
Table 1.
Prospective test: Absolute binding free energy predictions
| Structure | ΔGb,exp (Kcal/mol) a | ΔG b,calc (Kcal/mol) b | RMSD (Å) c | PDB ID | |
|---|---|---|---|---|---|
| n-phenylglycinonitrile |  |
−5.52 ± 0.18 | −5.63 ± 0.38 | 0.87 | 2RBO |
| 2-nitrothiophene | 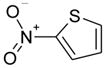 |
−4.85 ± 0.25 | −5.73 ± 0.13 | 1.09(A); 2.86(B) | 2RBN |
| thieno[3,2-b]thiophene | 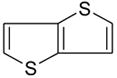 |
−4.67 ± 0.17 | −6.8 ± 0.23 | 0.73(A) 0.44(B) | 3HUQ |
| 4,5,6,7-tetrahydroindole | 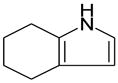 |
−4.61 ± 0.09 | −5.4 ± 0.45 | 0.66 & 1.78** | 3HUA |
| benzyl acetate | 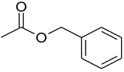 |
−4.48 ± 0.16 | −1.31 ± 0.44 | >10 | 3HUK |
| nitrosobenzene | 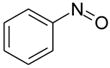 |
weak | −5.55 ± 0.23 | 3.24(A), 3.22(B) | 3HU9 |
| 2-ethoxy-3,4-dihydro-2h-pyran | 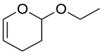 |
weak | −3.8 ± 0.18 | 4.35 | 3HTG |
| 4-chloro-1h-pyrazole |  |
weak | −7.86 ± 0.12 | 2.07 | 3HTF |
| (E)-thiophene-2-carboxaldoxime | NB | −2.30 ± 0.09 | --- | --- | |
| 1-phenylsemicarbazide | 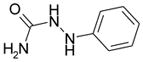 |
NB | 0.45 ± 0.24 | --- | --- |
| o-benzylhydroxylamine | 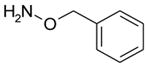 |
NB | −2.58 ± 0.13 | --- | --- |
| 1-2-hydroxyethylpyrrole | 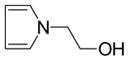 |
NB | −5.72 ± 0.12 | --- | --- |
| phenylhydrazine | 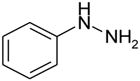 |
NB | −2.51 ± 0.44 | --- | --- |
Compound names of mispredictions are in bold italics.
A
Free energy of binding determined by ITC at 10°C. For ligands designated weak binding was established by Tm upshift but ΔGb could not be determined; NB = nonbinder, ΔTm ≈ 0°C at concentrations between 1–10mM.
B
Calculated free energy of binding.
C
RMSD of predicted ligand geometry to experimentally observed crystal pose. If multiple ligand orientations were present in the crystal (designated A and B) only the best RMSD to the prediction is reported.
**
indicates RMSD for one crystal pose calculated to two predictions.