac contractile function, consistent with our previous report of cardiac excitation-contraction dysfunction under oxidant injury 19. The deleterious effects of DAHP may be explained by increased O2•− due to the uncoupled eNOS. BH4 depletion is known to prompt eNOS to produce O2•− instead of NO as a result of the theronine 497 rather than serine 1179 phosphorylation of eNOS 9, consistent with our finding of enhanced Thr497 phosphorylation. Our observation that L-NAME is capable of inhibiting the DAHP-elicited O2•− production further consolidated the presence of eNOS uncoupling in our current experimental setting. O2•− accumulation has been demonstrated to result in myocardial contractile dysfunction, rigor and cytosolic Ca2+ overload 27, 28. In addition, O2•− and other free radicals may disrupt intracellular Ca2+ homeostasis through inhibition of L-type Ca2+ currents, SR Ca2+ load, Na+-Ca2+ exchange and SR Ca2+ uptake 27, 28, en route to compromised cardiac contractility, contraction and relaxation duration consistent with the reduced fractional shortening, PS, ± dL/dt, prolonged TR90 and intracellular Ca2+ decay found in our study. TPS and resting intracellular Ca2+ were spared by DAHP treatment, indicating possible disparity in the sensitivity of cardiac contractile/Ca2+ regulating proteins to myocardial eNOS uncoupling. Prolonged duration of contraction and relaxation, delayed intracellular Ca2+ clearance and dampened intracellular Ca2+ release are considered hallmarks of cardiomyopathy in a number of disease states such as heart failure, hypertension, obesity, diabetes and oxidative stress 12, 13, 19, 23, 28, 29. The alterations in mechanical and intracellular Ca2+ properties observed in our study, although relatively small (~20%), were consistent with the degree change from failing human hearts 29, and were further supported by the apparent cardiac remodeling (changes in wall thickness and ESD) and focal structural damage (mitochondrial swelling and displacement of cardiac contractile filaments) following chronic DAHP treatment. Mitochondria, a main target for O2*−, exhibit mitochondrial permeability transition leading to mitochondrial swelling, cytochrome c release, loss of biogenesis and function in response to sustained O •− challenge 24. This is consolidated by our data of lost mitochondrial biogenesis, UCP2 and HSP90 as well as mitochondrial O2•− generation following DAHP treatment. Collectively, our data favored an essential role of BH4 and functional eNOS in the maintenance of cardiac functional and ultrastructural integrity under physiological conditions.
Perhaps the most interesting finding of our study is that cardiac-specific overexpression of MT prevents cardiac functional, ultrastructural, O2•− production, and protein disarray associated with DAHP treatment. MT failed to elicit any effect on the loss of total biopterin and BH4 although it mimicked BH4 supplementation-elicited protection against DAHP-induced detrimental effects on cardiomyocyte mechanical and intracellular Ca2+ properties. Given that MT alleviated DAHP-induced L-NAME-inhibitable intracellular O2•− production and theronine 497 phosphorylation of eNOS, it is plausible to speculate that the MT transgene may protect against DAHP-induced cardiac injury through counteracting “eNOS uncoupling”. Despite the low plasma BH4 levels, MT may efficiently scavenge the O2•− and other free radicals in cardiomyocytes thus prompting the “eNOS recoupling”. Administration of antioxidants such as L-ascorbic acid or the eNOS coupler folic acid has been shown to improve endothelial function through “eNOS dimerization or eNOS recoupling” 10, 30, 31. Furthermore, our data revealed that MT abrogated the DAHP-induced mitochondrial damage, mitochondrial O2•− production, loss of mitochondrial DNA, and downregulation of mitochondrial biogenesis factor PGC-1α and its downstream nuclear factors. Reduced mitochondrial biogenesis and downregulation of genes required for mitochondrial oxidative phosphorylation have been implicated in insulin resistance and type 2 diabetes 12, 32. PGC-1α promotes mitochondrial genes by enhancing levels of NRF1 and NRF2, alternatively called GA-binding protein (GABP), and facilitate GABP binding to regulatory regions of mtTFA promoter 25. It is reasonable to speculate that eNOS uncoupling-induced downregulation of PGC-1α, NRF1, NRF2 and mtTFA contributes to the loss of mitochondrial DNA and biogenesis. PGC-1α is crucial for normal heart function as PGC-1α knockout or deficiency results in compromised cardiac function 25. The precise mechanism responsible for loss of mitochondrial biogenesis under eNOS uncoupling is unknown although certain posttranslational oxidative modification may play a role 25. Our findings demonstrated the ability of MT to compensate for the DAHP-induced downregulation of HSP90. Reduction of the mitochondrial chaperon HSP90 has been implicated as an event secondary to uncoupling of eNOS, leading to mitochondrial dysfunction and dysregulated endothelial function associated with cardiovascular diseases 16. Our results suggest that the antioxidant protection against downregulated HSP90 but not mitochondrial UCP2 may contribute to the beneficial effect of MT on mitochondrial integrity. Last but not the least, nNOS and iNOS are constitutively expressed or pathologically induced, respectively, in cardiomyocytes. Both NOS isoforms participate in the modulation of myocardial functions including inotropy, relaxation, β-adrenergic responsiveness, and force-frequency relationship 33. Further study is warranted to elucidate the role other NOS isoforms in the antioxidant-offered beneficial effects in myocardial function in the state of eNOS uncoupling.
PERSPECTIVES
Our present work has provided evidence that eNOS uncoupling directly triggers cardiac ultrastructural and functional anomalies associated with mitochondrial oxidative and biogenesis damage. These results have consolidated a role of functional eNOS in the maintenance of cardiac physiology and more importantly, the therapeutic value of antioxidants against the eNOS uncoupling-induced cardiac pathology. Our data also indicated the therapeutic value of BH4 and GTP cyclohydrolase I in the management of cardiovascular dysfunction 10, 34. Furthermore, it prompts the speculation that eNOS uncoupling seen in the clinical conditions of vascular injuries such as hypertension and atherosclerosis may account, at least in part, for the increased cardiac events under these conditions. Therefore, eNOS recoupling may benefit both vascular anomalies and cardiac pathologies.
ACKNOWLEDGEMENTS
The authors greatly appreciated the technical assistance of Dr. Kurt Dolence and Bonnie Zhao from University of Wyoming College of Health Sciences as well as Donna Laturnus and Janice Audette from the University of North Dakota Department of Anatomy and Cell Biology. MT transgenic line was kindly provided by Dr. Paul N. Epstein from University of Louisville.
SOURCE OF FUNDING This work was supported in part by grants from the American Diabetes Association (7-08-RA-130), the American Heart Association Pacific Mountain Affiliate (#0355521Z), the NIH/NCRR University of Wyoming North Rockies Regional INBRE (P20 RR016474) to JR; American Diabetes Association (7-05-CD-02) to LC; NIH R01 GM077352, American Diabetes Association (7-08-RA-23), the American Heart Association (0855601G) to AFC. SJL was an awardee of the AHA Postdoctoral Fellowship 0720118Z.
Footnotes
DISCLOSURES None.
Reference List
- (1).Belge C, Massion PB, Pelat M, Balligand JL. Nitric oxide and the heart: update on new paradigms. Ann N Y Acad Sci. 2005;1047:173–182. doi: 10.1196/annals.1341.016. [DOI] [PubMed] [Google Scholar]
- (2).Massion PB, Balligand JL. Modulation of cardiac contraction, relaxation and rate by the endothelial nitric oxide synthase (eNOS): lessons from genetically modified mice. J Physiol. 2003;546:63–75. doi: 10.1113/jphysiol.2002.025973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Massion PB, Pelat M, Belge C, Balligand JL. Regulation of the mammalian heart function by nitric oxide. Comp Biochem Physiol A Mol Integr Physiol. 2005;142:144–150. doi: 10.1016/j.cbpb.2005.05.048. [DOI] [PubMed] [Google Scholar]
- (4).Ren J, Zhang X, Scott GI, Esberg LB, Ren BH, Culver B, Chen AF. Adenovirus gene transfer of recombinant endothelial nitric oxide synthase enhances contractile function in ventricular myocytes. J Cardiovasc Pharmacol. 2004;43:171–177. doi: 10.1097/00005344-200402000-00002. [DOI] [PubMed] [Google Scholar]
- (5).Oak JH, Cai H. Attenuation of Angiotensin II Signaling Recouples eNOS and Inhibits Nonendothelial NOX Activity in Diabetic Mice. Diabetes. 2007;56:118–126. doi: 10.2337/db06-0288. [DOI] [PubMed] [Google Scholar]
- (6).Hink U, Li H, Mollnau H, Oelze M, Matheis E, Hartmann M, Skatchkov M, Thaiss F, Stahl RA, Warnholtz A, Meinertz T, Griendling K, Harrison DG, Forstermann U, Munzel T. Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circ Res. 2001;88:E14–E22. doi: 10.1161/01.res.88.2.e14. [DOI] [PubMed] [Google Scholar]
- (7).Alp NJ, McAteer MA, Khoo J, Choudhury RP, Channon KM. Increased endothelial tetrahydrobiopterin synthesis by targeted transgenic GTP-cyclohydrolase I overexpression reduces endothelial dysfunction and atherosclerosis in ApoE-knockout mice. Arterioscler Thromb Vasc Biol. 2004;24:445–450. doi: 10.1161/01.ATV.0000115637.48689.77. [DOI] [PubMed] [Google Scholar]
- (8).Antoniades C, Shirodaria C, Warrick N, Cai S, de Bono J, Lee J, Leeson P, Neubauer S, Ratnatunga C, Pillai R, Refsum H, Channon KM. 5-methyltetrahydrofolate rapidly improves endothelial function and decreases superoxide production in human vessels: effects on vascular tetrahydrobiopterin availability and endothelial nitric oxide synthase coupling. Circulation. 2006;114:1193–1201. doi: 10.1161/CIRCULATIONAHA.106.612325. [DOI] [PubMed] [Google Scholar]
- (9).Lin MI, Fulton D, Babbitt R, Fleming I, Busse R, Pritchard KA, Jr., Sessa WC. Phosphorylation of threonine 497 in endothelial nitric-oxide synthase coordinates the coupling of L-arginine metabolism to efficient nitric oxide production. J Biol Chem. 2003;278:44719–44726. doi: 10.1074/jbc.M302836200. [DOI] [PubMed] [Google Scholar]
- (10).Forstermann U, Munzel T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation. 2006;113:1708–1714. doi: 10.1161/CIRCULATIONAHA.105.602532. [DOI] [PubMed] [Google Scholar]
- (11).Schulz E, Jansen T, Wenzel P, Daiber A, Munzel T. Nitric oxide, tetrahydrobiopterin, oxidative stress, and endothelial dysfunction in hypertension. Antioxid Redox Signal. 2008;10:1115–1126. doi: 10.1089/ars.2007.1989. [DOI] [PubMed] [Google Scholar]
- (12).Dong F, Li Q, Sreejayan N, Nunn JM, Ren J. Metallothionein prevents high-fat diet induced cardiac contractile dysfunction: role of peroxisome proliferator activated receptor gamma coactivator 1alpha and mitochondrial biogenesis. Diabetes. 2007;56:2201–2212. doi: 10.2337/db06-1596. [DOI] [PubMed] [Google Scholar]
- (13).Ye G, Metreveli NS, Ren J, Epstein PN. Metallothionein prevents diabetes-induced deficits in cardiomyocytes by inhibiting reactive oxygen species production. Diabetes. 2003;52:777–783. doi: 10.2337/diabetes.52.3.777. [DOI] [PubMed] [Google Scholar]
- (14).Mitchell BM, Dorrance AM, Webb RC. Phenylalanine improves dilation and blood pressure in GTP cyclohydrolase inhibition-induced hypertensive rats. J Cardiovasc Pharmacol. 2004;43:758–763. doi: 10.1097/00005344-200406000-00004. [DOI] [PubMed] [Google Scholar]
- (15).Lee KU, Lee IK, Han J, Song DK, Kim YM, Song HS, Kim HS, Lee WJ, Koh EH, Song KH, Han SM, Kim MS, Park IS, Park JY. Effects of recombinant adenovirus-mediated uncoupling protein 2 overexpression on endothelial function and apoptosis. Circ Res. 2005;96:1200–1207. doi: 10.1161/01.RES.0000170075.73039.5b. [DOI] [PubMed] [Google Scholar]
- (16).Sud N, Wells SM, Sharma S, Wiseman DA, Wilham J, Black SM. Asymmetric dimethylarginine inhibits HSP90 activity in pulmonary arterial endothelial cells: role of mitochondrial dysfunction. Am J Physiol Cell Physiol. 2008;294:C1407–C1418. doi: 10.1152/ajpcell.00384.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Kobayashi T, Hasegawa H, Kaneko E, Ichiyama A. Gastrointestinal serotonin: depletion due to tetrahydrobiopterin deficiency induced by 2,4-diamino-6-hydroxypyrimidine administration. J Pharmacol Exp Ther. 1991;256:773–779. [PubMed] [Google Scholar]
- (18).Zheng JS, Yang XQ, Lookingland KJ, Fink GD, Hesslinger C, Kapatos G, Kovesdi I, Chen AF. Gene transfer of human guanosine 5'-triphosphate cyclohydrolase I restores vascular tetrahydrobiopterin level and endothelial function in low renin hypertension. Circulation. 2003;108:1238–1245. doi: 10.1161/01.CIR.0000089082.40285.C3. [DOI] [PubMed] [Google Scholar]
- (19).Ren J, Privratsky JR, Yang X, Dong F, Carlson EC. Metallothionein alleviates glutathione depletion-induced oxidative cardiomyopathy in murine hearts. Crit Care Med. 2008;36:2106–2116. doi: 10.1097/CCM.0b013e31817bf925. [DOI] [PubMed] [Google Scholar]
- (20).Dong F, Zhang X, Ren J. Leptin regulates cardiomyocyte contractile function through endothelin-1 receptor-NADPH oxidase pathway. Hypertension. 2006;47:222–229. doi: 10.1161/01.HYP.0000198555.51645.f1. [DOI] [PubMed] [Google Scholar]
- (21).Zhao H, Joseph J, Fales HM, Sokoloski EA, Levine RL, Vasquez-Vivar J, Kalyanaraman B. Detection and characterization of the product of hydroethidine and intracellular superoxide by HPLC and limitations of fluorescence. Proc Natl Acad Sci USA. 2005;102:5727–5732. doi: 10.1073/pnas.0501719102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Song Y, Du Y, Prabhu SD, Epstein PN. Diabetic Cardiomyopathy in OVE26 Mice Shows Mitochondrial ROS Production and Divergence Between In Vivo and In Vitro Contractility. Rev Diabet Stud. 2007;4:159–168. doi: 10.1900/RDS.2007.4.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Relling DP, Esberg LB, Fang CX, Johnson WT, Murphy EJ, Carlson EC, Saari JT, Ren J. High-fat diet-induced juvenile obesity leads to cardiomyocyte dysfunction and upregulation of Foxo3a transcription factor independent of lipotoxicity and apoptosis. J Hypertens. 2006;24:549–561. doi: 10.1097/01.hjh.0000203846.34314.94. [DOI] [PubMed] [Google Scholar]
- (24).Munzel T, Sinning C, Post F, Warnholtz A, Schulz E. Pathophysiology, diagnosis and prognostic implications of endothelial dysfunction. Ann Med. 2008;40:180–196. doi: 10.1080/07853890701854702. [DOI] [PubMed] [Google Scholar]
- (25).Handschin C, Spiegelman BM. The role of exercise and PGC1alpha in inflammation and chronic disease. Nature. 2008;454:463–469. doi: 10.1038/nature07206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Kolinsky MA, Gross SS. The mechanism of potent GTP cyclohydrolase I inhibition by 2,4-diamino-6-hydroxypyrimidine: requirement of the GTP cyclohydrolase I feedback regulatory protein. J Biol Chem. 2004;279:40677–40682. doi: 10.1074/jbc.M405370200. [DOI] [PubMed] [Google Scholar]
- (27).Esberg LB, Ren J. The oxygen radical generator pyrogallol impairs cardiomyocyte contractile function via a superoxide and p38 MAP kinase-dependent pathway: protection by anisodamine and tetramethylpyrazine. Cardiovasc Toxicol. 2004;4:375–384. doi: 10.1385/ct:4:4:375. [DOI] [PubMed] [Google Scholar]
- (28).Goldhaber JI, Qayyum MS. Oxygen free radicals and excitation-contraction coupling. Antioxid Redox Signal. 2000;2:55–64. doi: 10.1089/ars.2000.2.1-55. [DOI] [PubMed] [Google Scholar]
- (29).von LD, Bruns S, Walther S, Kogler H, Pieske B. Insulin causes [Ca2+]i-dependent and [Ca2+]i-independent positive inotropic effects in failing human myocardium. Circulation. 2005;111:2588–2595. doi: 10.1161/CIRCULATIONAHA.104.497461. [DOI] [PubMed] [Google Scholar]
- (30).Forstermann U. Janus-faced role of endothelial NO synthase in vascular disease: uncoupling of oxygen reduction from NO synthesis and its pharmacological reversal. Biol Chem. 2006;387:1521–1533. doi: 10.1515/BC.2006.190. [DOI] [PubMed] [Google Scholar]
- (31).Moat SJ, Clarke ZL, Madhavan AK, Lewis MJ, Lang D. Folic acid reverses endothelial dysfunction induced by inhibition of tetrahydrobiopterin biosynthesis. Eur J Pharmacol. 2006;530:250–258. doi: 10.1016/j.ejphar.2005.11.047. [DOI] [PubMed] [Google Scholar]
- (32).Boirie Y. Insulin regulation of mitochondrial proteins and oxidative phosphorylation in human muscle. Trends Endocrinol Metab. 2003;14:393–394. doi: 10.1016/j.tem.2003.09.002. [DOI] [PubMed] [Google Scholar]
- (33).Seddon M, Shah AM, Casadei B. Cardiomyocytes as effectors of nitric oxide signalling. Cardiovasc Res. 2007;75:315–26. doi: 10.1016/j.cardiores.2007.04.031. [DOI] [PubMed] [Google Scholar]
- (34).Du YH, Guan YY, Alp NJ, Channon KM, Chen AF. Endothelium-specific GTP cyclohydrolase I overexpression attenuates blood pressure progression in salt-sensitive low-renin hypertension. Circulation. 2008;117:1045–1054. doi: 10.1161/CIRCULATIONAHA.107.748236. [DOI] [PubMed] [Google Scholar]