

Abstract
Aromatase inhibitors (AIs) are important drugs to treat estrogen receptor α (ERα) positive post-menopausal breast cancer patients. However, development of resistance to AIs has been observed. We examined whether the heat shock protein 90 (HSP90) inhibitor 17-(dimethylaminoethylamino)-17-demethoxygeldanamycin (17-DMAG), can inhibit the growth of AI-resistant breast cancers and the mechanisms by which 17-DMAG affects proliferation. AI-responsive MCF-7aro and AI-resistant LTEDaro breast epithelial cells were used in this study. We observed that 17-DMAG inhibited proliferation in both MCF-7aro and LTEDaro cells in a dose-dependent manner. 17-DMAG induced apoptosis and G2 cell cycle arrest in both cell lines. Although inhibition of HSP90 decreased the levels of ERα, the ERα transcriptional activity was not affected when cells were treated with 17-DMAG together with estradiol. Moreover, detailed mechanistic studies suggested that 17-DMAG inhibits cell growth via degradation of HSP90 client proteins AKT and Her2. Collectively, results from this study provide data to support that HSP90 inhibitors may be an effective therapy to treat AI-resistant breast cancers and that improved efficacy can be achieved by combined use of an HSP90 inhibitor and an AKT inhibitor.
Keywords: HSP90 inhibitors, 17-DMAG, aromatase inhibitors, breast cancer
Introduction
Aromatase is the enzyme that converts androgen into estrogen. Estrogen is known to play an important role in breast cancer growth through its activation of the estrogen receptor α (ERα). Activated ERα can then translocate into the nucleus where it can bind to estrogen response elements on various gene promoters and subsequently transactivate these genes, which are involved in promoting tumor cell growth 1. Tamoxifen, an antagonist of ERα, and aromatase inhibitors (AI) [anastrozole, letrozole, and exemestane] which inhibit the synthesis of estrogens, have been effective therapies to combat estrogen-dependent breast cancer 2-4. However, resistance to these inhibitors has been observed. How resistance to these therapies develops as well as the mechanisms of how these cells survive and proliferate in the presence of these inhibitors are not completely understood. Studies of AI and tamoxifen resistance have revealed an important role of the ERα in the acquisition of resistance. In the long-term estrogen deprived (LTEDaro) cells, a model of AI resistance, and other AI resistant cells, ERα was found to be constitutively active 5. Moreover, it is thought that this ERα activity is dependent on growth factor pathway signaling which is responsible for activation and influencing the levels of ERα in AI-resistant breast cancers in a ligand-independent manner 6, 7. Growth factor-upregulated kinases can phosphorylate ERα and activate it, leading to transcriptional activation of target genes and signaling pathways involved in growth 6-12.
Heat shock proteins (HSPs) are chaperone proteins which correctly fold and assist proteins in the active, correct conformations. They are involved in stress response and also in assembly and transportation across different cell compartments 13, 14. HSP90 is the most abundant protein in cells, comprising about 1-2% of the total soluble cytosolic protein 15. HSPs are expressed in normal cells, but are overexpressed in cancer cells 16. Many HSP client proteins are involved in processes such as proliferation, apoptosis, and cell cycle progression 17, 18. It is not surprising that cancer cells exploit these HSPs to correctly fold client proteins to further the growth and survival of the cancer cells. Due to the importance of these HSPs in the growth and survival of cancer cells, inhibitors against these proteins have been developed. Early version of inhibitors against HSP90 include geldanamycin and its synthetic derivative, 17-aminoallyl, 17-demethoxygeldanamycin (17-AAG) 19-22. Due to their toxicity and low solubility, another HSP90 inhibitor, 17-(dimethylaminoethylamino)-17-demethoxygeldanamycin (17-DMAG), was developed. 17-DMAG displays better water-solubility and oral bioavailability and has been tested in Phase I clinical trials for treatment of metastatic or unresectable tumors, or lymphomas 23, 24. While HSP90 is expressed in both normal and cancer cells, HSP90 inhibitors display preference for cancerous cells 25. Furthermore, due to the wide range of protein targets HSP90 affects, its inhibitors can be useful for treating cancer, such as AI resistant breast cancers which are thought to rely heavily on growth factor signaling pathways, which include many HSP90 client proteins. However, to date, the efficacy of 17-DMAG on AI therapy resistant breast cancers has not been examined. The purpose of this study is to determine whether 17-DMAG may be used as a therapy to treat AI-resistant breast cancers and to study how it alters molecular properties of AI-resistant breast cancer cells. Here, we show that nanomolar concentrations of 17-DMAG can inhibit both AI-responsive and AI-resistant breast cancer cell growth by inducing apoptosis as well as arresting the cell cycle at the G2 phase. We also demonstrate that 17-DMAG has no effect on the ERα transcriptional activity in the AI-responsive and AI-resistant breast cells in the presence of hormone. This suggests that the mechanism of 17-DMAG inhibition of breast cell proliferation functions independently of the ERα signaling pathway. Instead, we found that HSP90 client proteins, AKT and Her2, are involved in the growth of both AI-responsive and AI-resistant breast cancer cell lines, suggesting that 17-DMAG mediated inhibition of growth may result from inhibition of these signaling pathways. The data suggests that HSP90 inhibitors will be a suitable therapy for breast cancers which have developed resistance to current AI therapies.
Materials and Methods
Cell lines and cell culture
The human MCF-7 breast epithelial derived cell lines MCF-7aro and LTEDaro were previously generated in this lab and have been reported 5, 26. Human mammary epithelial cells (HMEC) (Lonza, Walkersville, MD) were cultured in MEGM media supplemented with bovine pituitary extract, human epidermal growth factor, hydrocortisone, gentamicin sulfate and amphotericin B, and insulin.
Reagents and antibodies
17-DMAG was from the Drug Synthesis and Chemistry Branch, Developmental Therapeutics Program, National Cancer Institute (Bethesda, MD) and dissolved in DMSO. Exemestane was from Pharmacia & Upjohn S.p.A. (Milano, Italy). Triciribine was from Cayman Chemical (Ann Arbor, MI) and AG 825 was from Calbiochem (San Diego, CA). MTT compound was from Sigma-Aldrich (Milwaukee, WI). The antibody to HSP90 was from Stressgen Bioreagents (Victoria, British Columbia, Canada); the antibodies to phospho-Her2 and Her2 were from Upstate (Temecula, CA); antibodies to phospho-ERα Ser118, cleaved PARP, cyclin D1, and AKT were from Cell Signaling Technology (Danvers, MA); antibody to ERα was from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA); antibody to GAPDH was from Chemicon (Danvers, MA).
Cell Proliferation Assay
MCF-7aro cells were hormone-deprived for one day prior to plating. MCF-7aro, LTEDaro, and HMEC cells were cultured in 96 well plates at a concentration of 1.5-4×103 cells/well (100μl/well). Cells were treated with DMSO, or inhibitor for up to three days and MCF-7aro cells were additionally treated with 1nM testosterone. Cell viability was assessed by the MTT assay. Briefly, the MTT powder was dissolved in cell culture media to a concentration of 0.5mg/ml. 150μl of MTT reagent was added to each well and incubated for 1 hour at 37°C, 5% CO2. DMSO was added to solubilize the formazan product and formazan absorbance was measured at 570nm on a SpectraMax M5 microplate reader (Molecular Devices, Sunnyvale, CA). Three replicates were used for each measurement and the mean and standard deviation values were calculated.
Apoptosis Analysis
Cells were plated at a density of 5×105 cells per 60mm dish. After 24 hours, either DMSO or 100nM 17-DMAG was added to the dishes, in addition to 1nM testosterone for MCF-7aro cells, and the cells were cultured for 48 or 72 hours. To detect apoptotic cell death, DNA fragmentation was detected using Cell Death Detection ELISAPLUS (Roche Applied Science, Indianapolis, IN) according to the manufacturer’s instructions.
Cell Cycle Analysis
Cells were plated on 100cm dishes at a density of 1×106 cells/dish. DMSO or 100nM 17-DMAG was added to the dishes the following day, in addition to 1nM testosterone for MCF-7aro cells. At each time point, 2×106 cells were collected, resuspended in 1ml of cold PBS, and fixed by the addition of 4ml of -20°C absolute ethanol. The fixed cells were resuspended in 500μl of PBS/0.1% BSA and 100μl of 200μg/ml DNase-free, RNaseA was added. After incubation at 37°C for 30 minutes, 50μl of 1mg/ml propidium iodide was added to the cells and allowed to incubate for at least 1 hour before analysis by a CyAn ADP 9 color flow cytometer (Dako Inc., Carpinteria, CA).
ER Functional Assays
MCF-7aro and LTEDaro cells were plated in 12-well plates at 1×105 cells/well. The following day, each well was transfected with 1μg of pGL3-(ERE)3 reporter using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). After 5 hours, transfection media was replaced with media ± 1nM E2, and 100nM 17-DMAG. After 48 hours, the firefly luciferase activity was measured using the luciferase assay system (Promega, Madison, WI) and a TD-20/20 Luminometer (Turner Designs, Sunnyvale, CA). Luciferase activity was normalized to protein concentration.
Western Blot Analysis
Cell monolayers were washed twice with ice-cold phosphate-buffer saline and then lysed. The protein concentration was quantified by Bradford’s Method. Equal amounts of protein were resolved by SDS-PAGE, transferred to nitrocellulose membrane (Bio-Rad, Hercules, CA), and detected by using SuperSignal West Pico Chemiluminescent Substrate (Pierce Biotechnology, Rockport, IL).
Statistical Analysis
The synergistic and antagonistic effect of the combination of 17-DMAG and Triciribine or AG 825 were analyzed using Calcusyn (Biosoft, Cambridge, UK), a software program based on the Chou-Talalay method 27.
Results
LTEDaro is a model of aromatase inhibitor resistance
MCF-7aro cells were generated in this laboratory as a model of aromatase-positive and ER-positive breast cancer 26. MCF-7aro cells were continuously cultured in steroid-depleted media to generate LTEDaro cells 5. Therefore, through the conversion to E2 by aromatase, testosterone is needed for the proliferation of MCF-7aro cells, but LTEDaro cells grow well in the absence of testosterone. To demonstrate that the LTEDaro cells are a model of AI resistance, we treated both MCF-7aro and LTEDaro cells with 1μM Exemestane, an AI, and measured cell proliferation. MCF-7aro cells treated with 1μM Exemestane showed significant growth inhibition after 72 hours of treatment (Fig. 1). This marked sensitivity was not observed in the LTEDaro cells, which displayed growth levels similar to the DMSO vehicle control. This result confirms that MCF-7aro cells are AI-sensitive, whereas LTEDaro are AI-resistant. The LTEDaro cells have also been shown to be resistant to tamoxifen and two non-steroidal AIs, letrozole and anastrozole 5. Clinically, lack of cross-resistance among different AIs is typically observed, i.e., patients will respond partially to a second AI after acquiring resistance to the first AI. Therefore, we believe the LTEDaro cell line is representative of the last stage of resistance since these cells no longer respond to any of the AIs. Thus, LTEDaro is valuable to test for new drugs that can overcome AI resistance.
Figure 1.
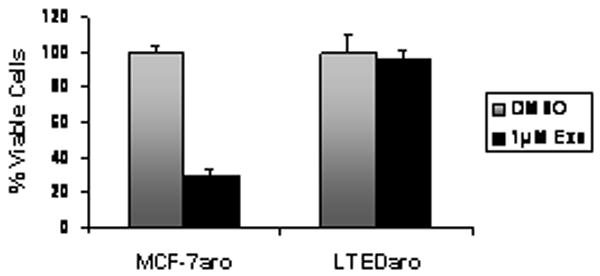
Effect of the Aromatase Inhibitor Exemestane on LTEDaro and MCF-7aro cell proliferation. Cells were treated with 1nM testosterone, DMSO, or 1μM Exemestane for 72 hours. Cell viability was assessed by the MTT assay.
17-DMAG inhibits cell growth in AI-resistant epithelial breast cancer cells
It has been demonstrated that breast cancer cells which become AI resistant, no longer depend on estrogen production by aromatase for growth. Instead, the cells shift their dependence on growth factor signaling pathways for promoting AI resistant breast cancer growth 5. We reasoned that HSP90 inhibitors might be effective in suppressing the proliferation of AI resistant cells since many members of growth factor signaling pathways are HSP90 clients. To determine whether AI responsive and AI resistant cells are susceptible to the HSP90 inhibitor, 17-DMAG, cell proliferation assays were conducted. 17-DMAG inhibited hormone-independent LTEDaro and hormone-dependent MCF-7aro cell growth in a dose dependent manner at nanomolar concentrations as well as a time dependent manner. At low doses (5-15nM) of 17-DMAG, up to 70% growth inhibition was observed in both cell lines. Moreover, by 72 hours treatment with 100nM 17-DMAG, both LTEDaro (Fig. 2A) and MCF-7aro cells (Fig. 2B) were almost completely growth inhibited compared to the DMSO control. Furthermore, 17-DMAG inhibited growth of exemestane-resistant cells in a dose and time dependent manner (data not shown). Normal human mammary epithelial cells (HMEC) were treated with 17-DMAG to test the selectivity of the drug for cancerous cells. The drug was ineffective at low doses (5-15nM) and growth inhibition was evident only at higher doses (≥20nM) (Fig. 2C). These results confirmed that proliferation of AI resistant breast cancer cells can be attenuated by HSP90 inhibitors. Moreover, at lower doses, 17-DMAG is well tolerated by normal cells, but it can still inhibit cancer cell growth.
Figure 2.
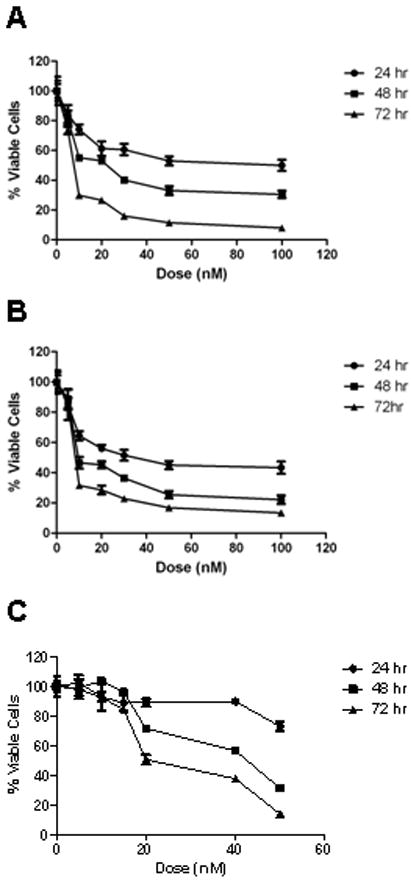
Effect of 17-DMAG on A) LTEDaro, B) MCF-7aro, and C) HMEC cell proliferation. Cells were treated with DMSO, or 17-DMAG for 24, 48, or 72 hours. Cell viability was assessed by the MTT assay.
17-DMAG induces apoptosis and a G2 phase arrest
The inhibition of cell proliferation could possibly occur due to either induced apoptosis, an arrest in the cell cycle, or both. To determine whether the 17-DMAG induced inhibition of cell growth was due to apoptosis, we decided to detect DNA fragmentation resulting from treatment with 100nM 17-DMAG. A two-to-four-fold increase in the levels of DNA fragmentation was observed in both cell lines after 48 and 72 hours treatment (Fig. 3A,B). This result confirms that apoptosis is induced by inhibition of HSP90.
Figure 3.
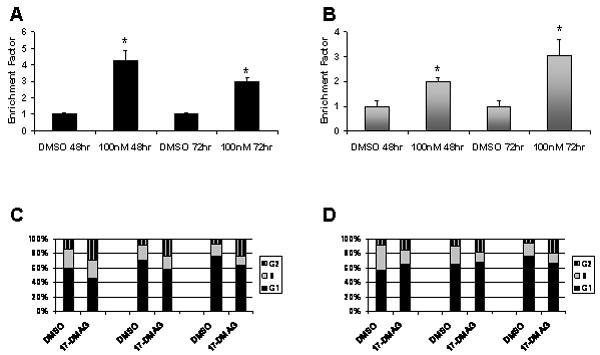
Induction of apoptosis and G2-M phase arrest by 17-DMAG. The Cell Death Detection ELISAPLUS kit was used to quantify the number of A) LTEDaro and B) MCF-7aro cells undergoing apoptosis in the presence of either DMSO or 100nM 17-DMAG for 48 or 72 hours. *, P < 0.05 by Student’s t-test when comparing against the DMSO control. Bars, SE. C) LTEDaro and D) MCF-7aro cells were treated with either DMSO or 100nM 17-DMAG for 24, 48, or 72 hours. After each time point, cells were stained with propidium iodide and analyzed by flow cytometry.
Next, to determine whether, in addition of apoptosis, 17-DMAG treatment causes alterations of the cell cycle, cells were treated with 100nM 17-DMAG and analyzed by flow cytometry to measure the population of cells in each phase of the cell cycle. LTEDaro cells treated with 17-DMAG showed significantly higher percentage (2-3 fold) of cells in G2 compared to DMSO treated cells (Fig. 3C). Similarly, MCF-7aro cells treated with 17-DMAG displayed a decreasing population of cells in S phase and an increase in the number of cells in G2 with each day of treatment (Fig. 3D). These results indicate that 17-DMAG arrests cells at the G2-M phase transition.
17-DMAG mediated inhibition of growth does not target the estrogen receptor α pathway
Our proliferation, apoptosis and cell cycle studies revealed that 17-DMAG is effective on both hormone dependent and independent cell lines in a similar fashion. This suggests that the mechanism by which 17-DMAG inhibits growth does not involve ERα. To confirm this hypothesis, we analyzed the effect of 17-DMAG on ERα levels and activity. Total ERα levels decreased with 17-DMAG treatment in a dose and time dependent manner (Fig. 4A,B). These results indicate that ERα is degraded with 17-DMAG treatment and confirm that ERα is an HSP90 client protein. Next, we examined whether 17-DMAG can inhibit the ERα transcriptional activity, a result of constitutive ligand-independent ERα phosphorylation in hormone independent cells or a result of ligand activation of ERα in hormone dependent cells. We transfected both LTEDaro and MCF-7aro cells with a reporter plasmid encoding three ERE sequences, in tandem, upstream of the firefly luciferase gene. After transfection, the cells were treated with media containing either DMSO or 17-DMAG, along with or without 1nM E2. Our analysis revealed that 17-DMAG abolished ligand-independent ERα activity in LTEDaro cells, as well as the basal ERα activity in MCF-7aro cells, compared to the DMSO control (Fig. 4C,D). The basal ERα activity was high in the LTEDaro cells and was not affected by the treatment of 1nM E2 (Fig. 4C). However, co-treatment with E2 and 17-DMAG was unable to completely abolish the ERα transcriptional activity (Fig. 4C). In MCF-7aro cells, as expected, E2 stimulated transcriptional activation of ERα (Fig. 4D). Surprisingly, treatment with both E2 and 17-DMAG further enhanced the transcriptional activity of the MCF-7aro cells. These results show that while 17-DMAG can abolish ERα transcriptional activity in the absence of hormone, it is unable to inhibit this transcriptional activity in the presence of ligand. Additional studies by western blot analysis corroborate these results. Basal phosphorylation of ERα at S118 was observed in DMSO and 1nM E2 treated LTEDaro cells (Supplementary Fig. S1). Phosphorylation was abolished by 17-DMAG and total levels of ERα also decreased indicating degradation by 17-DMAG treatment. However, phosphorylation was restored by cotreatment with 1nM E2 and 100nM 17-DMAG (Supplementary Fig. S1). Phosphorylation of ERα at S118 was detected in MCF-7aro cells treated with 1nM E2, but was not detected with DMSO or 100nM 17-DMAG treatment. In addition, total ERα was degraded by 17-DMAG treatment. These results confirm that ERα is an HSP90 client protein in both hormone dependent and independent cells. However, 17-DMAG does not affect ERα activity in the presence of ligand, confirming that 17-DMAG mediated inhibition of growth does not occur by targeting of the ERα pathway.
Figure 4.
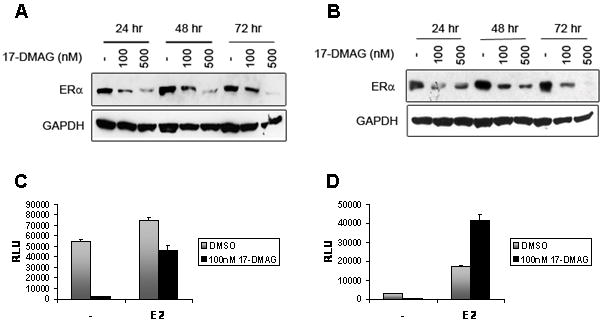
ERα protein levels and activity in the LTEDaro and MCF-7aro cell lines after 17-DMAG treatment. A) LTEDaro and B) MCF-7aro cells were treated with either DMSO or 17-DMAG for 24, 48, or 72 hours. MCF-7aro cells were additionally treated with 1nM testosterone. ERα protein expression was determined by Western Blot. The PGL3-(ERE)3 reporter plasmid was transiently transfected into C) LTEDaro and D) MCF-7aro cell lines. Both cell lines were treated with either DMSO or 100nM 17-DMAG, and with or without 1nM E2 for 48 hours. Data (mean ± SE) is representative of 3 independent experiments performed in triplicate.
Inhibition of HSP90 alters protein expression in epithelial breast cancer cells
To understand the molecular mechanisms of growth defect in 17-DMAG treated cells and to identify other pathways which may be affected by 17-DMAG treatment, we performed Western blot analysis to detect levels of HSP90 and its client protein expression. HSP90 expression was similar in both MCF-7aro and LTEDaro whole cell lysates (Fig. 5A,B). To analyze whether apoptotic markers were activated, we checked the expression of poly (ADP-ribose) polymerase [PARP], which is cleaved due to caspase-3 and -7 activations 28. Levels of cleaved PARP increased with increasing concentration of 17-DMAG and with time, indicating induction of apoptosis resulting from 17-DMAG treatment and confirming our data that 17-DMAG induces apoptosis (Fig. 3A,B). In addition, Cyclin D1 levels also decreased with increasing concentration, indicating that fewer cells are in the G1 phase of the cell cycle (Fig. 5A,B). These results also corroborate our cell-cycle analysis (Fig. 3C,D), which indicate an increased population of cells in the G2 phase after 17-DMAG treatment.
Figure 5.
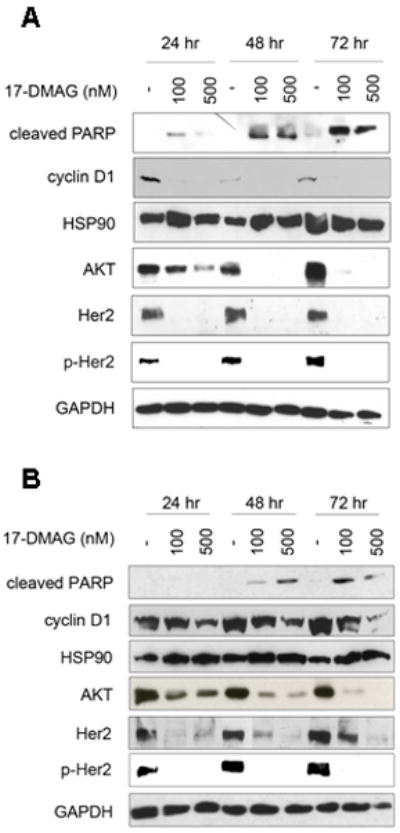
Activity of 17-DMAG in LTEDaro and MCF-7aro breast cancer cells. A) LTEDaro and B) MCF-7aro cells were treated with either DMSO or 17-DMAG for 24, 48, or 72 hours. MCF-7aro cells were additionally treated with 1nM testosterone. Cleaved PARP, cyclin D1, HSP90, AKT, Her2, p-Her2 and GAPDH protein expression were determined by Western Blot.
To identify other pathways which may be affected by 17-DMAG treatment we examined the expression levels of HSP90 client proteins after 17-DMAG treatment. We determined the levels of AKT, phospho-Her2, and Her2, proteins which have growth promoting and pro-survival effects 29-31. Client proteins AKT, Her2, as well as the phosphorylated, activated form of Her2 protein levels decreased with increasing dose of 17-DMAG treatment and in a time-dependent manner in the LTEDaro (Fig. 5A) and MCF-7aro (Fig. 5B) cell lines. These results confirm that AKT and Her2 are targeted by 17-DMAG.
AKT and Her2 pathways are important for cell growth in the MCF-7aro and LTEDaro cell lines
Our western blot data suggested that the pathways by which AKT and Her2 function may be targeted by 17-DMAG. To determine whether these pathways play an important role in the growth of the cells, we treated both LTEDaro and MCF-7aro cells with the AKT inhibitor Triciribine and the Her2 inhibitor AG 825 and measured cell proliferation. Triciribine and AG 825, individually, inhibited both LTEDaro and MCF-7aro cell growth in a dose dependent manner and were similarly potent in both cell lines (Supplementary Fig. S2A,S2B). Inhibition of growth by the AKT or Her2 inhibitors indicates the importance of these proteins for cell growth in the AI-responsive and resistant cell lines and suggests that these pathways may be targeted by 17-DMAG.
To test this hypothesis, we treated both LTEDaro and MCF-7aro cells with 17-DMAG, along with Triciribine or AG 825 and measured growth of these cells. Triciribine, 17-DMAG, and the combination of both inhibited LTEDaro cell growth in a dose-dependent manner (Supplementary Fig. S3A). Interestingly, when both inhibitors were used in combination, statistical analysis indicated synergistic reduction in cell growth (Table 1; Supplementary Fig. S3B). Similarly, the effect of 17-DMAG on MCF-7aro cells was synergistically enhanced with increasing concentration of Triciribine (Table 1; Supplementary Fig. S3C,S3D).
Table 1.
Affected fractions and combination indices with 17-DMAG and Triciribine or AG825, in combination, on LTEDaro or MCF-7aro at 48 hrs
| Cell line | 17-DMAG (nM/L) | Triciribine (nM/L) | AG825 (uM/L) | FA | Cl |
|---|---|---|---|---|---|
| LTEDaro | 15 | 50 | 0 | 0.6343 | 0.708 |
| 15 | 100 | 0 | 0.7167 | 0.523 | |
| 30 | 50 | 0 | 0.7757 | 0.819 | |
| 30 | 100 | 0 | 0.7754 | 0.823 | |
| MCF-7aro | 15 | 50 | 0 | 0.503 | 0.976 |
| 15 | 100 | 0 | 0.555 | 1.041 | |
| 30 | 50 | 0 | 0.69 | 0.621 | |
| 30 | 100 | 0 | 0.72 | 0.621 | |
| LTEDaro | 15 | 0 | 30 | 0.4517 | 1.992 |
| 15 | 0 | 50 | 0.8226 | 1.197 | |
| 30 | 0 | 30 | 0.6337 | 1.951 | |
| 30 | 0 | 50 | 0.9295 | 0.958 | |
| MCF-7aro | 15 | 0 | 30 | 0.3311 | 1.677 |
| 15 | 0 | 50 | 0.5651 | 1.166 | |
| 30 | 0 | 30 | 0.4839 | 1.459 | |
| 30 | 0 | 50 | 0.6692 | 1.066 | |
Abbreviation: FA, affected fraction; Cl, combination index
Cell growth was inhibited with increasing concentration of Her2 inhibitor, AG 825, 17-DMAG, as well as the combination of both in LTEDaro and MCF-7aro cell lines (Supplementary Fig. S4A,S4C). While increasing concentrations of both inhibitors resulted in increased suppression of growth, statistical analysis of these results indicated that these inhibitors work in an antagonistic manner in both cell lines (Table 1; Supplementary Fig. S4B,S4D). These results demonstrate that both AKT and Her2 pathways play a role in the growth of AI-responsive and resistant breast cancer cells, and both these pathways are targeted by 17-DMAG. However, since AKT inhibitor, Triciribine, and 17-DMAG function synergistically, they can be used in a combinatorial manner to treat AI-resistant breast cancer.
Discussion
AIs are currently first-line therapies for the treatment of post-menopausal ER-positive breast cancer patients. However, acquired resistance to the inhibitors does develop. In the clinical setting, patients who develop resistance to AIs do not respond to current available endocrine therapies. This creates a need for new therapies or treatment regiments which can be used to treat these AI-resistant breast cancer patients. HSP90 inhibitors have been demonstrated to be effective at inhibiting the growth of various different cancers, including breast cancer. 17-AAG, an early generation HSP90 inhibitor, has been tested on hormone-refractory breast cancers and several tyrosine kinase inhibitor-resistant cancers and was shown to be quite effective at inhibiting cell proliferation 32 as well as inducing apoptosis 33, 34. The HSP90 inhibitor, 17-DMAG has been reported to inhibit proliferation and angiogenesis in pancreatic and gastric cancer by interfering with growth-factor signaling 35, 36.
AI-resistant breast cancers do not rely on hormone-mediated signaling, but growth-factor signaling is important for their growth. It has been found that the ERα can be phosphorylated and activated in a ligand-independent manner. This activation is mainly due to the cross-talk between the ERα and growth-factor signaling pathways, such as IGF-1R and ErbB2-mediated signaling pathways. Since these growth factor signaling proteins are important for resistance to AIs and are also HSP90 client proteins, we were interested in studying whether the HSP90 inhibitor 17-DMAG would be an effective mode of treatment for AI- resistant breast cancer.
In this study, 17-DMAG is demonstrated to effectively suppress cell growth of both the AI-responsive MCF-7aro cells and the AI-resistant LTEDaro cells. The dosages used are in a sub-micromolar range, demonstrating that the inhibitor is very potent in these cells. A previous report indicated that HSP90 inhibitors display specificity towards cancerous cells 25. Our data shows that the inhibitor is ineffective at low doses in normal breast epithelial cells. This is expected since noncancerous cells do not overexpress HSP90 as cancerous cells do (Supplementary Fig. S5). Thus, cancerous cells would be more susceptible to the effects of the inhibitor at low doses. However, at higher doses, an excess of the inhibitor would affect all HSP90 client proteins necessary for growth, even in normal cells. This suggests that at a certain concentration range, 17-DMAG could inhibit cancer cell growth, without affecting normal cells.
Further analysis as to how treatment with 17-DMAG leads to inhibition of growth indicated DNA fragmentation. Confirmation of 17-DMAG mediated induction of apoptosis was observed by Western blotting for cleaved PARP, which facilitates cellular disassembly and is an indicator of cells undergoing apoptosis. 17-DMAG treatment also led to a decrease in the levels of HSP90 client proteins. Her2 protein levels decreased as well as its active phosphorylated state. Following the decreased Her2 levels, the downstream AKT protein levels also decreased. AKT plays an important role in the suppression of apoptosis and promotes growth. AKT has been shown to suppress apoptosis through the phosphorylation of proapoptotic proteins, transcription factors, and regulators of transcription factors, which results in blockage of transcriptional repression of proapoptotic genes and survival 37-39. Her2 and AKT proteins promote growth and decreased levels of these proteins would facilitate apoptosis in the cell.
In addition to inducing apoptosis, 17-DMAG also causes cell cycle arrest at the G2-M phase. This phenomenon has been observed in some studies 40, while others have detected arrest at the G1-S phase 40, 41, depending on the cell line. The levels of HSP90 client proteins may differ in each cell line, thus, treatment with the inhibitor may affect certain proteins more readily than others, which may explain the differences amongst the cell lines. Cyclin D1 is needed for the progression from G1 to S phase. While Cyclin D1 is not an HSP90 client protein, it is indirectly regulated by client proteins. One study has described a PI3K/AKT regulatory mechanism to control Cyclin D1 expression post-translationally 42. AKT degradation by 17-DMAG treatment might affect Cyclin D1 expression. Decreased Cyclin D1 expression might allow cells to bypass the G1-S checkpoints and continue through the cell cycle in an unregulated manner. We observed a reduction in the Cyclin D1 levels after 17-DMAG treatment in both MCF-7aro and LTEDaro cells, suggesting this could be a mechanism for higher number of cells in G2 stage. Our results demonstrate that 17-DMAG inhibits cell proliferation by blocking cells at G2 stage, while also inducing cell death of the remaining cells by inhibiting the signaling of growth promoting pathways.
17-DMAG treatment also degrades the HSP90 client protein ERα, which is implicated in AI resistance 5. Notably, total ERα protein levels and its ligand-independent phosphorylation are elevated in LTED cells compared to the AI-responsive MCF-7 cells 6, 9. However, our data shows that 17-DMAG affects both hormone dependent MCF-7aro and hormone independent LTEDaro cells in a similar fashion. This suggests that 17-DMAG does not inhibit both cell lines by targeting of the ERα pathway. ERα phosphorylation and activity were unaffected when cells were treated with 17-DMAG in the presence of E2. These results indicate that the presence of hormone has a protective effect against the inhibition of ERα transcriptional activation by 17-DMAG in both the LTEDaro and MCF-7aro cell lines. A mechanism described to link transcription and degradation by the ubiquitin-mediated proteolysis may explain the results 43. In the MCF-7aro cells, binding of the E2 ligand to ERα signals activation of ERα and dissociation from the HSP90 complex, thereby bypassing 17-DMAG mediated degradation of ERα. ERα can then induce transcriptional activation. However, ERα is ubiquitinated and targeted for degradation simultaneously as RNA polymerase II elongates transcription through a transcription-coupled degradation mechanism. In the LTEDaro cells, the ERα is not stimulated by E2, however, it may retain its ability to bind. Thus, hormone binding may prevent ERα from being degraded with 17-DMAG treatment, thereby restoring its activity. Despite this postulated mechanism, how ERα transcriptional activity is elevated in the MCF-7aro cells with 17-DMAG and hormone treatment is unclear. More studies are required to fully understand this effect. While ERα is an HSP90 client protein, the reporter assay results confirm that the ERα pathway is not a target by which 17-DMAG inhibits cell growth.
Our results demonstrate that 17-DMAG also targets growth factor pathways to inhibit breast cancer cell proliferation. We observed degradation of both AKT and Her2 by 17-DMAG treatment in LTEDaro and MCF-7aro cells. Additionally, AKT inhibitor Triciribine and Her2 inhibitor AG 825, alone, were able to inhibit growth of both LTEDaro and MCF-7aro cells. Moreover, we found that AG 825, along with 17-DMAG functions in an antagonistic manner while Triciribine and 17-DMAG work in a synergistic manner in both cell lines. Our data demonstrates that a combination of both 17-DMAG and an AKT inhibitor may be more effective than 17-DMAG alone in the treatment of AI-resistant breast cancers.
Aromatase inhibitors are specific and effective inhibitors for treating AI responsive breast cancer. It is breast cancers which have developed resistance to AIs that no good treatment is available. Our results show that the AI-resistant LTEDaro cells and the AI-sensitive MCF-7aro cells are growth inhibited by the HSP90 inhibitor 17-DMAG. 17-DMAG treatment displays potent efficacy in inhibiting breast cancer cell growth. It displays selectivity towards cancer cells at lower doses and displays some cytotoxicity towards normal cells only at higher doses. Currently, HSP90 inhibitors are continually being developed for improved potency and selectivity and these drugs remain to be tested. However, the importance of this study is to demonstrate the utility of HSP90 inhibitors as a potential therapy for the treatment of AI resistant breast cancers.
The major finding from this study is that HSP90 inhibitors may be used as a therapy to treat AI-resistant breast cancers. Their ability to target multiple client proteins involved in the different growth-promoting signaling pathways makes HSP90 inhibitors a valuable and promising novel treatment option for AI-resistant breast cancer patients. This study provides a clinical basis for future clinical trials using HSP90 inhibitors in AI-resistant breast cancers. In addition, treatment regimens using both an HSP90 inhibitor and a signal transduction inhibitor may also be considered for improved efficacy.
Supplementary Material
Acknowledgments
We thank Dr. George Somlo for his assistance with acquiring 17-DMAG and Dr. Tim O’Connor for providing the human mammary epithelial cells.
Grant support: This work was supported by California Breast Cancer Research Program 13GB-0157 to Cynthie Wong and NIH grants CA44735 and ES08258 to Shiuan Chen
Footnotes
Potential conflicts of interest: None.
References
- 1.Nilsson S, Makela S, Treuter E, et al. Mechanisms of estrogen action. Physiological reviews. 2001;81:1535–65. doi: 10.1152/physrev.2001.81.4.1535. [DOI] [PubMed] [Google Scholar]
- 2.Systemic treatment of early breast cancer by hormonal, cytotoxic, or immune therapy. 133 randomised trials involving 31,000 recurrences and 24,000 deaths among 75,000 women. Early Breast Cancer Trialists’ Collaborative Group. Lancet. 1992;339:1–15. [PubMed] [Google Scholar]
- 3.Geisler J, Haynes B, Anker G, Dowsett M, Lonning PE. Influence of letrozole and anastrozole on total body aromatization and plasma estrogen levels in postmenopausal breast cancer patients evaluated in a randomized, cross-over study. J Clin Oncol. 2002;20:751–7. doi: 10.1200/JCO.2002.20.3.751. [DOI] [PubMed] [Google Scholar]
- 4.Geisler J, King N, Anker G, et al. In vivo inhibition of aromatization by exemestane, a novel irreversible aromatase inhibitor, in postmenopausal breast cancer patients. Clin Cancer Res. 1998;4:2089–93. [PubMed] [Google Scholar]
- 5.Masri S, Phung S, Wang X, et al. Genome-wide analysis of aromatase inhibitor-resistant, tamoxifen-resistant, and long-term estrogen-deprived cells reveals a role for estrogen receptor. Cancer research. 2008;68:4910–8. doi: 10.1158/0008-5472.CAN-08-0303. [DOI] [PubMed] [Google Scholar]
- 6.Martin LA, Farmer I, Johnston SR, Ali S, Marshall C, Dowsett M. Enhanced estrogen receptor (ER) alpha, ERBB2, and MAPK signal transduction pathways operate during the adaptation of MCF-7 cells to long term estrogen deprivation. The Journal of biological chemistry. 2003;278:30458–68. doi: 10.1074/jbc.M305226200. [DOI] [PubMed] [Google Scholar]
- 7.Campbell RA, Bhat-Nakshatri P, Patel NM, Constantinidou D, Ali S, Nakshatri H. Phosphatidylinositol 3-kinase/AKT-mediated activation of estrogen receptor alpha: a new model for anti-estrogen resistance. The Journal of biological chemistry. 2001;276:9817–24. doi: 10.1074/jbc.M010840200. [DOI] [PubMed] [Google Scholar]
- 8.Kato S, Endoh H, Masuhiro Y, et al. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science (New York, NY) 1995;270:1491–4. doi: 10.1126/science.270.5241.1491. [DOI] [PubMed] [Google Scholar]
- 9.Sabnis GJ, Jelovac D, Long B, Brodie A. The role of growth factor receptor pathways in human breast cancer cells adapted to long-term estrogen deprivation. Cancer research. 2005;65:3903–10. doi: 10.1158/0008-5472.CAN-04-4092. [DOI] [PubMed] [Google Scholar]
- 10.Martin MB, Franke TF, Stoica GE, et al. A role for Akt in mediating the estrogenic functions of epidermal growth factor and insulin-like growth factor I. Endocrinology. 2000;141:4503–11. doi: 10.1210/endo.141.12.7836. [DOI] [PubMed] [Google Scholar]
- 11.Bunone G, Briand PA, Miksicek RJ, Picard D. Activation of the unliganded estrogen receptor by EGF involves the MAP kinase pathway and direct phosphorylation. The EMBO journal. 1996;15:2174–83. [PMC free article] [PubMed] [Google Scholar]
- 12.Joel PB, Smith J, Sturgill TW, Fisher TL, Blenis J, Lannigan DA. pp90rsk1 regulates estrogen receptor-mediated transcription through phosphorylation of Ser-167. Molecular and cellular biology. 1998;18:1978–84. doi: 10.1128/mcb.18.4.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hartl FU. Molecular chaperones in cellular protein folding. Nature. 1996;381:571–9. doi: 10.1038/381571a0. [DOI] [PubMed] [Google Scholar]
- 14.Lindquist S, Craig EA. The heat-shock proteins. Annual review of genetics. 1988;22:631–77. doi: 10.1146/annurev.ge.22.120188.003215. [DOI] [PubMed] [Google Scholar]
- 15.Lai BT, Chin NW, Stanek AE, Keh W, Lanks KW. Quantitation and intracellular localization of the 85K heat shock protein by using monoclonal and polyclonal antibodies. Molecular and cellular biology. 1984;4:2802–10. doi: 10.1128/mcb.4.12.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ralhan R, Kaur J. Differential expression of Mr 70,000 heat shock protein in normal, premalignant, and malignant human uterine cervix. Clin Cancer Res. 1995;1:1217–22. [PubMed] [Google Scholar]
- 17.Burrows F, Zhang H, Kamal A. Hsp90 activation and cell cycle regulation. Cell cycle (Georgetown, Tex) 2004;3:1530–6. doi: 10.4161/cc.3.12.1277. [DOI] [PubMed] [Google Scholar]
- 18.Beliakoff J, Whitesell L. Hsp90: an emerging target for breast cancer therapy. Anti-cancer drugs. 2004;15:651–62. doi: 10.1097/01.cad.0000136876.11928.be. [DOI] [PubMed] [Google Scholar]
- 19.Whitesell L, Mimnaugh EG, De Costa B, Myers CE, Neckers LM. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:8324–8. doi: 10.1073/pnas.91.18.8324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sausville EA, Tomaszewski JE, Ivy P. Clinical development of 17-allylamino, 17-demethoxygeldanamycin. Current cancer drug targets. 2003;3:377–83. doi: 10.2174/1568009033481831. [DOI] [PubMed] [Google Scholar]
- 21.Schulte TW, Neckers LM. The benzoquinone ansamycin 17-allylamino-17-demethoxygeldanamycin binds to HSP90 and shares important biologic activities with geldanamycin. Cancer chemotherapy and pharmacology. 1998;42:273–9. doi: 10.1007/s002800050817. [DOI] [PubMed] [Google Scholar]
- 22.Supko JG, Hickman RL, Grever MR, Malspeis L. Preclinical pharmacologic evaluation of geldanamycin as an antitumor agent. Cancer chemotherapy and pharmacology. 1995;36:305–15. doi: 10.1007/BF00689048. [DOI] [PubMed] [Google Scholar]
- 23.Egorin MJ, Lagattuta TF, Hamburger DR, et al. Pharmacokinetics, tissue distribution, and metabolism of 17-(dimethylaminoethylamino)-17-demethoxygeldanamycin (NSC 707545) in CD2F1 mice and Fischer 344 rats. Cancer chemotherapy and pharmacology. 2002;49:7–19. doi: 10.1007/s00280-001-0380-8. [DOI] [PubMed] [Google Scholar]
- 24.Shadad FN, Ramanathan RK. 17-dimethylaminoethylamino-17-demethoxygeldanamycin in patients with advanced-stage solid tumors and lymphoma: a phase I study. Clinical lymphoma & myeloma. 2006;6:500–1. doi: 10.3816/CLM.2006.n.034. [DOI] [PubMed] [Google Scholar]
- 25.Kamal A, Thao L, Sensintaffar J, et al. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. 2003;425:407–10. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- 26.Zhou DJ, Pompon D, Chen SA. Stable expression of human aromatase complementary DNA in mammalian cells: a useful system for aromatase inhibitor screening. Cancer research. 1990;50:6949–54. [PubMed] [Google Scholar]
- 27.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Advances in enzyme regulation. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 28.Cohen GM. Caspases: the executioners of apoptosis. The Biochemical journal. 1997;326:1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nicholson RI, McClelland RA, Robertson JF, Gee JM. Involvement of steroid hormone and growth factor cross-talk in endocrine response in breast cancer. Endocrine-related cancer. 1999;6:373–87. doi: 10.1677/erc.0.0060373. [DOI] [PubMed] [Google Scholar]
- 30.Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes & development. 1999;13:2905–27. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 31.Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nature reviews. 2005;5:341–54. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- 32.Beliakoff J, Bagatell R, Paine-Murrieta G, Taylor CW, Lykkesfeldt AE, Whitesell L. Hormone-refractory breast cancer remains sensitive to the antitumor activity of heat shock protein 90 inhibitors. Clin Cancer Res. 2003;9:4961–71. [PubMed] [Google Scholar]
- 33.Zsebik B, Citri A, Isola J, Yarden Y, Szollosi J, Vereb G. Hsp90 inhibitor 17-AAG reduces ErbB2 levels and inhibits proliferation of the trastuzumab resistant breast tumor cell line JIMT-1. Immunology letters. 2006;104:146–55. doi: 10.1016/j.imlet.2005.11.018. [DOI] [PubMed] [Google Scholar]
- 34.Radujkovic A, Schad M, Topaly J, et al. Synergistic activity of imatinib and 17-AAG in imatinib-resistant CML cells overexpressing BCR-ABL--Inhibition of P-glycoprotein function by 17-AAG. Leukemia. 2005;19:1198–206. doi: 10.1038/sj.leu.2403764. [DOI] [PubMed] [Google Scholar]
- 35.Lang SA, Moser C, Gaumann A, et al. Targeting heat shock protein 90 in pancreatic cancer impairs insulin-like growth factor-I receptor signaling, disrupts an interleukin-6/signal-transducer and activator of transcription 3/hypoxia-inducible factor-1alpha autocrine loop, and reduces orthotopic tumor growth. Clin Cancer Res. 2007;13:6459–68. doi: 10.1158/1078-0432.CCR-07-1104. [DOI] [PubMed] [Google Scholar]
- 36.Lang SA, Klein D, Moser C, et al. Inhibition of heat shock protein 90 impairs epidermal growth factor-mediated signaling in gastric cancer cells and reduces tumor growth and vascularization in vivo. Molecular cancer therapeutics. 2007;6:1123–32. doi: 10.1158/1535-7163.MCT-06-0628. [DOI] [PubMed] [Google Scholar]
- 37.Datta SR, Dudek H, Tao X, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–41. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 38.Cardone MH, Roy N, Stennicke HR, et al. Regulation of cell death protease caspase-9 by phosphorylation. Science (New York, NY) 1998;282:1318–21. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- 39.Brunet A, Bonni A, Zigmond MJ, et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–68. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 40.Gossett DR, Bradley MS, Jin X, Lin J. 17-Allyamino-17-demethoxygeldanamycin and 17-NN-dimethyl ethylene diamine-geldanamycin have cytotoxic activity against multiple gynecologic cancer cell types. Gynecologic oncology. 2005;96:381–8. doi: 10.1016/j.ygyno.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 41.Robles AI, Wright MH, Gandhi B, et al. Schedule-dependent synergy between the heat shock protein 90 inhibitor 17-(dimethylaminoethylamino)-17-demethoxygeldanamycin and doxorubicin restores apoptosis to p53-mutant lymphoma cell lines. Clin Cancer Res. 2006;12:6547–56. doi: 10.1158/1078-0432.CCR-06-1178. [DOI] [PubMed] [Google Scholar]
- 42.Muise-Helmericks RC, Grimes HL, Bellacosa A, Malstrom SE, Tsichlis PN, Rosen N. Cyclin D expression is controlled post-transcriptionally via a phosphatidylinositol 3-kinase/Akt-dependent pathway. The Journal of biological chemistry. 1998;273:29864–72. doi: 10.1074/jbc.273.45.29864. [DOI] [PubMed] [Google Scholar]
- 43.Muratani M, Tansey WP. How the ubiquitin-proteasome system controls transcription. Nat Rev Mol Cell Biol. 2003;4:192–201. doi: 10.1038/nrm1049. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.