

Abstract
Background
Of the five human Alpha-class glutathione transferases, expression of hGSTA5 has not been experimentally documented, even though in silico the hGSTA5 sequence can be assembled into a mRNA and translated. The present work was undertaken to determine whether hGSTA5 is functional.
Methods
Human K562 cells were transfected with the hGSTA5 gene driven by the CMV promoter, and hGSTA5 cDNA was recovered from mature mRNA by reverse transcription. The cDNA was used in bacterial and eukaryotic protein expression systems. The resulting protein, after purification by glutathione affinity chromatography where appropriate, was tested for glutathione transferase activity.
Results
Human K562 cells transfected with the hGSTA5 gene under control of a CMV promoter produced a fully spliced mRNA which, after reverse transcription and expression in E. coli, yielded a protein that catalyzed the conjugation of the lipid peroxidation product 4-hydroxynonenal to glutathione. Similarly, transfection of human HEK-293 cells with the hGSTA5 gene driven by the CMV promoter led to an elevated 4-hydroxynonenal-conjugating activity in the cell lysate. In addition, translation of hGSTA5 cDNA in a cell-free eukaryotic system gave rise to a protein with 4-hydroxynonenal-conjugating activity.
Conclusions
hGSTA5 can be processed to a mature mRNA which is translation-competent, producing a catalytically active enzyme.
General Significance
Because a functional gene would not be maintained in the absence of selective pressure, we conclude that the native hGSTA5 promoter is active but has a spatially or temporally restricted expression pattern, and/or is expressed only under specific (patho)physiological conditions.
Keywords: glutathione transferase, Alpha class glutathione transferase, 4-hydroxynonenal, 4-HNE, lipid peroxidation, pseudogene
1. Introduction
Enzymes that catalyze detoxification reactions usually form large families which arise by gene duplication followed by specialization of the individual genes for related but non-identical functions. Such divergence in substrate specificity may be driven by the need to detoxify a wide range of compounds, including xenobiotics not previously encountered by the organism, or by adoption of some of the enzymes for biosynthetic purposes. In either case, only genes whose products contribute to the evolutionary fitness of the organism will be subject to selection and will be maintained. Genes that provide little or no competitive advantage will, in the absence of selective pressure, accumulate random mutations in both coding and regulatory sequences, and will become pseudogenes.
Genes coding for the Alpha class of the human glutathione transferase (GST) family [see refs. 1,2 for classification and overview of GSTs] are clustered on chromosome 6p12 [3]. This cluster contains five genes (hGSTA1 - hGSTA5) and seven pseudogenes [4]. Of the five Alpha-class genes, the expression patterns and functions of four are well characterized [reviewed in refs. 5,6]. In contrast, hGSTA5 is designated as a gene rather than a pseudogene only because it has an undamaged coding sequence. No transcript or protein gene product of hGSTA5 has been experimentally identified in human material [1,4,7, and present work], and, consequently, no functional characterization of the hypothetical hGSTA5-5 protein was possible. A lack of catalytic and other activities of hGSTA5-5 would preclude its evolutionary selection, and may indicate that hGSTA5 is in fact a pseudogene. Therefore, to clarify the gene-versus-pseudogene status of hGSTA5, we investigated its potential functionality. Our results demonstrate that the hGSTA5 transcript can form, at least under control of a heterologous promoter, and that it is correctly spliced and translated. Moreover, the resulting protein has distinct catalytic properties.
2. Materials and methods
2.1. Cloning of the hGSTA5 gene
A 9,021-bp fragment of the hGSTA5 gene was amplified from human genomic DNA using the Expand Long Template PCR System (Roche Diagnostics, Indianapolis, IN). The sense primer 1F (see Table 1 for this and all other primers used in this work) encompassed the beginning of the open reading frame which is located in exon 2. A mutation was introduced in position -2 relative to the initiator ATG to create a Nhe I recognition site. This change does not significantly alter the functionality of the Kozak context of the initiation codon [8]. The antisense primer 1R, located at 8 – 38 bp downstream of the translation termination codon in exon 7, was mutated to introduce a Kpn I recognition site (Table 1). The following cycling protocol was used: 92°C for 2 min, followed by 10 cycles of 92°C for 10 s, 60°C for 30 s, 68°C for 8 min, followed by 20 cycles of 92°C for 10 s, 60°C for 30 s, 68°C for 8 min +20 s/cycle, followed by 8 min at 68°C. The blunt-ended PCR product was inserted into pCR-Blunt II-TOPO (Invitrogen, Carlsbad, CA) and excised using Nhe I and Kpn I. Finally, the resulting 9,007-bp hGSTA5 genomic fragment was inserted between the Nhe I and Kpn I sites of pcDNA 3.1(-) (Invitrogen), resulting in plasmid hGSTA5(genomic)/pcDNA 3.1(-).
Table 1.
Sequences of PCR primers used in the present work
| Primer name | Primer sequence |
|---|---|
| 1F | 5′-TCCAGGAGACTgctagcATGGCAGAGAAGC |
| 2F | 5′-TcatatgGCAGAGAAGCCCAAGCTCCACTACTC |
| 3F | 5′-GGCTGCAGCTGGAGTAGAGTTG |
| 4F | 5′-CAAGACTACCTTGTTGGCAACAAGCTGAGCT |
| 5F | 5′-CTTTTCTACTACGTGGAAGAGCTTGACTCG |
| 1R | 5′-ATTggtaccGCATGTTCTTGGCCTCCATAGC |
| 2R | 5′-AggatccTCTGGCATGTTCTTGGCCTCCATAGCTGCT |
| 3R | 5′-AGCAGAGGGAAGCTGGAGATAAGACTC |
Restriction enzyme recognition sites not present in the native sequence but introduced in the primer are shown in lower-case letters. Residues mutated to introduce these restriction sites are in bold. Regions of primers that correspond to protein coding sequences are underlined. Names of forward and reverse primers contain the letter F and R, respectively.
2.2. Generation of hGSTA5 cDNA by cell-mediated production of hGSTA5 mRNA followed by RT-PCR
Human K562 cells were transiently transfected with hGSTA5(genomic)/pcDNA 3.1(-). After 48 hr, total RNA was prepared using TRI Reagent (Molecular Research Center, Cincinnati, OH). After DNase I (Invitrogen) digestion, the first cDNA strand was synthesized with Superscript II (Invitrogen) and oligo(dT) according to the manufacturer's protocol. PCR was performed with the Expand Long Template PCR System (Roche) using the following cycling conditions: 92°C for 2 min, followed by 35 cycles of 92°C for 10 s, 60°C for 30 s, 68°C for 1 min, followed by 7 min at 68°C. To amplify a cDNA fragment that includes the entire open reading frame, the forward primer 2F (which spans the translation initiation codon and introduces an Nde I restriction site that includes the initiator ATG) was paired with the reverse primer 2R which is located immediately downstream of the two consecutive translation stop codons of the coding sequence and introduces a Bam HI recognition site. The resulting PCR product was gel-purified and cloned into the pGEM-T Easy plasmid (Promega). For diagnostic purposes, additional amplifications was performed using primers 5F and 2R (Table 1) which are located in exons 6 and 7, respectively.
2.3. Generation of a hGSTA5 bacterial expression vector
To liberate the cDNA insert from hGSTA5/pGEM-T Easy, the plasmid was digested with Bam HI followed by a partial digestion with Nde I (the cDNA contains an internal Nde I site). The resulting fragment was subcloned into pET-30a(+) (Novagen/EMB, Gibbstown, NJ) previously digested with the same two restriction enzymes, yielding plasmid hGSTA5(cDNA)/pET-30a(+). The identity of the insert was confirmed by sequencing.
2.4. Bacterial expression and purification of hGSTA5
Escherichia coli BL21-CodonPlus-RIL competent cells (Stratagene/Agilent, La Jolla, CA) were transformed with hGSTA5(cDNA)/pET-30a(+). For expression, a 500-ml culture was grown at 37°C and was induced at A600 = 0.6 with 1 mM isopropyl-1-thio-β-D-galactopyranoside (IPTG). After an additional 4 h at 37°C, the bacterial cells were harvested by centrifugation and frozen at -20° C until use. For purification and solubilization of inclusion bodies, the Protein Refolding Kit (cat. no. 70123-3REF, Novagen) was used. In brief, the frozen bacterial pellet was sonicated in 20 mM Tris-HCl, pH 7.5, 10 mM EDTA, 1% Triton X-100. Following centrifugation at 12,000 × g for 30 min, the supernatant was discarded, and the pellet was subjected to two additional cycles of Triton X-100 extraction. The final pellet consisting of enriched inclusion bodies was resuspended at 10 mg of wet weight/ml of 50 mM CAPS, pH 11, 0.3% N-lauroylsarcosine, incubated for 30 min at room temperature to solubilize aggregated proteins, and centrifuged at 10,000 × g for 10 min. Refolding was achieved by dialysis of the supernatant against refolding buffer (15 mM Tris-HCl, pH 8.0, 10 mM DTT, 5 mM EDTA, 5 mM PMSF, 20% glycerol) for 24 h at 4°C, followed by dialysis for 48 h against 20 mM potassium phosphate, pH 7.0, 2 mM EDTA, 1.4 mM 2-mercaptoethanol, and centrifugation at 10,000 × g for 10 min. The supernatant was loaded onto a glutathione agarose column for GST purification [9].
2.5. In vitro synthesis of hGSTA5-5 in an eukaryotic cell-free system
The synthesis of hGSTA5-5 was performed with the EasyXpress Insect Kit II (Qiagen, Valencia, CA) according to manufacturer's protocol using hGSTA5(cDNA)/pET-30a(+) plasmid as the template. This plasmid contains a T7 promoter that is required for in vitro transcription using the insect cell-free system.
2.6. Enzyme activities
The determination of the enzymatic properties of hGSTA5-5 with a panel of substrates was performed as described by us previously [10].
2.7. Attempts to identify hGSTA5 transcript in human cells
RNA was isolated from the human cell lines K562, HuH-7 and HEK-293 using TRI Reagent (Molecular Research Center). The DNase I-treated RNA served as the template for reverse-transcription primed with a mixture of oligo(dT) and random hexamers. In separate experiments, PCR was performed using three different DNA polymerases and the following primer pairs: 5F/2R, 2F/2R, 3F/3R, and 4F/3R. Except for 2F and 2R, all primers have 3′ ends that, among human Alpha-class GSTs, are unique for hGSTA5 (Fig. 1).
Fig. 1.
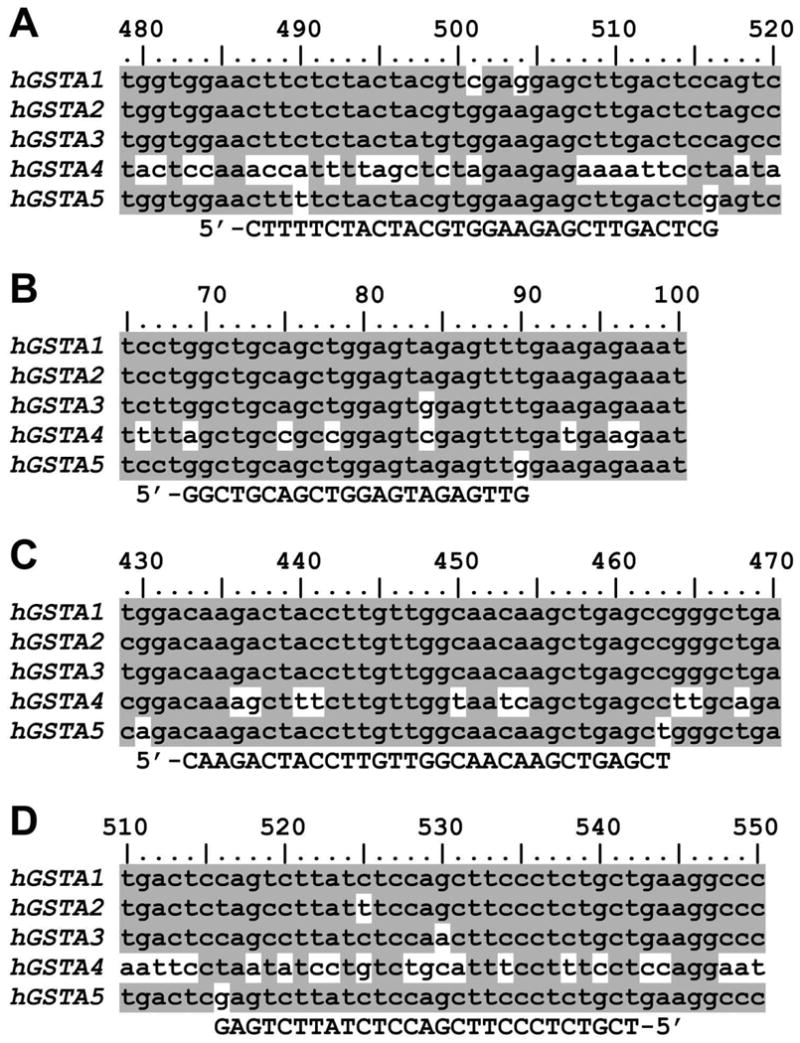
Specificity of primers used for the identification of hGSTA5 transcript by RT-PCR. The PCR primers (shown in the bottom line of each panel) were designed so that they do not recognize targets other than Alpha-class GSTs (as shown by a BLAST search against the human genome; not shown), and that, among human Alpha class sequences, their 3′ end are unique for hGSTA5. A partial alignment of all human Alpha-class GSTs is depicted; nucleotides unique for only one of the five sequences are shown on white background, whereas nucleotides shared by two or more sequences are on shaded background. The numbering is relative to the translation initiation site. Panel A: primer 5F; panel B: 3F; panel C: 4F; panel D: 3R (the primer sequences are listed in Table 1).
3. Results and Discussion
3.1 Transcription of the hGSTA5 gene
The hGSTA5 gene contains 7 exons and 6 introns, with the translation initiation and termination sites located in exons 2 and 7, respectively. A 9-kb region of the gene, starting at the translation initiation codon and ending 34 bp downstream of the translation stop codon, was cloned into the pcDNA 3.1(-) vector. Thus, the native promoter of the hGSTA5 gene was replaced with the CMV promoter supplied by the vector. When transiently transfected with this construct, K562 cells expressed the hGSTA5 transcript, as detected by reverse-transcription (RT) using oligo(dT) followed by PCR amplification with hGSTA5-specific primers. Two such amplifications were carried out. For diagnostic purposes, forward and reverse primers located in exons 6 and 7, respectively, were used (see Methods for details). The expected 223-bp product was observed in amplification reactions using cDNA from two independently transformed batches of K562 cells (Fig. 2, lanes 1 and 2), confirming the presence of a specific hGSTA5 transcript. In addition, a band at 1.1 kb was observed, likely derived either from traces of remaining genomic DNA or from unspliced mRNA, both of which would result in an amplicon of 1,111 bp. A parallel preparative amplification (see Methods for details) was carried out to generate a fragment containing the full-length hGSTA5 open reading frame for subsequent expression studies. The expected 713-bp amplification product was obtained, along with several additional bands, in reactions on cDNAs from both batches of transfected K562 cells (Fig. 2, lanes 3 and 4). The identity of this fragment was confirmed by sequencing (see below). That the transcript was derived from the transfected construct and not from the endogenous hGSTA5 gene present in human K562 cells is indicated by our inability, in parallel experiments, to generate a hGSTA5 cDNA from untransfected K562 cells. This result is in agreement with literature reporting that no hGSTA5 transcript was found in human tissues [4,7].
Fig. 2.
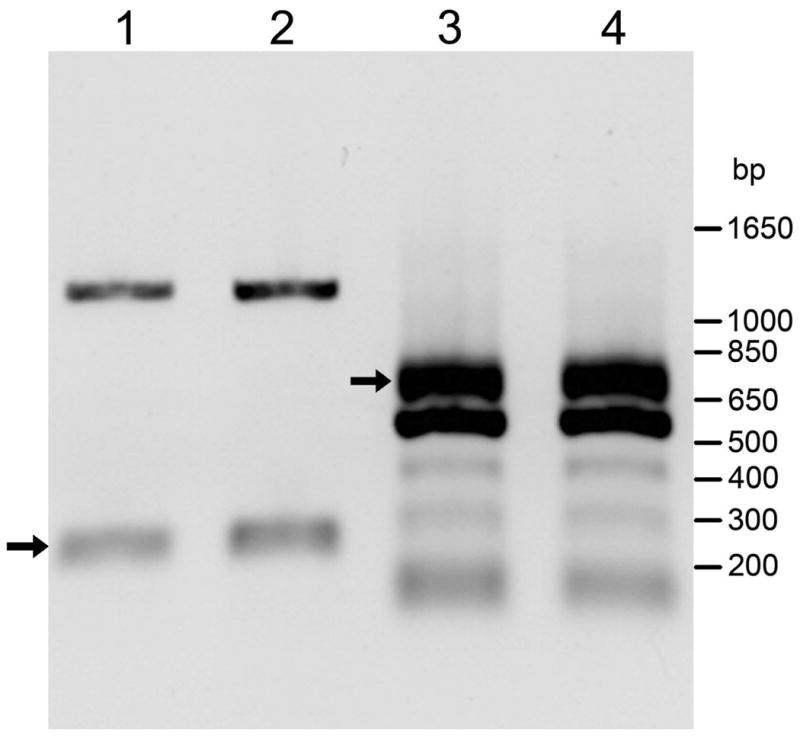
RT-PCR products from RNA isolated from K562 cells transfected with hGSTA5(genomic)/pcDNA 3.1(-). Lanes 1 and 3 are derived from one transfection, and lanes 2 and 4, from a second, independent replicate transfection. Lanes 1 and 2: PCR amplification with sense primer 5F and reverse primer 2R. Lanes 3 and 4: PCR amplification with sense primer 2F and reverse primer 2R. hGSTA5-specific cDNA products are marked by arrows.
The identification of a hGSTA5 transcript in transfected K562 cells depends critically on the specificity of the PCR reaction. Therefore, primer 5F, used for the diagnostic PCR to demonstrate that hGSTA5 mRNA has formed (Fig. 2, lanes 1 and 2), was designed such that its 3′ nucleotide uniquely matches hGSTA5 but none of the other human Alpha-class GSTs (Fig. 1A). BLAST searches against the human genome additionally verified that neither 5F nor the reverse primer 2R recognize targets other than Alpha class GSTs. For the amplification of the complete open reading frame, constraints on the positions of the primers somewhat limited the specificity, and indeed, more than one product was formed (Fig. 2, lanes 3 and 4). However, hGSTA5 was readily identifiable by restriction mapping (not shown), and was conclusively confirmed by sequencing (see below).
3.2. Nucleotide sequence analysis of the hGSTA5 gene product
After subcloning into pET-30a(+), the RT-PCR product containing the full-length hGSTA5 open reading frame was sequenced. The sequence fully matched that of the predicted hGSTA5 transcript (NCBI, locus NM_153699) except for position 163 of the open reading frame which is G in the NCBI reference sequence but was A in our hGSTA5 clone, resulting in Val-55 in the reference sequence and Ile-55 in our clone. This missense change is one of three polymorphisms affecting the amino acid sequence that are reported for hGSTA5 in the NCBI Single Nucleotide Polymorphism database (build 130). The perfect sequence agreement between our cDNA clone and one of the known hGSTA5 alleles conclusively identifies the clone, and proves that the hGSTA5 gene carries no mutations that would prevent its transcription and processing by human cells to a fully spliced, polyadenylated mature mRNA.
3.3. Expression of the hGSTA5 gene product in a bacterial system
The coding sequence of the hGSTA5 mRNA is complete and does not contain any obvious features that would prevent its translation. To verify experimentally that the hGSTA5-5 protein can be indeed synthesized, we used the hGSTA5 cDNA both in prokaryotic and eukaryotic protein expression systems. In E. coli, the expression of hGSTA5-5 was robust but most of the protein was sequestered in the insoluble fraction (inclusion bodies; Fig. 3A). Various interventions have been shown to minimize aggregation of proteins expressed in E. coli. These methods include growing bacteria at temperatures lower than 37°C, using low concentrations of IPTG for induction, using different strains of bacteria, co-expression with chaperones or with thioredoxin, applying osmotic pressure, using growth media other than LB, and others [11-18]. In our hands, however, none of these methods had a significant effect on hGSTA5-5 solubility. Therefore, the bacterially expressed, aggregated hGSTA5-5 was solubilized and refolded prior to affinity purification. This approach yielded an essentially pure, soluble hGSTA5-5 protein (Fig. 3B).
Fig. 3.
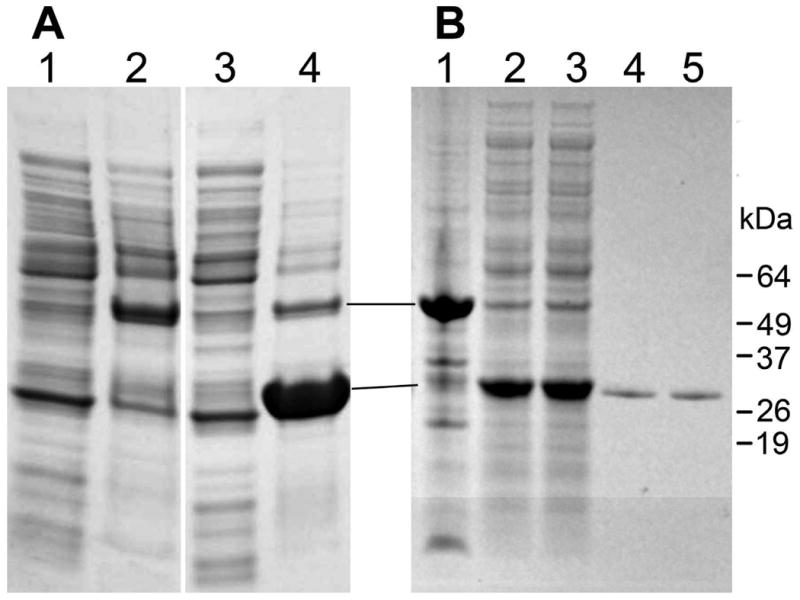
Solubilization, refolding, and purification of hGSTA5-5 from inclusion bodies after expression in bacteria. Panel A: E. coli were transformed with hGSTA5(cDNA)/pET-30a(+) and cultured as described in the Methods section. The bacterial lysate was centrifuged and resulting fractions were resolved on a NuPAGE 4-12% Bis-Tris gel using MES running buffer (Novex/Invitrogen). Lanes 1 and 2: supernatant and pellet, respectively, from uninduced cells; lanes 3 and 4: supernatant and pellet, respectively, from cells induced with IPTG. Panel B: Inclusion bodies were isolated from E. coli that was transformed with hGSTA5(cDNA)/pET-30a(+) and induced with IPTG. The inclusion bodies were solubilized, and the proteins were refolded and dialyzed as described in the Methods section. The dialysis retentate was centrifuged, resulting in pellet (lane 1) and supernatant (lanes 2,3) fractions. The supernatants were subjected to affinity chromatography on glutathione agarose, yielding purified protein (lanes 4,5). Material shown in lanes 2,4 and lanes 3,5, respectively, is derived from two independent solubilization/refolding experiments. All fractions of panel B were resolved on a NuPAGE 4-12% Bis-Tris gel using MOPS running buffer (Novex/Invitrogen). Please note that in the gel system used in this experiment, hGSTA5-5 was observed to migrate at a molecular weight that is higher than calculated.
3.4. Functional characterization of bacterially expressed hGSTA5-5
Purified hGSTA5-5 was enzymatically active (Table 2). This demonstrates that hGSTA5 mRNA is translationally competent, as it gives rise to an intact, functional protein. The specific activities of hGSTA5-5 with several commonly used GST substrates were moderate (Table 2). This could reflect the intrinsic properties of the protein, and perhaps indicate a specialized function involving a hypothetical substrate that was not tested, but could be also caused by incomplete refolding of the enzyme after solubilization of inclusion bodies. Thus, ratios of activities for the various substrates are more informative than absolute values. hGSTA5-5 is considerably more active with 4-hydroxynonenal (4-HNE) and trans-2-nonenal than with the GST model substrate 1-chloro-2,4-dinitrobenzene (CDNB). This suggests a functional role of hGSTA5-5 in the conjugation of electrophilic α,β-unsaturated carbonyl compounds, often derived from the peroxidation of polyunsaturated fatty acids. Higher activity for 4-HNE than for CDNB is characteristic for hGSTA4-4 [19] and for murine mGSTA4-4 [20,21], i.e., for prototypical enzymes of a sub-class of Alpha-class GSTs that are specialized for the conjugation of electrophilic lipid peroxidation products [2,6]. Invertebrates also express GSTs with similar properties. For example, little or no activity for CDNB but considerable activity for 4-HNE is characteristic for the Caenorhabditis elegans gst-10 gene product [22], as well as for Drosophila melanogaster Sigma-class GSTS1-1 [10] and several Delta and Epsilon-class GSTs [23]. As discussed elsewhere [24,25], the existence of 4-HNE-conjugating GSTs in phylogenetically distant species suggests a physiological need for this activity. Indeed, it is likely that all aerobic organisms that contain polyunsaturated fatty acids in their membranes must be able to metabolize 4-HNE and similar compounds. hGSTA5-5 appears to be part of a group of enzymes catalyzing this reaction.
Table 2.
Catalytic activities of hGSTA5-5 expressed in E. coli a
| Substrate | Specific activity (μmol/mg · min)b | |
|---|---|---|
| Purification 1 | Purification 2 | |
| 1-chloro-2,4-dinitrobenzene | 0.55 ± 0.07 | 0.64 ± 0.06 |
| 4-hydroxynonenal | 7.63 ± 0.35 | 7.32 ± 0.45 |
| trans-nonenal | 3.67 ± 0.52 | 3.58 ± 0.67 |
| acrolein | 0.11 ± 0.05 | 0.05 ± 0.03 |
| crotonaldehyde | ND | ND |
| cumene hydroperoxide | ND | ND |
a Activity was measured after solubilization/refolding and purification by affinity chromatography.
b Results from two independent bacterial expression and purification experiments are shown. Data are given as means ± standard deviation. ND: not detectable.
3.5. Predicted protein sequence of hGSTA5 and its functional consequences
The catalytic properties of hGSTA5-5 may be considered in the context of its sequence. The predicted amino acid sequence of the hGSTA5 subunit, in alignment with the sequences of the remaining four human Alpha-class GSTs, is shown in Fig. 4. The key tyrosine residue that participates in the catalytic cycle, Y9, is present in hGSTA5, consistent with the finding that the protein is enzymatically active. With one exception, all amino acids involved in binding and activation of glutathione [the G-site; refs. 26,27], are also preserved. The G-site residues are marked with asterisks in Fig. 4. In a major deviation from other Alpha-class GSTs, hGSTA5 has no arginine in position 15; this residue is replaced by serine. In the R15 residue, which is present in almost all Alpha-class GSTs, the ε nitrogen atom is involved in the activation of the nucleophilic substrate, glutathione [26,28]. In hGSTA1-1, mutagenesis of R15 leads to a substantial loss of activity [28]. Therefore, the substitution of R15 by serine may account for the moderate overall activity of hGSTA5-5 (Table 2).
Fig. 4.

Predicted amino acid sequence of hGSTA5 and its alignment with other human Alpha-class GSTs. The predicted sequence of hGSTA5 is shown in bold type. Underneath that sequence, the remaining human Alpha-class GSTs are listed, with amino acid identities to hGSTA5 shown as dots. The line labeled “A1–A4”, above the hGSTA5 sequence, contains a consensus sequence of hGSTA1, A2, A3, and A4 in which only those residues are shown that are identical in these four proteins. Amino acid residues involved in binding of glutathione (the G-site) are marked with asterisks [on the basis of hGSTA1; ref. 27]. Horizontal lines indicate the three regions in hGSTA1 and hGSTA4 that determine the binding of the electrophilic substrate (the H-site) [29].
The binding site for electrophilic/hydrophobic substrates, the H-site, of Alpha-class GSTs is composed of three regions of the protein [29], shown in Fig. 4 by horizontal lines. Interestingly, these regions within the hGSTA5 subunit have a greater similarity to hGSTA1 than to hGSTA4 (Fig. 4), even though of all the compounds tested, 4-HNE was the best substrate of hGSTA5-5 (see preceding section). In particular, hGSTA5 lacks G12 and Y212; these positions are occupied by alanine and serine, respectively, thus conforming to the hGSTA1 sequence (Fig. 4). G12 and Y212 have been determined to be important for a high catalytic efficiency of hGSTA4-4 toward 4-HNE [30,31], a finding consistent with a proposed reaction mechanism in which the lack of a side chain in G12 permits Y212 to assume a conformation conducive to catalysis [31]. Thus, the experimentally determined enzymatic activity of hGSTA5-5 toward 4-HNE, and especially the high ratio of activities for 4-HNE versus CDNB (Table 2), cannot be readily rationalized by the reaction mechanism postulated for hGSTA4-4 [31]. This suggests that hGSTA5-5 may have acquired the ability to conjugate 4-HNE independently of hGSTA4-4, and that the two enzymes utilize distinct catalytic mechanisms to facilitate the same reaction. A possible convergent evolution of hGSTA4-4 and hGSTA5-5 could thus resemble the already discussed example of invertebrate GSTs able to conjugate 4-HNE.
The sequence in Fig. 4 labeled as “A1–A4” shows residues that are identical in hGSTA1, A2, A3, and A4. hGSTA5 deviates from this human Alpha-class consensus in only four positions: 15, 30, 110, and 144. The lack of R15 in hGSTA5 has been already discussed, and the change in position 30 is a conservative substitution. However, the lack of a helix-breaking proline in position 110 may affect the overall conformation of the protein, and presence of arginine in position 144, which is on the surface of the protein, will affect surface charge. Such changes, even at positions remote from the active site, could influence the catalytic properties of the protein.
3.6. Expression of the hGSTA5 gene product in eukaryotic systems
In addition to bacteria, we expressed hGSTA5-5 in two eukaryotic systems. First, HEK-293 cells were transfected with plasmid hGSTA5(genomic)/pcDNA 3.1(-). A GST pool was purified from these cells as well as from untransfected control HEK-293. The specific activity for 4-HNE of the GST fraction increased by approximately 60% as a result of transfection, whereas the specific activity for CDNB remained virtually unchanged (Table 3). This indicates that the protein expressed in transfected cells was catalytically active, and that its activity for 4-HNE was greater than that for CDNB. These functional properties match those observed for bacterially expressed hGSTA5-5. To rule out the possibility that the activity increment in transfected cells is caused not by the transgene but by shifts in the expression of endogenous genes, hGSTA5-5 was synthesized in vitro using a cell-free insect transcription/translation system. The 4-HNE-conjugating activity was elevated in crude transcription/translation reactions supplemented with hGSTA5 cDNA, as compared with reactions containing control plasmids (Table 4). Material pooled from four in vitro reactions was subjected to affinity purification of GSTs on glutathione agarose. The resulting material had a specific activity (Table 4) that was statistically not different from that of bacterially expressed, homogenous hGSTA5-5. These results show that enzymatically active hGSTA5-5 can be synthesized in eukaryotic systems.
Table 3.
Catalytic activities of total GST fractions obtained from human HEK-293 cells transfected with hGSTA5(genomic)/pcDNA 3.1(-) and from untransfected control HEK-293 cells
| Cells | Specific activity (μmol/mg · min) | |
|---|---|---|
| 4-HNE | 1-chloro-2,4-dinitrobenzene | |
| Experiment 1 | ||
| HEK-293 | 1.32 ± 0.06 | 19.13 ± 0.05 |
| HEK-293/hGSTA5 | 2.13 ± 0.11 | 20.36 ± 0.08 |
| Experiment 2 | ||
| HEK-293 | 1.32 ± 0.52 | |
| HEK-293/hGSTA5 | 2.77 ± 0.83 | |
Total GST fractions were obtained by glutathione agarose affinity chromatography.
Table 4.
Supplementation of an in vitro transcription/translation system with hGSTA5 cDNA results in increased catalytic activity for 4-HNE conjugation
| Plasmid | Specific activity of 4-HNE conjugation (μmol/mg · min) |
|---|---|
| Insert-free pET-30a(+) | 0.076 ± 0.007 |
| Control plasmid a | 0.079 ± 0.009 |
| hGSTA5(cDNA)/pET-30a(+) | |
| Reaction 1 b | 0.133 ± 0.002 |
| Reaction 2 b | 0.114 ± 0.027 |
| Reaction 3 b | 0.102 ± 0.010 |
| Reaction 4 b | 0.106 ± 0.015 |
| hGSTA5(cDNA)/pET-30a(+); protein product purified c | 4.89 ± 1.67 |
a Control plasmid supplied with the Qiagen EasyXpress Insect Kit II.
b Four independent in vitro protein synthesis reactions were carried out and assayed for 4-HNE-conjugating activity without purification.
c Pooled material from four independent reactions (see preceding note) was purified by glutathione agarose affinity chromatography.
3.7. Is hGSTA5-5 present in cultured human cells or in normal human tissues?
As already mentioned, no hGSTA5 transcript has been found in human tissues [1,4,7]. In agreement with these results, we were unable to identify hGSTA5 mRNA by RT-PCR in two human cancer cell lines: K562 (chronic myelogenous leukemia) and HuH-7 (hepatocarcinoma), and in HEK-293 (human embryonic kidney cells, transformed with adenovirus 5) using primers selective for hGSTA5. Similarly, no unequivocal identification of hGSTA5-5 protein has been reported in the literature. A novel GST subunit, isolated from human pancreas, was named hGSTA5 [32]. However, the masses of this subunit, between 25,566 and 25,571 kDa in three individuals [32], were lower than the predicted mass of hGSTA5, with either Ile or Val in position 55, and with and without the initiator Met. In the absence of sequence data, it is not possible to classify the GST subunit reported in [32], but its mass indicates that it is not hGSTA5. Expression of hGSTA5-5 in human liver and pancreas has been reported in an on-line human protein database (www.humanproteinpedia.org) on the basis of mass spectrometry data. However, each of the peptides given as evidence for hGSTA5-5 is also part of several other GSTs belonging to the Alpha and Omega classes, and could be derived from these well-characterized proteins, some of them very abundant.
The lack of conclusive evidence in favor of hGSTA5 expression in human tissues appears to be at odds with the seemingly intact open reading frame of the hGSTA5 gene, a finding that led to the classification of hGSTA5 as a gene rather than a pseudogene. However, not all legitimate sequences encoding a particular protein are equivalent, and silent mutations may affect splicing, mRNA stability and translatability, and even protein folding [33,34]. Nevertheless, our results show that the apparent lack of naturally occurring hGSTA5-5 is not caused by the accumulation of exonic silent mutations: the hGSTA5 gene, if experimentally driven by an exogenous promoter, gives rise to a functional, enzymatically active protein, demonstrating that all intermediate steps are unimpaired.
Given the experimentally demonstrated functionality of the hGSTA5 gene, several possibilities may be considered to explain the absence of measurable hGSTA5 transcript or hGSTA5-5 protein in human tissues. One option is that the hGSTA5 promoter is inactive. This would mean that hGSTA5 is after all a pseudogene, in spite of an intact coding sequence. We consider this an unlikely possibility because, without expression and function of the gene product, there would be no selective pressure to prevent the accumulation of random mutations. However, our observation that the gene is in principle functional demonstrates that no detrimental mutations are present in hGSTA5. An inactive promoter would be compatible with a functional gene only if an evolutionarily very recent mutation greatly diminished the contribution of the gene product to Darwinian fitness. In such case, it is conceivable that, by chance, genetic drift inactivated the promoter before disabling the coding sequence. The already discussed absence of R15 in hGSTA5 could be the result of a recent R15S mutation likely to decrease the catalytic activity of an Alpha-class GST [26,28] and release the gene from the constraints of selection pressure. Whereas it would be appealing to propose that we are observing the evolutionary transition of a gene to a pseudogene, the transient nature of a state in which the promoter is inactive but the gene functional renders this scenario unlikely. Experimentally, an intrinsic lack of activity of the hGSTA5 promoter is not verifiable because a negative assay result may also signify a failure to supply a necessary but unknown combination of transcription factors and coactivators. For this reason, we did not study the function of the hGSTA5 promoter.
Alternatively, the hGSTA5 promoter could be functional but highly specialized and active only in particular tissues, at some developmental stages, or under specific physiological or pathological conditions. Although such selectivity would make it difficult to identify a gene product, we consider it the most likely explanation that is consistent with our experimental findings. Further work will be needed to define the physiological or pathological circumstances under which such restricted expression of hGSTA5-5 may occur, and to determine whether the major role of hGSTA5-5 is the conjugation of 4-HNE, an activity characterized by us in the present work, or whether the protein has other, hitherto unknown functions.
Acknowledgments
This work was supported in part by National Institutes of Health grants R01 AG028088 and ES007804 (to P.Z.). P.Z. is a recipient of a VA Research Career Scientist Award. We thank Xinhua Ji for discussion.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mannervik B, Board PG, Hayes JD, Listowsky I, Pearson WR. Nomenclature for mammalian soluble glutathione transferases. Meth Enzymol. 2005;401:1–8. doi: 10.1016/S0076-6879(05)01001-3. [DOI] [PubMed] [Google Scholar]
- 2.Zimniak P, Singh SP. Families of glutathione transferases. In: Awasthi YC, editor. Toxicology of glutathione transferases. CRC Press; Boca Raton, FL: 2006. pp. 11–26. [Google Scholar]
- 3.Board PG, Webb GC. Isolation of a cDNA clone and localization of human glutathione S-transferase 2 genes to chromosome band 6p12. Proc Natl Acad Sci USA. 1987;84:2377–2381. doi: 10.1073/pnas.84.8.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morel F, Rauch C, Coles B, Le Ferrec E, Guillouzo A. The human glutathione transferase alpha locus: genomic organization of the gene cluster and functional characterization of the genetic polymorphism in the hGSTA1 promoter. Pharmacogenetics. 2002;12:277–286. doi: 10.1097/00008571-200206000-00003. [DOI] [PubMed] [Google Scholar]
- 5.Hayes JD, Flanagan JU, Jowsey IR. Glutathione transferases. Annu Rev Pharm Toxicol. 2005;45:51–88. doi: 10.1146/annurev.pharmtox.45.120403.095857. [DOI] [PubMed] [Google Scholar]
- 6.Zimniak P. Substrates and reaction mechanisms of GSTs. In: Awasthi YC, editor. Toxicology of glutathione transferases. CRC Press; Boca Raton, FL: 2006. pp. 71–101. [Google Scholar]
- 7.Coles BF, Kadlubar FF. Human alpha class glutathione S-transferases: genetic polymorphism, expression, and susceptibility to disease. Meth Enzymol. 2005;401:9–42. doi: 10.1016/S0076-6879(05)01002-5. [DOI] [PubMed] [Google Scholar]
- 8.Kozak M. Initiation of translation in prokaryotes and eukaryotes. Gene. 1999;234:187–208. doi: 10.1016/s0378-1119(99)00210-3. [DOI] [PubMed] [Google Scholar]
- 9.Simons PC, Vander Jagt DL. Purification of glutathione S-transferases from human liver by glutathione-affinity chromatography. Anal Biochem. 1977;82:334–341. doi: 10.1016/0003-2697(77)90169-5. [DOI] [PubMed] [Google Scholar]
- 10.Singh SP, Coronella JA, Beneš H, Cochrane BJ, Zimniak P. Catalytic function of Drosophila melanogaster glutathione S-transferase DmGSTS1-1 (GST-2) in conjugation of lipid peroxidation end products. Eur J Bioch. 2001;268:2912–2923. doi: 10.1046/j.1432-1327.2001.02179.x. [DOI] [PubMed] [Google Scholar]
- 11.Kapust RB, Waugh DS. Escherichia coli maltose-binding protein is uncommonly effective at promoting the solubility of polypeptides to which it is fused. Protein Sci. 1999;8:1668–1674. doi: 10.1110/ps.8.8.1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hartman J, Daram P, Frizzell RA, Rado T, Benos DJ, Sorscher EJ. Affinity purification of insoluble recombinant fusion proteins containing glutathione-S-transferase. Biotechnol Bioeng. 1992;39:828–832. doi: 10.1002/bit.260390805. [DOI] [PubMed] [Google Scholar]
- 13.Kagawa N, Cao Q. Osmotic stress induced by carbohydrates enhances expression of foreign proteins in Escherichia coli. Arch Biochem Biophys. 2001;393:290–296. doi: 10.1006/abbi.2001.2516. [DOI] [PubMed] [Google Scholar]
- 14.Yasukawa T, Kanei-Ishii C, Maekawa T, Fujimoto J, Yamamoto T, Ishii S. Increase of solubility of foreign proteins in Escherichia coli by coproduction of the bacterial thioredoxin. J Biol Chem. 1995;270:25328–25331. doi: 10.1074/jbc.270.43.25328. [DOI] [PubMed] [Google Scholar]
- 15.Ghosh S, Rasheedi S, Rahim SS, Banerjee S, Choudhary RK, Chakhaiyar P, Ehtesham NZ, Mukhopadhyay S, Hasnain SE. Method for enhancing solubility of the expressed recombinant proteins in Escherichia coli. Biotechniques. 2004;37:418–420. 422–413. doi: 10.2144/04373ST07. [DOI] [PubMed] [Google Scholar]
- 16.Weickert MJ, Doherty DH, Best EA, Olins PO. Optimization of heterologous protein production in Escherichia coli. Curr Opin Biotechnol. 1996;7:494–499. doi: 10.1016/s0958-1669(96)80051-6. [DOI] [PubMed] [Google Scholar]
- 17.Widersten M. Heterologous expression in Escherichia coli of soluble active-site random mutants of haloalkane dehalogenase from Xanthobacter autotrophicus GJ10 by coexpression of molecular chaperonins GroEL/ES. Protein Expr Purif. 1998;13:389–395. doi: 10.1006/prep.1998.0913. [DOI] [PubMed] [Google Scholar]
- 18.Galloway CA, Sowden MP, Smith HC. Increasing the yield of soluble recombinant protein expressed in E. coli by induction during late log phase. Biotechniques. 2003;34:524–530. doi: 10.2144/03343st04. [DOI] [PubMed] [Google Scholar]
- 19.Hubatsch I, Ridderstrom M, Mannervik B. Human glutathione transferase A4-4: an Alpha class enzyme with high catalytic efficiency in the conjugation of 4-hydroxynonenal and other genotoxic products of lipid peroxidation. Biochem J. 1998;330:175–179. doi: 10.1042/bj3300175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Singh SP, Janecki AJ, Srivastava SK, Awasthi S, Awasthi YC, Xia SJ, Zimniak P. Membrane association of glutathione S-transferase mGSTA4-4, an enzyme that metabolizes lipid peroxidation products. J Biol Chem. 2002;277:4232–4239. doi: 10.1074/jbc.M109678200. [DOI] [PubMed] [Google Scholar]
- 21.Nanduri B, Hayden JB, Awasthi YC, Zimniak P. Amino acid residue 104 in an alpha-class glutathione S-transferase is essential for the high selectivity and specificity of the enzyme for 4-hydroxynonenal. Arch Biochem Biophys. 1996;335:305–310. doi: 10.1006/abbi.1996.0511. [DOI] [PubMed] [Google Scholar]
- 22.Ayyadevara S, Engle MR, Singh SP, Dandapat A, Lichti CF, Beneš H, Shmookler Reis RJ, Liebau E, Zimniak P. Lifespan and stress resistance of Caenorhabditis elegans are increased by expression of glutathione transferases capable of metabolizing the lipid peroxidation product 4-hydroxynonenal. Aging Cell. 2005;4:257–271. doi: 10.1111/j.1474-9726.2005.00168.x. [DOI] [PubMed] [Google Scholar]
- 23.Sawicki R, Singh SP, Mondal AK, Beneš H, Zimniak P. Cloning, expression, and biochemical characterization of one Epsilon-class (GST-3) and ten Delta-class (GST-1) glutathione S-transferases from Drosophila melanogaster, and identification of additional nine members of the Epsilon class. Biochem J. 2003;370:661–669. doi: 10.1042/BJ20021287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ayyadevara S, Dandapat A, Singh SP, Siegel ER, Shmookler Reis RJ, Zimniak L, Zimniak P. Life span and stress resistance of Caenorhabditis elegans are differentially affected by glutathione transferases metabolizing 4-hydroxynon-2-enal. Mech Ageing Dev. 2007;128:196–205. doi: 10.1016/j.mad.2006.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zimniak P. Detoxification reactions: relevance to aging. Ageing Res Rev. 2008;7:281–300. doi: 10.1016/j.arr.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sinning I, Kleywegt GJ, Cowan SW, Reinemer P, Dirr HW, Huber R, Gilliland GL, Armstrong RN, Ji X, Board PG, Olin B, Mannervik B, Jones TA. Structure determination and refinement of human alpha class glutathione transferase A1-1, and a comparison with the mu and pi class enzymes. J Mol Biol. 1993;232:192–212. doi: 10.1006/jmbi.1993.1376. [DOI] [PubMed] [Google Scholar]
- 27.Grahn E, Novotny M, Jakobsson E, Gustafsson A, Grehn L, Olin B, Madsen D, Wahlberg M, Mannervik B, Kleywegt GJ. New crystal structures of human glutathione transferase A1-1 shed light on glutathione binding and the conformation of the C-terminal helix. Acta Cryst. 2006;62D:197–207. doi: 10.1107/S0907444905039296. [DOI] [PubMed] [Google Scholar]
- 28.Bjornestedt R, Stenberg G, Widersten M, Board PG, Sinning I, Jones TA, Mannervik B. Functional significance of arginine 15 in the active site of human alpha glutathione transferase A1-1. J Mol Biol. 1995;247:765–773. doi: 10.1016/s0022-2836(05)80154-8. [DOI] [PubMed] [Google Scholar]
- 29.Nilsson LO, Gustafsson A, Mannervik B. Redesign of substrate-selectivity determining modules of glutathione transferase A1-1 installs high catalytic efficiency with toxic alkenal products of lipid peroxidation. Proc Natl Acad Sci USA. 2000;97:9408–9412. doi: 10.1073/pnas.150084897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bjornestedt R, Tardioli S, Mannervik B. The high activity of rat glutathione transferase 8-8 with alkene substrates is dependent on a glycine residue in the active site. J Biol Chem. 1995;270:29705–29709. doi: 10.1074/jbc.270.50.29705. [DOI] [PubMed] [Google Scholar]
- 31.Bruns CM, Hubatsch I, Ridderstrom M, Mannervik B, Tainer JA. Human glutathione transferase A4-4 crystal structures and mutagenesis reveal the basis of high catalytic efficiency with toxic lipid peroxidation products. J Mol Biol. 1999;288:427–439. doi: 10.1006/jmbi.1999.2697. [DOI] [PubMed] [Google Scholar]
- 32.Coles BF, Anderson KE, Doerge DR, Churchwell MI, Lang NP, Kadlubar FF. Quantitative analysis of interindividual variation of glutathione S-transferase expression in human pancreas and the ambiguity of correlating genotype with phenotype. Cancer Res. 2000;60:573–579. [PubMed] [Google Scholar]
- 33.Chamary JV, Parmley JL, Hurst LD. Hearing silence: non-neutral evolution at synonymous sites in mammals. Nat Rev Genet. 2006;7:98–108. doi: 10.1038/nrg1770. [DOI] [PubMed] [Google Scholar]
- 34.Tsai CJ, Sauna ZE, Kimchi-Sarfaty C, Ambudkar SV, Gottesman MM, Nussinov R. Synonymous mutations and ribosome stalling can lead to altered folding pathways and distinct minima. J Mol Biol. 2008;383:281–291. doi: 10.1016/j.jmb.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]