

Abstract
Salmonella are closely related to commensal Escherichia coli but have gained virulence factors enabling them to behave as enteric pathogens. Less well studied are the similarities and differences that exist between the metabolic properties of these organisms that may contribute toward niche adaptation of Salmonella pathogens. To address this, we have constructed a genome scale Salmonella metabolic model (iMA945). The model comprises 945 open reading frames or genes, 1964 reactions, and 1036 metabolites. There was significant overlap with genes present in E. coli MG1655 model iAF1260. In silico growth predictions were simulated using the model on different carbon, nitrogen, phosphorous, and sulfur sources. These were compared with substrate utilization data gathered from high throughput phenotyping microarrays revealing good agreement. Of the compounds tested, the majority were utilizable by both Salmonella and E. coli. Nevertheless a number of differences were identified both between Salmonella and E. coli and also within the Salmonella strains included. These differences provide valuable insight into differences between a commensal and a closely related pathogen and within different pathogenic strains opening new avenues for future explorations.
Salmonella is a major cause of human and animal enteric disease. Salmonella consists of two species, bongori and enterica, and the latter can be further divided into subspecies (I-VI). The majority of human and animal infections are caused by S. enterica subspecies I, of which Salmonella typhimurium and Salmonella enteritidis are the most prevalent causes of human inflammatory gastroenteritis, often referred to as food poisoning (1). The recent availability of genome sequences of bacterial pathogens, including Salmonella, provides an opportunity to interrogate these organisms using a systems biology approach. By contrasting the genotype-phenotype relationship of pathogens such as Salmonella against closely related commensals such as an Escherichia coli K12 insights can be revealed into how these pathogens have adapted to their environmental niche(s). Salmonella and E. coli K12 share ∼85% of their genome (2–6). DNA microarray and genome sequencing studies have highlighted regions of the genome that are conserved between these closely related bacteria and those that are different. Many of the differences are attributable to the acquisition of virulence factors, although a significant proportion of their genome codes is for metabolic genes (2–8).
A genome scale model consists of a stoichiometric reconstruction of all reactions known to act in the metabolism of an organism along with a set of accompanying constraints on the flux of each reaction in the system (9, 10). These models define the organism's global metabolic space, network structural properties, and flux distribution potential (9, 10). Therefore constraint-based models can help predict cellular phenotypes given particular environmental conditions. Genome scale models have been useful in understanding the metabolic properties of a variety of organisms including E. coli, Bacillus subtilis, Pseudomonas putida, and Lactobacillus (9–12). Genome scale models can be validated in various ways such as continuous culture experiments, substrate utilization assays, specific gene mutations, and isotopic carbon measurements. The high through-put phenotype microarray (PM)3 system that is available through Biolog (Hayward, CA) is ideal to use for substrate utilization assays as it provides a comprehensive large-scale phenotyping technology to assess gene function at the cellular level (13).
The aim of this work was to construct a Salmonella genome scale model. The model highlights the similarities and differences between pathogenic bacteria such as S. typhimurium and S. enteritidis and the commensal E. coli K12 laboratory strains. The model was validated using the PM system and literature-derived (i.e. bibliomic) information. The substrate utilization assays also highlighted current knowledge gaps that will require further experimental data that can be used in the future for refining and extending the model.
EXPERIMENTAL PROCEDURES
Phenotype MicroArray (Biolog)
E. coli MG1655 was obtained from ATCC (700926), S. typhimurium LT2 (ACTCC 700220), S. typhimurium DT104 (NCTC 13348), and S. enteritidis PT4 (NCTC 13349) were obtained from the Sanger Center (Wellcome Trust Genome Campus, Hinxton, Cambridge). The four bacterial strains were analyzed using the OmniLog Phenotype MicroArray technology provided by Biolog (13) that allows high throughput substrate utilization screening of bacterial cells and included 191 sole carbon sources, 95 sole nitrogen sources, 59 sole phosphorus sources, and 35 sole sulfur sources. All fluids, reagents and PM panels were supplied by Biolog and used according to the manufacturer's instructions. Briefly, bacterial strains were cultured for 16 h on Luria-Bertani agar plates at 37 °C. Cells were picked from the agar surface with a sterile cotton swab and suspended in 10 ml of inoculating fluid (IF-0), and the cell density was adjusted to 85% transmittance (T) on a Biolog turbidimeter. The minimal media inoculating fluid (IF-0) contained 100 mm NaCl, 30 mm triethanolamine-HCl (pH 7.1), 5.0 mm NH4Cl, 2.0 mm NaH2PO4, 0.25 mMNa2SO4, 0.05 mm MgCl2, 1.0 mm KCl, 1.0 μm ferric chloride, and 0.01% tetrazolium violet (13). Before the addition to PM microtiter plates, bacterial suspensions were further diluted into 12 ml of IF-0 (per plate) in the relevant inoculating fluid. The carbon source for PM3 and -4 experiments that measure nitrogen, phosphorus, sulfur, and peptide utilization was 20 mm sodium succinate and 2 μm ferric citrate. Substrate utilization was measured via the reduction of a tetrazolium dye forming a purple formazan (supplied by Biolog) and is indicative of active cellular respiration at 37 °C. Formazan formation was monitored at 15-min intervals for 26 h. Kinetic data were analyzed with OmniLog-PM software. Each experiment was performed at least twice per strain. The list of compounds and the ability of each strain to utilize these substrates are detailed in the supplemental files AbuOunSuppl-01 to AbuOunSuppl-04). It must be noted that although tetrazolium dye reduction is indicative of cellular respiration, it may occur independent of cell growth (cell multiplication/formation of biomass) in some cases (13, 14).
Model Construction
The S. typhimurium metabolic model was constructed starting with the core gene data set present in E. coli (7) and the annotated genome of S. typhimurium LT2. We then made use of the recently published iAF1260 metabolic model of E. coli MG1655 (ECO/K12) (15) to identify the reactions and to establish the gene-protein reaction associations that could be directly incorporated into the initial Salmonella model. Protein homology searches were used to identify additional genes and reactions to append to the model. These reactions were added to the model after evaluating charge and elemental balancing. A list of reactions, gene associations, exchange fluxes, and metabolites included in Salmonella model construction are detailed in supplemental file AbuOunSuppl-05.
Generation of the Biomass Equation
The biomass equation was generated by accounting for as many as possible of the constituents that form the cellular biomass of S. typhimurium. We started with the biomass equation from the core biomass equation from the E. coli iAF1260 metabolic model (15). Amino acid utilization was incorporated as charged, and uncharged tRNA molecules in the biomass equation was incorporated as reactants and products, respectively.
Generation of a Computations-ready Model
Testing the metabolic model using optimization-based approaches requires the definition of a number of sets and parameters. Sets: I, {i} = set of metabolites; J = {j} = set of reactions; JR ⊆ J = set of reversible reactions; IE ⊆ metabolites that can cross cell boundaries (either direction); IF ⊆ I = metabolites present in growth medium. Parameters: Sij = stoichiometric matrix; LBj = lower bound of flux of reaction j; UBj = upper bound of flux of reaction j. Variable: vj = flux of reaction j; upper and lower bounds; UBj and LBj, were chosen as not to exclude any physiologically relevant metabolic flux values. The upper bound for all reaction fluxes was set to 1000. The lower bound was set equal to 0 for irreversible reactions and to −1000 for reversible reactions. The non-growth associated ATP maintenance limit was set to LBATPM = 8.4 per g of dry weight/h. The maximum transport rate into the cell was 20 mmol of per g of dry weight/h (i.e. LBj = −20) for any source exchange reactions (15).
Using the principle of stoichiometric analysis along with the application of a pseudo-steady-state hypothesis to the intracellular metabolites (16), an overall flux balance can be written as follows.
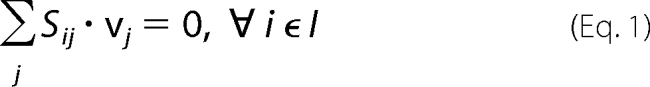 |
When constructing the model, we also generated the gene-protein reaction associations that link the ORFs to the reactions that are catalyzed by their gene products using standard conventions (17).
Analysis and Restoration of Network Connectivity
Once a mathematical representation of the metabolic model was generated, we first determined using GapFind (18) which metabolites could not be produced (i.e. cannot carry any net influx) given the availability of all substrates supported by the model. Much of the results obtained through GapFill (18) was subsumed by GrowMatch (19) (described briefly below), but blocked metabolites known to be present in S. typhimurium (e.g. adenosylcobalamin) were unblocked using GapFill.
Model Adjustment Using in Vivo Phenotypes
We tested the in silico growth predictions of the S. typhimurium metabolism network by examining the flux of the biomass equation. Given a particular growth environment, we solved the following formulation: maximize vbiomass, subject to Equations 1 and 2,
We simulated a particular growth environment by setting LBj = −20 for only the reactions associated with the components in the medium. This formulation was solved for each source condition using CPLEX Version 11 accessed within the General Algebraic Modelling System modeling environment to directly compare with the Biolog PM panels. During these comparisons oxygen uptake was permitted in both experiments and simulations, and the presence or absence of growth was tabulated for both experiment and model.
We next applied the GrowMatch method to reconcile inconsistencies between in silico and in vivo growth predictions (19). Full details of the method are given in the referenced work, but the salient points of method are briefly described here. To this end, we first classified growth predictions as “Grow” for those with vbiomassmax greater than zero and “NoGrow” otherwise. We then sorted growth prediction inconsistencies with the in vivo data into two categories; (a) Grow/NoGrow, Grow/NoGrow if the in silico model predicts growth, whereas there is no observed growth in vivo and (b) No Grow/Grow, if the in silico model predicts no growth in contrast with observed in vivo growth. In Grow/NoGrow mutants, the model over-predicts the metabolic capabilities of the organism. GrowMatch automatically restores consistency in these cases by suppressing reaction activities to prevent in silico growth (i.e. by identifying erroneously added reactions or missing regulation). Conversely, in No Grow/Grow cases, the model under-predicts the metabolic capabilities of the organism. GrowMatch restores consistency in these mutants by adding functionalities that ensure in silico growth consistent with in vivo data. The reaction source databases used by GrowMatch in this work were the KEGG (20) and MetaCyc (21) databases. In all cases, GrowMatch operates so as not to perturb any correct growth predictions. Although the focus in (19) was on gene essentiality using a single growth medium, the GrowMatch procedure is directly applicable to other growth phenotypes as used in the present work. As in the earlier steps of the model generation, the addition of reactions was carefully monitored, and we associated ORFs with these reactions where possible.
RESULTS
Reconstruction of Metabolic Network
The general principles of the metabolic reconstruction process have been previously outlined (15, 22). We followed a recently published reconstruction process that makes use of semiautomated tools during the series of successive refinements (23). Briefly, this process involves 1) identification of biotransformations using homology searches, 2) assembly of reaction sets into a genome scale metabolic model, 3) network connectivity analysis and restoration, and 4) evaluation and improvement of model performance when compared with in vivo growth phenotypes; details are available in the supplemental Excel file AbuOunSuppl-06.
During the first step, the S. typhimurium metabolic model was initially constructed using the core gene data set also present in E. coli genes (7) and the annotated genome of LT2 (5). We then used these homologies to generate a list of those genes present in the current E. coli metabolic reconstruction iAF1260 model (15). We used iAF1260 to identify the reactions and to establish the gene-protein reaction associations that could be directly incorporated into the initial Salmonella model. We also added all non-gene associated reactions from iAF1260 that were required for aerobic growth on glucose. Homology searches from protein databases and use of the KEGG (20) and MetaCyc (21) databases were also used to identify additional genes and reactions present in S. typhimurium. The model was tested using optimization based approaches, and gene-protein reaction associations that link the ORFs to the reactions that are catalyzed by their gene products were also generated. GapFind and GapFill (18) were used to unblock in the model metabolites known to be present in S. typhimurium (see “Experimental Procedures”).
Comparison of the homologous genes in Salmonella with those present in iAF1260 identified 842 ORFs that could be directly incorporated into the Salmonella model. A breakdown of the predicted functionality of the ORFs using COGs (Cluster of Orthologous Genes (24)) showed that the classes most represented were involved with transport and metabolism of various compounds such as amino acid, carbohydrate, lipid, and nucleotide, although genes involved in cell wall, envelope, or membrane biogenesis and energy production and conversion were also present (Fig. 1). Therefore, after the reconstruction process, model iMA945 incorporated 945 ORFs, of which 103 were unique to Salmonella. This could be further broken down into proteins, protein complexes, and isoenzymes. The model incorporated 1964 reactions and 1036 metabolites, which again were subdivided into various classes (see Table 1, model details are provided in supplemental file AbuOunSuppl-05).
FIGURE 1.

A, a Venn diagram that showsthe homology overlap between the S. typhimurium model iMA945 and the E. coli model iAF1260. B, classification of the ORFs included in iMA945 grouped into COG functional categories. The length of each bar represents the number of genes in each COG that is included in the model. The percent assigned to each class refers to the coverage of the total number in the genome accounted for in the model. Some of the ORFs do not currently have a COG functional category assignment (here represented as N/A). Note that although each ORF is only counted once within each COG functional category, some ORFs have multiple COG category assignments.
TABLE 1.
Properties of SalmonellaiMA945 model
| Included genes | 945 |
| Proteins | 810 |
| Protein complexes | 129 |
| Isozymes | 233 |
| Reactions | 1964 |
| Metabolic reactions | 1267 |
| Transport reactions | 697 |
| Gene-protein reaction associations | |
| Gene-associated (metabolic transport) | 1722 |
| Spontaneous | 29 |
| Non-gene-associated (metabolic transport) | 213 |
| Exchange reactions | 335 |
| Metabolites | 1036 |
| Cytoplasmic | 937 |
| Periplasmic | 445 |
| Extracellular | 330 |
Validation of iMA945 Using Substrate Utilization Patterns
The OmniLog PM technology system (Biolog) was used to validate the model whereby utilization of 191 carbon sources, 95 nitrogen sources, 59 phosphorus sources, and 35 sulfur sources were compared with simulations of iMA945 grown in silico in minimal medium with the respective compounds as the sole carbon, nitrogen, phosphorus, and sulfur sources (Table 2 and supplemental file AbuOunSuppl-06). For example, nitrogen source utilization was determined in the presence of sodium succinate/ferric citrate as the carbon source, sodium dihydrogen phosphate as the phosphorous source, and sodium sulfate as the sulfur source. In the case of phosphorus source utilization, sodium succinate/ferric citrate, ammonium chloride, and sodium sulfate were included as sources of carbon, nitrogen, and sulfur, respectively. In silico growth yields were calculated using the biomass composition and growth-associated and non-growth-associated energy maintenance factors taken from the E. coli model (15).
TABLE 2.
Validation of iMA945 metabolic model using substrate assays
E, experimental; C, computational; G, growth; NG, no growth; T, true; F, false; P, positive; N, negative; NAN, not a number (division by zero). Accuracy = (TP + TN)/(TP + TN + FP + FN); sensitivity = TP/(TP + FN); specificity = TN/(TN + FP).
| Source | Substrates | Overall agreement | Compounds with exchange reactions | Agreement |
Total accuracy | Disagreement |
Sensitivity | Specificity | ||
|---|---|---|---|---|---|---|---|---|---|---|
| E-G C-G TP | E-NG C-NG TN | E-NG C-G FP | E-G C-NG FN | |||||||
| % | % | % | ||||||||
| Carbon | 191 | 147 | 92 | 65 | 14 | 85.9 | 12 | 1 | 98.5 | 53.8 |
| Nitrogen | 95 | 83 | 66 | 35 | 20 | 83.3 | 7 | 4 | 89.7 | 74.1 |
| Phosphorous | 59 | 41 | 29 | 28 | 0 | 96.6 | 0 | 1 | 96.6 | NAN |
| Sulfur | 35 | 18 | 14 | 5 | 4 | 64.3 | 0 | 5 | 50.0 | 100.0 |
| Total | 380 | 289 | 201 | 133 | 38 | 85.1 | 19 | 11 | 92.4 | 66.7 |
The model was able to predict growth with 147 of the 191 carbon, 83 of the 95 nitrogen, 41 of the 59 phosphorous, and 18 of the 35 sulfur compounds correctly. In general, the total accuracy, sensitivity, and specificity were well above the standards established for metabolic genome reconstructions (see Table 2). In fact, a comparison of the iMA945 model with in silico and substrate utilization data from iAF1260 (15) reveals higher percentage total accuracy and specificity for carbon and nitrogen growth in iMA945, although they were similar for phosphorus and sulfur. For examples, comparison of predicted growth yields using the in silico predictions show compounds such as d-malic acid as a carbon source or l-tryptophan as a nitrogen source results in no growth for Salmonella, whereas maltose as a carbon source or adenosine as nitrogen source provides adequate in silico growth yields (see the supplemental data). The PM data match these predictions (see supplemental data). These results indicate that even though E. coli biomass composition was taken to predict in silico growth yields because of a lack of Salmonella-specific experimental information, the resulting predictions were congruous with in vivo experiments. However, in the future the model should be validated with Salmonella-specific biomass composition to evaluate the “true” energy cost to the living and growing Salmonella cells using different substrates.
Metabolic Differences between Salmonella and E. coli K12 and within Salmonella Strains
Both the model and the PM data helped us to identify metabolic differences between Salmonella and E. coli K12. Of the 379 conditions tested there were only 19 conditions under which E. coli K12 was unable to utilize substrates that Salmonella were able to utilize and 17 conditions in which E. coli K12 was able to utilize substrates that all three Salmonella strains included in our study were unable to utilize. These results allude to a common evolutionary pathway by which these enteric bacteria have evolved, and the fact that Salmonella and E. coli share ∼85% of their genomes (7, 25) is also reflected in conservation of many of their metabolic properties. For the majority of differences seen in substrate utilization between the two organisms we were able to assign corresponding genes and explanations (Table 3). The most common factor was due to the presence of operons or genes in Salmonella that were absent from E. coli and vice versa. Examples of genes/operons that are present in Salmonella but are absent from E. coli K12 include the apeE gene, pdu operon, rtl gene, and the hpa operon that confer the ability for Salmonella to utilize Tween 40 or 80, 1–2-propanediol, adonitol/ribitol, and p-hydroxyphenylacetic acid, respectively. Genomic regions/genes that are present in E. coli but absent in Salmonella included ebgAC genes, als operon, and tnaAB, which confer the ability for E. coli to utilize lactulose, β-d-allose, and l-tryptophan, respectively (Table 3). There were several instances where the most plausible explanation for the differences seen in substrate utilization was given, e.g. phosphocholine, taurocholic acid, or taurine utilization. These explanations require verification in the future by either complementing Salmonella strains with the respective genes or operon or by mutating the genetic pathway in E. coli.
TABLE 3.
Metabolic differences between E. coli and Salmonella
Phenotypic differences were compared with the predicted growth, and where possible a plausible explanation has been given for these differences. E, experimental; C, computational.
| Source | Compound | Model abbreviation | E. coli MG1655 (E)a | E. coli MG1655 (C)a | Salmonella (E)ab | Salmonella (C)ab | Gene/mutation | Explanation |
|---|---|---|---|---|---|---|---|---|
| Carbon | l-Proline | EX_pro-L | 0 | 1 | 1 | 1 | glnP | Mutation in glnP, the glutamine-binding protein, results in the inability to grow on proline (29) |
| Carbon | l-Glutamic acid | EX_glu-L | 0 | 1 | 1 | 1 | glnP | Mutation in glnP results in an inability to grow on l-glutamate (29) |
| Carbon | Dulcitol/galactitol | EX_galt | 0 | 1 | 1 | 1 | kba | Many E. coli K12 strains harbor a thermolabile-specific aldolase involved in galactitol degradation (51) |
| Carbon | l-Asparagine | EX_asn-L | 0 | 1 | 1 | 1 | ansB | FNR i.e. anaerobiosis-dependent expression of ansB (l-asparagine II) occurs in E. coli but not Salmonella (37) |
| Carbon | d-Glucosaminic acid | 0 | 1 | – | – | |||
| Carbon | 1,2-Propanediol | EX_12ppd-S | 0 | 1 | 1 | 1 | pduCDEGHPQW | The pdu operon is present in Salmonella but absent in E. coli (38, 52) |
| Carbon | Tween 40 | 0 | – | 1 | – | apeE | ApeE, an outer membrane esterase present in Salmonella, allows hydrolysis of naphthyl esters (53, 54) | |
| Carbon | Tween 80 | 0 | – | 1 | – | apeE | ApeE, an outer membrane esterase present in Salmonella, allows hydrolysis of naphthyl esters (53, 54) | |
| Carbon | Adonitol/ribitol | 0 | – | 1 | – | rtlBAC | Genes for ribitol catabolism missing in E. coli K12 (55, 56) | |
| Carbon | Citric acid | EX_cit | 0 | 1 | 1 | 1 | citAB (tcuABC) | The cit/tcu operon is absent in most E. coli but enables citrate utilization in Salmonella (57) |
| Carbon | Tricarballylic acid | 0 | – | 1 | – | citAB (tcuABC) | The cit/tcu operon, absent in E. coli, is also responsible for metabolism of the structurally related tricarballylic acid (58) | |
| Carbon | p-Hydroxy phenyl acetic cid | EX_4hphac | 0 | – | 1 | 1 | hpaBC, hpaGEDFHI, hpaR, hpaA, and hpaX | The hpa operon required for catabolism of 4-HPA is present in Salmonella but missing in E. coli K12 strains (39) |
| Carbon | m-Hydroxy phenyl acetic acid | 0 | – | 1 | – | hpaBC, hpaGEDFHI, hpaR, hpaA, and hpaX | The hpa operon required for catabolism of 4-HPA is present in Salmonella but missing in E. coli K12 (39) | |
| Carbon | d-Psicose | 0 | – | 1 | – | – | – | |
| Carbon | 2-Deoxy-d-ribose | 0 | – | 1 | – | deoQKPX | The deoQKPX operon, absent from E. coli, is required for the uptake, phosphorylation, and regulation of 2-deoxy-d-ribose utilization (59) | |
| Carbon | d-Tagatose | 0 | – | 1 | – | tagKHT | The presence of tag genes in the gat (galactitol) operon enables degradation of d-tagatose in Salmonella | |
| Carbon | d-Tartaric acid | 0 | – | 1 | – | – | – | |
| Carbon | Tyramine | EX_tym | 0 | 0 | 1 | 1 | hpa operon | In Salmonella, tyramine utilization as a carbon source results from its oxidative deamination by the cell membrane-bound tyramine oxidase (TynA/MaoA) to 4-HPA, requiring the hpa operon, which is absent from E. coli K12 (60) |
| Carbon | Lactulose | 1 | – | 0 | – | ebgAC | The ebg operon required for lactulose utilization is absent in Salmonella (61, 62) | |
| Carbon | d-Malic acid | EX_mal-D | 1 | 1 | 0 | 0 | yeaU/ttuC | Tartrate dehydrogenase, yeaU or ttuC, required for the breakdown of d-malate, is present in E. coli (22, 63) but absent from Salmonella |
| Carbon | l-Galactonicacid-γ-lactone | EX_galctn-L | 1 | 1 | 0 | 1 | YjjLMN | Growth dependent on the yjj operon (22), which is only partially present in S. typhimurium LT2 |
| Carbon | d-Galacturonic acid | EX_galur | 1 | 1 | 0 | 1 | exuT-uxaCA and uxaB | Genes required for the hexauronate catabolism exuT-uxaCA and uxaB (64) are absent in S. typhimurium |
| Carbon | β-d-Allose | EX_all-D | 1 | 1 | 0 | 0 | alsRBACEK | The β-d-allose utilization genes found in E. coli (65, 66) are missing from the Salmonella-sequenced genomes including S. typhimurium |
| Carbon | 3-0-β-d-Galactopyranosyl-d-arabinose | 1 | – | 0 | – | – | – | |
| Carbon | β-Methyl-d-galactoside | 1 | – | 0 | – | ebg and lac systems | This is probably due to absence of the ebg and lac system in Salmonella | |
| Nitrogen | Tyramine | EX_tym | 0 | 1 | 1 | 1 | maoB/tyramine | In E. coli, tyramine concentration and maoB regulate its utilization (31) |
| Nitrogen | N-Acetyl-d-mannosamine (ManNAc) | EX_acmana | 0 | 1 | 1 | 1 | mlc and nanR | Mlc and NanR regulators repress expression of the manXYZ-encoded transporter for ManNac uptake and nan genes for its utilization, resulting in very slow growth in E. coli K12 (67) |
| Nitrogen | l-Tryptophan | EX_trp-L | 1 | 1 | 0 | 0 | tnaAB | The l-tryptophan inducible tryptophanase utilized by E. coli (68) is missing in Salmonella |
| Nitrogen | Cytosine | EX_csn | 1 | 1 | 0 | 0 | codAB and nac | E. coli contains the codAB genes required for cytosine uptake and deamination regulated by Nac (69), which are absent in S. typhimurium |
| Phosphorus | Carbamyol phosphate | 1 | – | 0 | – | yeaI, phoA, and ynbD | Three phosphatases (YeaI, PhoA, YnbD), which may enable E. coli to cleave phosphorous from phosphate esters, are absent in Salmonella (70) | |
| Phosphorus | Phosphoryl choline | 1 | – | 0 | – | betT | BetT, the high affinity choline transporter present in E. coli (71), is absent in S. typhimurium, which also has the three phosphatases missing | |
| Sulfur | Taurocholic acid | 1 | – | 0 | – | ssuEADCB or tauABC tauD | The two sulfonate systems present in E. coli are absent in Salmonella and may be responsible for utilization of this compound | |
| Sulfur | Taurine | EX_taur | 1 | 1 | 0 | 0 | tauABC tauD | The taurine uptake and utilization operon from E. coli (72, 73) is absent in Salmonella |
| Sulfur | Hypotaurine | 1 | – | 0 | – | tauABC tauD | Hypotaurine is probably utilized by the same operon in E. coli | |
| Sulfur | Butane sulfonic acid | EX_butso3 | 1 | 1 | 0 | 0 | ssuEADCB | This operon is present in E. coli under regulation of CysB and Cbl and is required for butane sulfonic acid utilization (74–76) |
| Sulfur | 2-Hydroxyethane sulfonic acid | EX_isetac | 1 | 1 | 0 | 0 | ssuEADCB | The ssu operon is also involved in 2-hydroxyethane sulfonic acid utilization in E. coli |
| Sulfur | Methane sulfonic acid | EX_mso3 | 1 | 1 | 0 | 0 | ssuEADCB | The ssu operon is also involved in methane sulfonic acid utilization in E. coli |
a0 indicates no respiration was observed when grown in compound, 1 indicates respiration was observed when grown in compound, and–indicates that no data are available.
b Collated Biolog data are from S. typhimurium LT2, DT104 and S. enteritidis PT4.
Comparative genomic hybridization microarray have shown that strains from S. enterica subspecies I have a large portion of their genome conserved (7); therefore, it was not unexpected that of the compounds tested, only six showed differences in substrate utilization between the Salmonella strains. Differences included the inability to utilize l-histidine, l- and meso-tartaric acid, d-tagatose, glyoxylic acid, and d-saccharic acid (see the supplemental data). Interrogation of the S. typhimurium LT2 genome indicated that the hutU gene, involved in histidine metabolism (26, 27), harbored a frameshift mutation and, hence, is a pseudogene. This gene mutation is not present in S. typhimurium definitive type (DT) 104 or the S. enteritidis phage type (PT) 4 strain genome sequence (Sanger Institute), which unlike LT2, are both able to utilize histidine. S. typhimurium DT104 strains are missing a group of genes (STM0517-STM0529) involved in allantoin utilization (28) that is also involved in glyoxylic acid utilization.4 Again, these genes are present in both LT2 and PT4 strains, which unlike DT104 are able to utilize this substrate. S. enteritidis PT4 is unable to catabolize d-tagatose probably because of the absence of the tag operon (tagKHT) responsible for d-tagatose utilization in the genome sequence of S. enteritidis PT4; this operon is present in the both S. typhimurium strains, which are able to utilize d-tagatose. PT4 was unable to utilize d-saccharic acid, whereas DT104 was unable to utilize both levo and meso-tartaric acid, and LT2 was unable to utilize l-tartaric acid. These differences were not readily explainable and will require future experimental work. Further experimental work using strains from different Salmonella serovars and phage types will also inform whether a large core of metabolic properties in Salmonella are conserved and the Salmonella model iMA945 can be adapted to different serovars and definitive or phage types by including strain specific differences such as those identified in this study. In the future conditions such as osmolarity and pH, which are important stress responses that are likely to be involved in niche specific adaptation, should also be included.
Discrepancies and Future Refinement of the Model
Several discrepancies were identified between substrate utilization and model predictions for both the E. coli (iAF1260) and Salmonella (iMA945) models (see the supplemental data; Table 2). For many of these an adequate explanation was not available from the literature (Table 3). For example, the model predicts growth of E. coli MG1655 on both l-proline and l-glutamic acid, but the PM data shows no substrate utilization in the presence of either compound. These data also match with Feist et al. (15) and could be due to recent point mutations acquired by strain MG1655 in glnP. GlnP is essential for glutamate and proline transport and catabolism (29) and requires further exploration in the future. Tyramine used as a nitrogen source was another compound that showed discrepancy between substrate utilization data from both this data set and Feist et al. (15) and the model. The breakdown products of tyramine include ammonia, which should be utilizable as a nitrogen source in both Salmonella and E. coli (30). It has been shown for E. coli that both the presence of the MaoB regulator and tyramine concentration in the medium is essential for monoamine oxidase (MaoA) activity (31); hence, the inability of MG1655 to utilize tyramine may be an indication of insufficient tyramine in the PM medium used for these studies, although these factors seem not to have affected the ability of LT2 to utilize tyramine as a nitrogen source. This could indicate differences in regulation of tyramine metabolism in E. coli and Salmonella. There were several substrates for which an in silico pathway and, hence, the predicted growth yield was not available for either iAF1260 or iMA945 or for the strain-specific differences seen within Salmonella. As more information becomes available on the metabolism and regulation involved in utilization of some of the substrates highlighted here, it can be included in future iterations of the models to help in model refinement and to increase its accuracy.
DISCUSSION
Salmonella and E. coli are closely related bacterial species that have diverged from each other about a 100 million years ago (32). Since their divergence, Salmonella have become a pathogen for humans and animals, whereas E. coli largely remains a commensal (2, 5). The ability for Salmonella to survive within the host and cause disease has been attributed to the acquisition of specific virulence factors by horizontal gene transfer such as the Salmonella pathogenicity islands (8, 33, 34). These have been studied intensively over the past decade, and currently up to 10 genomic regions have been identified as Salmonella pathogenicity islands (25). However, metabolic differences that have enabled Salmonella to adapt to its specific niche are less well studied but may be key in understanding how this pathogen evolved. Therefore, the aim of this study was to construct a Salmonella genome scale metabolic model to identify similarities and differences between E. coli K12, which for this discussion we consider to be representative of a commensal strain, and Salmonella, which may give clues of its adaptability to a specific niche. There are significant differences between E. coli pathogenic types,5 and perhaps similar approaches to those described here can be used to gain clues of their adaptation to specific niches too.
Genome scale models can be used to characterize metabolic resource allocation, experimentally testable predictions of cell phenotype, to elucidate metabolic network evolution scenarios and to design experiments that most effectively reveal genotype-phenotype relationships (9, 35). In this work comparison of the Salmonella genome scale model, based on that constructed for the E. coli strain MG1655 (15), revealed that ∼90% of ORFs/genes included in the model overlap between the two organisms. This is similar to the ∼85% gene overlap found from comparative genomic hydridization microarray data between MG1655 and S. enterica subspecies I (sspI) strains that comprise the S. enterica sspI core genome (7), highlighting their evolutionary similarity and synteny in their genomes. Further exploration and verification of model iMA945 using phenotypic data gathered using the OmniLog PM system (Biolog) revealed differences in utilization of carbon, nitrogen, phosphorous, and sulfur substrates between the three Salmonella strains and E. coli.
Our data showed more differences in the utilization of carbon compounds rather than nitrogen, sulfur, or phosphorous, which is contrary to the common understanding that the catabolic repertoire for carbohydrate utilization is largely the same in E. coli and S. typhimurium (36). However, this may be because of inclusion of a larger number of carbon compounds than nitrogen, phosphorous, and sulfur in the list of substrates tested. Carbon compounds that were only utilizable by Salmonella included a diverse range from amino acids to sugar alcohols to napthyl esters and aromatic compounds. How these compounds help Salmonella to adapt to its niche will need to be investigated. Interestingly, often genes involved in catabolism of these compounds e.g. l-proline and l-asparagine, were present in E. coli K12 (29, 37), and the in silico model predicted growth, but probably because of differences in regulation and gene expression, only Salmonella strains showed utilization of these substrates in the system used in this study. In other instances such as catabolism of 4-HPA and 1,2-propanediol, catabolic gene cassettes had been recruited by Salmonella or lost by E. coli to enable utilization of the compound (38, 39). Interestingly, it is only E. coli K12 strains that are unable to catabolize 4-HPA, as E. coli B, C, and W are able to fully or partially degrade 4-HPA (39–41), indicating that even within the E. coli genus there are considerable metabolic differences between the well studied K12 strains and other E. coli present in nature.
Similarly, differences were also found in substrate utilization of the Salmonella strains included in this study. The S. typhimurium DT104 strain included in this study differs from the S. typhimurium LT2 strain in that the former strain has acquired an extrachromosomal genomic island (Salmonella Genomic Island I) that confers a penta-antibiotic resistance phenotype on this strains (42, 43). This strain has been implicated in human epidemic outbreaks in the past decade, and the presence of penta-antibiotic resistance makes it difficult to treat infections (44). Similarly, S. enteritidis is highly prevalent in human infections, usually transmitted through chickens or eggs (1, 45). Data from Health Protection Agency, UK data base have recorded more than 40,000 cases of human infections over the past 10 years in the UK because of S. enteritidis PT4.6 Therefore, although only a handful of differences were identified between the Salmonella strains, these differences may be significant in understanding how current pathogenic strains such as S. typhimurium DT104 and S. enteritidis PT4 have become highly successful in passing through the food chain and causing salmonellosis in humans in comparison to a largely laboratory-adapted Salmonella strain.
The developed Salmonella model provides a complementary resource to the recently published model by Raghunathan et al. (46). The data presented offer an experimentally robust method for the analysis of differences and similarities between Salmonella serovars of medical and veterinary importance (1). In addition, the use of succinate as the source of carbon to analysis the in silico and in vitro metabolism of nitrogen, phosphorus, and sulfur has the advantage of utilizing a core tricarboxylic acid cycle compound rather than a three-carbon compound, which can lead to auxiliary dissimilatory pathways (47). Also presented are the contributions of phosphorus and sulfur metabolism, thus providing a holistic view of the metabolome of Salmonella serovars. Inclusion of these substrates is an important factor to consider in such metabolic genome reconstructions, as these compounds are essential components of amino acids involved in several chemical reactions and are structural components of the bacterial cell such as phosphorus in nucleic acids, adenosine triphosphate, and cell membrane phospholipids.
For future refinement and improvement of the iMA945 model we will integrate within the model other cellular processes such as regulation, transcription, translation, and DNA replication, which place direct metabolic and energy demands on the metabolic network (17). In fact, Covert and co-workers (48) show that in an unregulated E. coli genome scale network model 83.6% of the predictions were correct, whereas in the regulated network model 91.4% of predictions were correct. In addition, genome-wide single gene deletion data have proven useful during the construction and curation of metabolic models (19). For E. coli, these have been performed in glucose (49) or glycerol (50) minimal media. Such experiments for the Salmonella serovars/strains discussed here would provide significant data for refining iMA945. Moreover, performing growth studies of the mutant library on different growth substrates could provide additional discernment into differences in metabolism of the epidemic Salmonella serovars or strains.
Supplementary Material
Acknowledgments
The Salmonella strains were part of a collaborative project with the Pathogen Sequencing Unit at Sanger Centre, Hinxton, UK.
This work was supported by United Kingdom Department of Environment, Food, and Rural Affairs Grant OZ0324 and the United States Department of Energy Grant DOE DE-FG02-05ER25684.

The on-line version of this article (available at http://www.jbc.org) contains supplemental data.
M. P. Saunders, unpublished data.
J. Hobman, personal communication.
C. Lane, Health Protection Agency Centre for Infections, personal communication.
- PM
- phenotype microarray
- COGs
- Cluster of Orthologous Genes
- DT
- definitive type
- PT
- phage type
- 4-HPA
- hydroxylphenylacetic acid
- ORF
- open reading frame.
REFERENCES
- 1.Rodrigue D. C., Tauxe R. V., Rowe B. (1990) Epidemiol. Infect. 105, 21–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blattner F. R., Plunkett G., 3rd, Bloch C. A., Perna N. T., Burland V., Riley M., Collado-Vides J., Glasner J. D., Rode C. K., Mayhew G. F., Gregor J., Davis N. W., Kirkpatrick H. A., Goeden M. A., Rose D. J., Mau B., Shao Y. (1997) Science 277, 1453–1462 [DOI] [PubMed] [Google Scholar]
- 3.Crosa J. H., Brenner D. J., Ewing W. H., Falkow S. (1973) J. Bacteriol. 115, 307–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McClelland M., Florea L., Sanderson K., Clifton S. W., Parkhill J., Churcher C., Dougan G., Wilson R. K., Miller W. (2000) Nucleic Acids Res. 28, 4974–4986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McClelland M., Sanderson K. E., Spieth J., Clifton S. W., Latreille P., Courtney L., Porwollik S., Ali J., Dante M., Du F., Hou S., Layman D., Leonard S., Nguyen C., Scott K., Holmes A., Grewal N., Mulvaney E., Ryan E., Sun H., Florea L., Miller W., Stoneking T., Nhan M., Waterston R., Wilson R. K. (2001) Nature 413, 852–856 [DOI] [PubMed] [Google Scholar]
- 6.McClelland M., Wilson R. K. (1998) Infect. Immun. 66, 4305–4312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anjum M. F., Marooney C., Fookes M., Baker S., Dougan G., Ivens A., Woodward M. J. (2005) Infect. Immun. 73, 7894–7905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Porwollik S., Boyd E. F., Choy C., Cheng P., Florea L., Proctor E., McClelland M. (2004) J. Bacteriol. 186, 5883–5898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Puchałka J., Oberhardt M. A., Godinho M., Bielecka A., Regenhardt D., Timmis K. N., Papin J. A., Martins dos Santos V. A. (2008) PLoS Comput Biol. 4, e1000210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reed J. L., Patel T. R., Chen K. H., Joyce A. R., Applebee M. K., Herring C. D., Bui O. T., Knight E. M., Fong S. S., Palsson B. O. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 17480–17484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ohl M. E., Miller S. I. (2001) Annu Rev. Med. 52, 259–274 [DOI] [PubMed] [Google Scholar]
- 12.Teusink B., Smid E. J. (2006) Nat. Rev. Microbiol. 4, 46–56 [DOI] [PubMed] [Google Scholar]
- 13.Bochner B. R., Gadzinski P., Panomitros E. (2001) Genome Res. 11, 1246–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bochner B. R. (2009) FEMS Microbiol. Rev. 33, 191–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feist A. M., Henry C. S., Reed J. L., Krummenacker M., Joyce A. R., Karp P. D., Broadbelt L. J., Hatzimanikatis V., Palsson B. Ø. (2007) Mol. Syst. Biol. 3, 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vallino J. J., Stephanopoulos G. (1993) Biotechnol. Bioeng. 41, 633–646 [PubMed] [Google Scholar]
- 17.Reed J. L., Vo T. D., Schilling C. H., Palsson B. O. (2003) Genome Biol. 4, R54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Satish Kumar V., Dasika M. S., Maranas C. D. (2007) BMC Bioinformatics 8, 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kumar V. S., Maranas C. D. (2009) PLoS Comput. Biol. 5, e1000308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kanehisa M., Araki M., Goto S., Hattori M., Hirakawa M., Itoh M., Katayama T., Kawashima S., Okuda S., Tokimatsu T., Yamanishi Y. (2008) Nucleic Acids Res. 36, D480–D484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caspi R., Foerster H., Fulcher C. A., Kaipa P., Krummenacker M., Latendresse M., Paley S., Rhee S. Y., Shearer A. G., Tissier C., Walk T. C., Zhang P., Karp P. D. (2008) Nucleic Acids Res. 36, D623–D631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reed J. L., Famili I., Thiele I., Palsson B. O. (2006) Nat. Rev. Genet 7, 130–141 [DOI] [PubMed] [Google Scholar]
- 23.Suthers P. F., Dasika M. S., Kumar V. S., Denisov G., Glass J. I., Maranas C. D. (2009) PLoS Comput Biol. 5, e1000285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tatusov R. L., Fedorova N. D., Jackson J. D., Jacobs A. R., Kiryutin B., Koonin E. V., Krylov D. M., Mazumder R., Mekhedov S. L., Nikolskaya A. N., Rao B. S., Smirnov S., Sverdlov A. V., Vasudevan S., Wolf Y. I., Yin J. J., Natale D. A. (2003) BMC Bioinformatics 4, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parkhill J., Dougan G., James K. D., Thomson N. R., Pickard D., Wain J., Churcher C., Mungall K. L., Bentley S. D., Holden M. T., Sebaihia M., Baker S., Basham D., Brooks K., Chillingworth T., Connerton P., Cronin A., Davis P., Davies R. M., Dowd L., White N., Farrar J., Feltwell T., Hamlin N., Haque A., Hien T. T., Holroyd S., Jagels K., Krogh A., Larsen T. S., Leather S., Moule S., O'Gaora P., Parry C., Quail M., Rutherford K., Simmonds M., Skelton J., Stevens K., Whitehead S., Barrell B. G. (2001) Nature 413, 848–852 [DOI] [PubMed] [Google Scholar]
- 26.Meiss H. K., Brill W. J., Magasanik B. (1969) J. Biol. Chem. 244, 5382–5391 [PubMed] [Google Scholar]
- 27.Smith G. R., Halpern Y. S., Magasanik B. (1971) J. Biol. Chem. 246, 3320–3329 [PubMed] [Google Scholar]
- 28.Matiasovicova J., Adams P., Barrow P. A., Hradecka H., Malcova M., Karpiskova R., Budinska E., Pilousova L., Rychlik I. (2007) Arch. Microbiol. 187, 415–424 [DOI] [PubMed] [Google Scholar]
- 29.Masters P. S., Hong J. S. (1981) J. Bacteriol. 147, 805–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Díaz E., Ferrández A., Prieto M. A., García J. L. (2001) Microbiol. Mol. Biol. Rev. 65, 523–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yamashita M., Azakami H., Yokoro N., Roh J. H., Suzuki H., Kumagai H., Murooka Y. (1996) J. Bacteriol. 178, 2941–2947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Doolittle R. F., Feng D. F., Tsang S., Cho G., Little E. (1996) Science 271, 470–477 [DOI] [PubMed] [Google Scholar]
- 33.Chan K., Baker S., Kim C. C., Detweiler C. S., Dougan G., Falkow S. (2003) J. Bacteriol. 185, 553–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ochman H., Lawrence J. G., Groisman E. A. (2000) Nature 405, 299–304 [DOI] [PubMed] [Google Scholar]
- 35.Feist A. M., Palsson B. Ø. (2008) Nat. Biotechnol. 26, 659–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mayer C., Boos W. (2005) Hexose/Pentose and Hexitol/Pentitol Metabolism. (Bock A. ed) www.ecosal.org, American Society for Microbiology, Washington, D. C. [Google Scholar]
- 37.Jennings M. P., Scott S. P., Beacham I. R. (1993) Mol. Microbiol. 9, 165–172 [DOI] [PubMed] [Google Scholar]
- 38.Price-Carter M., Tingey J., Bobik T. A., Roth J. R. (2001) J. Bacteriol. 183, 2463–2475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prieto M. A., Díaz E., García J. L. (1996) J. Bacteriol. 178, 111–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Burlingame R. P. (1983) The Degradation of 3-Phenylpropionic Acid by Escherichia coli Ph.D. thesis, University of Minnesota [Google Scholar]
- 41.Cooper R. A., Skinner M. A. (1980) J. Bacteriol. 143, 302–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boyd D., Peters G. A., Cloeckaert A., Boumedine K. S., Chaslus-Dancla E., Imberechts H., Mulvey M. R. (2001) J. Bacteriol. 183, 5725–5732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Doublet B., Boyd D., Mulvey M. R., Cloeckaert A. (2005) Mol. Microbiol. 55, 1911–1924 [DOI] [PubMed] [Google Scholar]
- 44.Threlfall E. J., Ward L. R., Rowe B. (1997) Euro. Surveill. 2, 81–84 [DOI] [PubMed] [Google Scholar]
- 45.Humphrey T. J., Baskerville A., Chart H., Rowe B., Whitehead A. (1992) Vet. Rec. 131, 386–388 [DOI] [PubMed] [Google Scholar]
- 46.Raghunathan A., Reed J., Shin S., Palsson B., Daefler S. (2009) BMC Syst. Biol. 3, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Unden G., Kleefeld A. (2004) in Aerobic and Anaerobic Growth (Bock A. ed) Chapter 3.4.1., www.ecosal.org, American Society for Microbiology, Washington, D. C. [Google Scholar]
- 48.Covert M. W., Knight E. M., Reed J. L., Herrgard M. J., Palsson B. O. (2004) Nature 429, 92–96 [DOI] [PubMed] [Google Scholar]
- 49.Baba T., Ara T., Hasegawa M., Takai Y., Okumura Y., Baba M., Datsenko K. A., Tomita M., Wanner B. L., Mori H. (2006) Mol. Syst. Biol. 2, 2006.0008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Joyce A. R., Reed J. L., White A., Edwards R., Osterman A., Baba T., Mori H., Lesely S. A., Palsson B. Ø., Agarwalla S. (2006) J. Bacteriol. 188, 8259–8271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lin E. C. (1996) in Escherichia coli and Salmonella: Cellular and Molecular Biology ( Neidhardt F. C. ed), 2nd Ed. pp. 307–342, American Society for Microbiology, Washington, D. C. [Google Scholar]
- 52.Chen P., Andersson D. I., Roth J. R. (1994) J. Bacteriol. 176, 5474–5482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carinato M. E., Collin-Osdoby P., Yang X., Knox T. M., Conlin C. A., Miller C. G. (1998) J. Bacteriol. 180, 3517–3521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Collin-Osdoby P., Miller C. G. (1994) Mol. Gen. Genet. 243, 674–680 [DOI] [PubMed] [Google Scholar]
- 55.Link C. D., Reiner A. M. (1983) Mol. Gen. Genet. 189, 337–339 [DOI] [PubMed] [Google Scholar]
- 56.Woodward M. J., Charles H. P. (1983) J. Gen. Microbiol. 129, 75–84 [DOI] [PubMed] [Google Scholar]
- 57.Reynolds C. H., Silver S. (1983) J. Bacteriol. 156, 1019–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lewis J. A., Horswill A. R., Schwem B. E., Escalante-Semerena J. C. (2004) J. Bacteriol. 186, 1629–1637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Christensen M., Borza T., Dandanell G., Gilles A. M., Barzu O., Kelln R. A., Neuhard J. (2003) J. Bacteriol. 185, 6042–6050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Parrott S., Jones S., Cooper R. A. (1987) J. Gen. Microbiol. 133, 347–351 [DOI] [PubMed] [Google Scholar]
- 61.Campbell J. H., Lengyel J. A., Langridge J. (1973) Proc. Natl. Acad. Sci. U.S.A. 70, 1841–1845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hall B. G. (1978) Genetics 90, 673–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tipton P. A., Peisach J. (1990) Biochemistry 29, 1749–1756 [DOI] [PubMed] [Google Scholar]
- 64.Nemoz G., Robert-Baudouy J., Stoeber F. (1976) J. Bacteriol. 127, 706–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kim C., Song S., Park C. (1997) J. Bacteriol. 179, 7631–7637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Poulsen T. S., Chang Y. Y., Hove-Jensen B. (1999) J. Bacteriol. 181, 7126–7130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Plumbridge J., Vimr E. (1999) J. Bacteriol. 181, 47–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stewart V., Yanofsky C. (1985) J. Bacteriol. 164, 731–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Muse W. B., Bender R. A. (1998) J. Bacteriol. 180, 1166–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhou L., Lei X. H., Bochner B. R., Wanner B. L. (2003) J. Bacteriol. 185, 4956–4972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lamark T., Styrvold O. B., Strøm A. R. (1992) FEMS Microbiol. Lett. 75, 149–154 [DOI] [PubMed] [Google Scholar]
- 72.Eichhorn E., van der Ploeg J. R., Kertesz M. A., Leisinger T. (1997) J. Biol. Chem. 272, 23031–23036 [DOI] [PubMed] [Google Scholar]
- 73.van der Ploeg J. R., Weiss M. A., Saller E., Nashimoto H., Saito N., Kertesz M. A., Leisinger T. (1996) J. Bacteriol. 178, 5438–5446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Iwanicka-Nowicka R., Hryniewicz M. M. (1995) Gene 166, 11–17 [DOI] [PubMed] [Google Scholar]
- 75.van der Ploeg J. R., Eichhorn E., Leisinger T. (2001) Arch. Microbiol. 176, 1–8 [DOI] [PubMed] [Google Scholar]
- 76.van der Ploeg J. R., Iwanicka-Nowicka R., Bykowski T., Hryniewicz M. M., Leisinger T. (1999) J. Biol. Chem. 274, 29358–29365 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.