

Abstract
Peptidomimetic imidazolidin-4-one derivatives of primaquine (imidazoquines) recently displayed in vitro activity against blood schizonts of a chloroquine-resistant strain of Plasmodium falciparum. Preliminary studies with a subset of such imidazoquines showed them to both block transmission of P. berghei malaria from mouse to mosquito and be highly stable towards hydrolysis at physiological conditions. This prompted us to have deeper insight into the activity of imidazoquines against both Plasmodia and Pneumocystis carinii, on which primaquine is also active. Full assessment of the in vivo transmission-blocking activity of imidazoquines, in vitro tissue-schizontocidal activity on P. berghei-infected hepatocytes, and in vitro anti-P. carinii activity is now reported. All compounds were active in these biological assays, with generally lower activity than the parent drug. However, imidazoquines’ stability against both oxidative deamination and proteolytic degradation suggest that they will probably have higher oral bioavailability and lower hematotoxicity than primaquine, which might translate into higher therapeutic indexes.
Keywords: bioavailability, gametocytocidal, malaria, pharmacokinetics, Pneumocystis, pneumocystosis, Plasmodium, primaquine, schizontocidal, transmission-blocking
Introduction
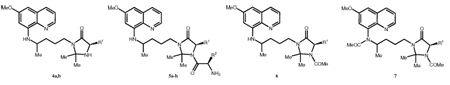
8-Aminoquinolines (8-AQ) are an important class of antimicrobial agents, with important roles in the treatment of malaria and Pneumocystis infection.1 Considering malaria, primaquine (1, Scheme 1) is still the only clinically available 8-AQ active against both gametocytes, responsible for disease transmission between the human host and the mosquito vector, and all exoerythrocytic forms (EEFs) of Plasmodia, including the liver hypnozoites responsible for relapses of vivax and ovale malarias.2–5 Primaquine (PQ) is also useful for the treatment of Pneumocystis infections5 caused by Pneumocystis jirovecii (formerly P. carinii f. sp. hominis6,7), which is a common cause of pneumonia in immunocompromised individuals and frequently the first serious illness encountered by HIV-infected patients.8,9,10,11 P. jirovecii also infects other immunocompromised individuals such as those undergoing cancer therapy and organ and bone marrow transplants.12 Unfortunately, PQ has low oral bioavailability, since it is rapidly metabolized to the inactive metabolite carboxyprimaquine (2, Scheme 1) by oxidative deamination13,14 and presents blood toxicity, namely hemolytic anemia after primary induction of methemoglobinemia.4,15–17 Hematotoxicity is greatest in those with deficiency in glucose-6-phosphate dehydrogenase (6GPD) and is further aggravated by the need of frequent administration of high PQ doses in order to compensate for its low oral bioavailability.1–5 We previously reported that N-acylation of PQ with amino acids or dipeptides (3, Scheme 1) leads to active structures not susceptible to oxidative deamination to 2, but sensitive to degradation by amino- and endo-peptidases18. We now set out to explore the effect of introducing an imidazolidin-4-one moiety in such N-acyl-PQ derivatives on the stability and bioactivity of the resulting compounds. Imidazolidin-4-one structures are often used to protect the N-terminal residues of peptides and peptidomimetics19–23 as well as to mimetize the amino acid proline.24 In this context, we prepared imidazolidin-4-ones 4 (Scheme 1)25 and found them to be promising therapeutic agents, given their significant activity in blocking the transmission of P. berghei malaria from mice to mosquitoes,26 remarkable stability in pH 7.4 buffer and human plasma,26,27 and overall activity profile comparable to that of the parent drug against Plasmodia and Pneumocystis carinii.28
Scheme 1.

Chemical structures for primaquine and relevant derivatives (see text)
In view of initial promising results, we undertook the N1-acylation of imidazolidin-4-ones 4 with amino acids, i.e., the preparation of structures 5 (Scheme 1),29 aimed at (i) fully suppressing hydrolysis of the imidazolidin-4-one ring through acylation of its N-1 nitrogen, (ii) increasing the compounds’ aqueous solubility by insertion of a basic amino group, and (iii) potentially increasing the compounds’ antimalarial activity, given the relevant role usually attributed to the basic amino group of PQ.18 A set of different structures 5, where the amino acids’ side chains (R1 and R2) were varied, was prepared in order to check for the influence of these substituents on compound properties. Preliminary biological evaluation of compounds 5 demonstrated blood-schizontocidal activity against a chloroquine-resistant strain of P. falciparum, as well as the ability to block transmission of P. berghei malaria between Balb/C mice and Anopheles stephensi mosquitoes.30 These results prompted us to further evaluate compounds 5 as antimalarial tissue-schizontocides and against P. carinii.
Results and Discussion
Chemistry
Compounds 4 were obtained and further N1-acylated by previously reported methods, through the synthetic route depicted on Scheme 2.29,30 Briefly, once prepared as previously described,29 imidazolidin-4-one precursors 4 were coupled to Nα-Boc-protected amino acids (BocAAOH) by means of a carbodiimide (diisopropylcarbodiimide, DIC) as condensation agent, in the presence of 1-hydroxybenzotriazole (HOBt) as auxiliary nucleophile. Boc was then removed from the condensation product by acidolysis with trifluoroacetic acid (TFA), yielding the target compounds 5 in good yields and with correct spectroscopic data. It is worth mentioning that bulky R1 side chains impair N-acylation of imidazolidin-4-ones 4, which limited the synthesis of structures 5 to those where R1 was either H or Me (respectively, glycine and alanine derivatives). Nonetheless, our previous work has shown that R1 substituents on imidazolidin-4-ones 4 (i) should be small (H, Me) for higher transmission blocking activities,2 and (ii) do not have a marked influence on compounds activity against both blood-stage P. falciparum or P. carinii.28 Bulky substituents on the imidazolidin-4-one’s C-2 carbon are also detrimental towards N-acylation, so only propanone-derived imidazolidin-4-ones are efficiently N-acylated.29 Nevertheless, insertion of larger substituents on the imidazolidin-4-one’s C-2, as those previously reported by us,26,28 would probably lead to a decreased water-solubility of final compounds 5. So, structural diversity of the latter was achieved by varying the second amino acid side chain (R2, see Table 1), in order to check for the influence of this substituent on compound’s bioactivity.
Scheme 2.

Synthetic route to imidazoquines 5 and their acetyl analogues 6 and 7. Reagents and conditions: (i) 1 equiv BocAAOH, 1.1 equiv DIC, 1.1 equiv HOBt, 1 equiv triethylamine (TEA), dichloromethane (DCM), 0 °C→rt; (ii) TFA, rt, 30% aq Na2CO3, extraction with CHCl3; (iii) excess propanone, 1 eq. TEA, molecular sieves, refluxing MeOH; (iv) 5 equiv BocAAOH, 5 equiv DIC/HOBt, 3 equiv TEA, DMF, -10 °C→rt, inert atmosphere; (v) TFA/DCM 30%, rt, followed by Na2CO3 aq 30% and extraction with DCM; (vi) 5 equiv Ac2O, 5 equiv DIC/HOBt, 3 equiv TEA, DMF, - 10 °C→rt, inert atmosphere; (vii) refluxing neat Ac2O (20 equiv).
Table 1.
Cytotoxicity, anti-Pneumocystis activity and antiplasmodial activity of compounds 1, 4a,b, 5a–h, 6 and 7 against blood-stage P. falciparum W2 and liver-stage P. berghei.
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compd | R1 | R2 | Activity against blood stage forms |
Activity against liver stage forms |
Anti-P. carinii activity |
Cytotoxicity (A549 cells)b |
||
| IC50 (µM) | IC50 (µM) | IC50 in µM (µg/mL)a | IC50 at 72 h in µM (µg/mL) |
|||||
| 24 h | 48 h | 72 h | ||||||
| 1 | - | - | 3.3 | 2 | 18.1 (4.7) | 8.5 (2.2) | 3.5 (0.9) | >100 |
| 4a | H | - | 9.1c | - | - | - | 23 (8.3)c | - |
| 4b | Me | - | >50c | - | - | - | 226 (83.7)c | - |
| 5a | H | H | 6.7 | 22 | 43.8 (18.1) | 37.5 (15.5) | 28.1 (11.6) | >100 |
| 5b | H | CH3 | 8.0 | 10 | 39.6 (16.9) | 18.7 (8.0) | 8.4 (3.6) | 7.8 |
| 5c | H | CHMe2 | 7.9 | 17 | 54.3 (24.7) | 20.2 (9.2) | 13.6 (6.2) | 5.5 |
| 5d | H | CH2CHMe2 | 6.3 | 24 | 31.1 (14.6) | 17.9 (8.4) | 10.9 (5.1) | 21.2 |
| 5e | H | CH(Me)Et | 8.7 | 13 | 27.5 (12.9) | 16.0 (7.5) | 9.8 (4.6) | 14.9 |
| 5f | H | (CH2)2SMe | 12.5 | 31 | 23.8 (11.6) | 13.1 (6.4) | 7.0 (3.4) | 7.9 |
| 5g | H | CH2Ph | 9.9 | 21 | 24.0 (12.1) | 18.5 (9.3) | 10.1 (5.1) | 9.1 |
| 5h | Me | H | 5.5 | 21 | Inactive | 75.4 (32.2) | 52.2 (22.3) | >100 |
| 6 | H | - | >10.0 | ND | Inactive | 101.7 (40.5) | 36.7 (14.6) | >100 |
| 7 | H | - | >10.0 | ND | Inactive | Inactive | Inactive | >100 |
The N-acetyl and N,N’-diacetyl imidazolidin-4-one derivatives 6 and 7, also included in the present study for comparison purposes, were prepared by reacting 4 (R1 = H, R2 = R3 = Me) with acetic anhydride (5 mol eq.), HOBt and DIC in N,N-dimethylformamide (DMF) and in refluxing neat acetic anhydride (20 mol eq.), respectively.29
Biology
Antiplasmodial activity and cytotoxicity
All compounds 5 were evaluated for in vitro antiplasmodial activity against the chloroquine-resistant P. falciparum strain W2 (Table 1). Compound toxicity was evaluated by assessing effects on the viability of A549 human adenocarcinoma epithelial lung cells. Inspection of the data in Table 1 allows the following observations:
the in vitro antiplasmodial activity of compounds 5 is not significantly affected by the nature of the amino acid residue at the imidazolidin-4-one’s N-1 atom. For example, the glycine derivative 5a (R2 = H) is equipotent to its leucine counterpart, 5d (R2 = i-Bu) and only ~two times more active than its methionine counterpart, 5f (R2 = CH2CH2SMe).
the activity of compounds 5 is also not significantly affected by the nature of the R1 substituent at the C-5 position of the imidazolidin-4-one moiety, as indicated by the similar IC50 values for compounds 5a and 5h. This result contrasts sharply with that of precursors 4, in which the incorporation of a methyl group at C-5 leads to complete loss of activity (4a vs 4b).28
acetylation of the imidazolidin-4-one’s N-1 atom leads to complete loss of activity (e.g. 6 vs 5a), suggesting that the presence of a basic amino group is a major requirement for antiplasmodial activity. This result is in line with previous reports on the importance of such group for the antimalarial activity of 8-aminoquinolines.18,31,32
in general, compounds 5 displayed greater cytoxicity than PQ against A549 cells, with the exception of 5a and 5h (i.e. the third most active and the most active compounds in the series, respectively), which showed no toxicity in this assay. Compound 5d also presented a favourable cytotoxicity/antiplasmodial ratio of 3.5.
Overall, these results suggest that the antiplasmodial activity of compounds 5 benefits from the N1-acylation of the imidazolidin-4-one ring with an amino acid. This fact may be partly due to the presence of the primary amino group brought by the inserted amino acid residue. This hypothesis is strengthened by the fact that N-acetylated derivatives 6 and 7 were found to be devoid of blood-schizontocidal activity.
Anti-Plasmodium liver stage activity
Given the tissue-schizontocidal activity of PQ and the fact that compounds 4 and 5 presented biological properties similar to those of the parent drug, it was predicted that the two sets of PQ imidazolidin-4-ones would be similarly active against liver forms of Plasmodia. To assess the ability of compounds 5 to inhibit the development of P. berghei schizonts in human hepatoma cells, we employed a recently described fluorescence activated cell sorting (FACS)-based method.33 This method is based on the measurement of the fluorescence of Huh-7 cells, a human hepatoma cell line, following infection with GFP-expressing P. berghei sporozoites. At a given time after infection, the percentage of parasitized cells is given by the percentage of GFP-positive events (Figure 1A). Since GFP expression is under the control of the house-keeping EF1α promoter, the extent of intracellular development is proportional to the number of GFP copies in the cell, measured as the intensity of fluorescence (Figure 1A). The hepatic anti-Plasmodial activity of compounds 5 was monitored by measuring infection of Huh-7 cells incubated with various concentrations of each, 48 hours after sporozoite addition. The presence of these compounds decreased the number of GFP positive Huh-7 cells to different extents when compared with control values, albeit less-so than the parent PQ (Figure S1 in Supporting Information). Most importantly, all compounds 5 displayed a marked, dose-dependent effect on parasite development (Figure 1B; Table 1). The IC50 values of all the compounds in terms of their ability to impair the development of P. berghei liver schizonts were determined following the process depicted in Figure 1C. Although IC50 values for compounds 5 were higher than that of PQ, several compounds displayed marked anti-plasmodial activity, with their ranking following the order 5b ≈ 5e < 5c < 5a ≈ 5g ≈ 5h < 5d < 5f.
Figure 1. Flow cytometry-based Plasmodium liver stage infection assays.

A, Representative dot plots (left) and histograms (right) of a control sample of Huh7 cells infected with GFP-expressing P.berghei parasites, analysed 48 hours after sporozoite addition; the GFP-positive cells, in a total of 50000 acquired events, corresponds to fraction of infected cells (left), while the geometric mean of the distribution of fluorescence intensities in the GFP-positive population corresponds to the intracellular development of the parasites (right). B, Dose-dependencies of parasite development in cells infected in the presence of increasing concentrations of the various compounds tested, relative to that of solvent-treated samples in the same experiment. C, Example of the calculation of the IC50 for inhibition of parasite development; left- representative histograms of the distribution of fluorescence intensities in the GFP-positive population of cells infected in the presence of different concentrations of compound and of the corresponding solvent control; centre- geometric means of fluorescence intensities relative to that of the solvent-treated control; right- sigmoidal curve of fluorescence intensities as a function of the logarithm of compound concentration; the example pertains the parent compound primaquine (green), with the solvent-treated control shown in red and the value of the IC50 shown as an orange sphere; only 4 out of 6 datapoints are shown to facilitate vieweing of the histogram lines.
Anti-Pneumocystis activity
The antifolate combination of trimethoprim and sulfamethoxazole (TMP-SMX) has been used for both prophylaxis and treatment of pneumocystosis,34,35 but development of microbial resistance and allergic reactions against the sulfa component often require the use of alternative therapies,32–34 among which is PQ, used in combination with clindamycin.35 Previous work suggests that there is a synchrony between the structure-activity relationships (SARs) presented by PQ-related drugs for malaria and pneumocystosis.34,37–39 Therefore, imidazolidin-4-ones 5 were evaluated against P. carinii in an ATP detection assay based on the release of bioluminescence driven by ATP in a luciferin-luciferase mediated reaction.40,41 All compounds, as well as PQ, reduced the ATP pools in P. carinii in a dose- and time-dependent manner. The maximal effect for compounds 5 was seen at 72 h with IC50 values ranging from 7 to 52 µM, i.e. from moderate to slight activity in the usual scoring scale42 while PQ presented an IC50 of 3.5 µM at 72 h (Table 2). Activity was not significantly affected by the nature of the side chain at the terminal amino acid residue, i.e. R2. For example, compounds 5b (R2 = Me), 5d (R2 = iso-Bu), 5e (R2 = sec-Bu), 5f (R2 = CH2CH2SMe) had less than 3-fold variation in IC50 values from compounds 5a to 5g. The N1-acetyl derivative 6, which is a desamino analogue of imidazoquines 5, though clearly much less active than the latter, did display some activity after 72 h. This result indicates that IC50 values decrease over time and that all compounds assayed inhibit P. carinii development after 72 h, with the exception of the N,N’-diacetyl derivative 7. These results suggest that PQ’s secondary amino group attached to the quinoline’s C-8 has a role in anti-pneumocystis activity. Remarkably, the N1-acyl-imidazolidin-4-ones 5 showed activity as soon as 48 h after initiation of incubation, which represents an improvement over most of their imidazolidin-4-one precursors 4 that were only visibly active after 72 h. Moreover, at 72 h, most compounds 5 were slightly more effective than their precursors 4, as illustrated by comparison of compound 4.2 (R1=R2=R4=CH3; IC50 at 72 h = 226 µM)28 with its N1-glycyl derivative 5h (IC50 at 72 h = 52 µM).
Table 2.
Effect of compounds 1 and 5a,b,e–h on the sporogonic development of Plasmodium berghei ANKA in Anopheles stephensi.
| Compd | Dose/µmol.kg−1 | Mean no. of oocysts per mosquito±SEMa |
% of infected mosquitoes |
|---|---|---|---|
| 1 | 10 | 3.5 ±1.2 | 40.0 |
| 50 | 0.2 ±0.2 | 2.9 | |
| 5a | 10 | 1.1 ±0.2 | 42.5 |
| 50 | 0.8 ±0.3 | 40.5 | |
| 5b | 10 | 9.7 ±2.5 | 67.9 |
| 50 | 1.3 ±0.3 | 40.1 | |
| 5e | 10 | 4.9 ±0.6 | 83.3 |
| 50 | 1.5 ±0.5 | 62.2 | |
| 5f | 10 | 1.2 ±0.4 | 47.1 |
| 50 | 3.8 ±1.4 | 26.4 | |
| 5g | 10 | 3.7 ±1.2 | 43.8 |
| 50 | 2.2 ±0.8 | 40.0 | |
| 5h | 10 | 3.3 ±1.1 | 62.0 |
| 50 | 1.8 ±0.5 | 37.5 | |
| Control | 0 | 8.2 ±1.2 | 72.1 |
standard error of the mean.
The relative positions of the two amino acid building blocks (i.e. switching between R1 and R2) also seems important, as shown by comparison of compound 5h with its isomer 5b (IC50 at 72 h = 8.4 µM).
Overall, these data show a certain degree of parallelism between the anti-plasmodial and anti-pneumocystis activities of 8-aminoquinolinic compounds, as N1-acylation of imidazolidin-4-ones 4 (to yield 5) seems to be beneficial both for the blood-schizontocidal and for the anti-pneumocystis activity of these compounds. However, the most efficient compounds against P. falciparum were not those that ranked the best against P. carinii.
In vivo transmission-blocking activity
The potential of compounds 5 to inhibit the sporogonic cycle of Plasmodia within the mosquito gut was studied using a model consisting of Balb/C mice infected with P. berghei and Anopheles stephensi mosquitoes, and compared to that of PQ.26 The activity was measured by the percentage of mosquitoes with oocysts and the mean number of oocysts per infected mosquito (Table 2). Although this model cannot specifically attribute the drug effect to either gametocytocidal or sporontocidal activity, it can clearly show if a compound is effective at interrupting the transmission of the infection to mosquitoes by interference with the cycle in these insects.26 Compounds were tested at 10 and 50 µmol.kg−1 and, although none of them was particularly good at decreasing the percentage of infected mosquitoes, all of them were active at effectively reducing the mean number of oocysts formed per infected mosquito. Moreover, while none of them was superior to PQ at the highest dose, at the lowest dose two of them (5g,h) were comparable to and two other (5a,f) were better than the parent drug. Compound 5a significantly reduced (P < 0.05) the sporogonic development of the parasite at both doses tested, while compound 5b was inactive at 10 µmol/kg. Interestingly, 5e was clearly active but did not rank the best of the set, in contrast to its relative activity against liver-stage P. berghei. Results also show that there is no correlation between transmission-blocking and anti-P.carinii activities, as previously noticed for imidazolidin-4-one precursors 4.28
Physicochemical properties and plasma stability
Several parameters were calculated for imidazolidin-4-ones 5 in order to gain some insight to their drug-likeness: molecular weight, HBD, HBA, logP, logS and Drug Score (see Table S1 on Supporting Information).43,44 All imidazoquines 5 have molecular weights below 500 Da (except for 5g that weighs 503 g/mol), less than five hydrogen bond donors (all compounds 5 have three HBD), less than ten hydrogen bond acceptors (all compounds 5 have five HBA) and estimated45,46 octan-1-ol/water partition coefficients (logP) below 5 (Table S1). While imidazolidin-4-one precursors 4a,b have slightly increased lipophilicities with respect to PQ, but at the expense of significantly decreased water-solubility, imidazoquines derived from Ala (5b,h) have only slightly reduced lipophilicity (logP respectively of 2.49 and 2.57, as compared to 2.76 for PQ) accompanied by a remarkable increment on their water-solubility (0.12 g/L as compared to 56.4 mg/L for PQ). Finally, drug scores were consistently higher for compounds 5 and 4a,b than for the parent compound. We also determined the stability of compounds 5 in 80% human plasma,26,27 which was found to be remarkably high, with no degradation detected after three days of incubation. Importantly, stability was not affected by the substitution pattern at the terminal amino acid or the imidazolidin-4-one moiety. Such stability had already been reported on previous studies involving some of these imidazoquines and their precursors 4.25–27,47,48 Overall, these results suggest that the oral availability of compounds 5 is unlikely to be limited by compound stability.
Conclusions
Imidazoquines, i.e., peptidomimetic derivatives of PQ with general structure 5, are stable compounds that preserve the overall bioactivity pattern of the parent drug. Due to blockage of the aliphatic amine of PQ by insertion of the peptidomimetic carrier, structures 5 are not vulnerable to oxidative deamination, which is the main metabolic process behind the low oral bioavailability of PQ. On the other hand, the use of a peptidomimetic instead of a dipeptide carrier makes compounds 5 stable to proteolytic degradation by action of amino- or endo-peptidases.
It is well established that the main problem of PQ, which is still the only drug in clinical use to treat hypnozoites of vivax and ovale malaria, is of metabolic nature, as metabolic transformations are behind both PQ’s low bioavailability and serious hematotoxicity, especially for people deficient in 6GPD. Consequently, even though most imidazoquines 5 were generally not as active as the parent PQ, their remarkable chemical and enzymatic stability, as well as preliminary data on their ADME properties, suggest that they are promising leads towards novel hydrolytically- and enzimatically-stable drugs with therapeutic indices superior to that of primaquine, useful against malaria and pneumocystosis.
Experimental Section
Chemistry
For compound synthesis, Nα-protected amino acids and Nα-protected dipeptides from Bachem (Switzerland) were used. Solvents were of p.a. quality, from Merck (VWR International, Portugal). When required, solvents were previously dried with pre-activated molecular sieves (4 Å) (Merck). Both thin layer chromatography (TLC) aluminium foil plates covered with silica 60 F254 (0.25mm) and silica-gel 60 (70–230 mesh ASTM) for preparative columm chromatography were also from Merck. Other chemicals (DIC, HOBt, TEA, TFA) were from Sigma-Aldrich.
The purity degrees of all compounds were checked to be higher than 95%, as determined by HPLC using a Merck Hitachi ELITE LaChrom equipped with an L-2130 pump, an L-2200 autosampler and an L-2455 diode-array detector. Samples were injected on a Merck Purospher STAR RP-18e 125 cm×4.6 mm (5µm) column equipped with a Merck Lichrocart pre-column (Merck, Germany). Analyses were run by either isocratic or gradient methods (see Supplementary Information for details) at a 1 mL/min flow rate. Chromatograms recorded at 265 nm are given in the Supporting Information.
NMR spectra of compounds dissolved in deuterated chloroform (CDCl3), containing tetramethysilane (TMS) as internal reference, were acquired on a Bruker AMX-300 spectrometer. Mass spectrometry (MS) was performed either by the matrix-assisted laser desorption ionization-time-of-flight (MALDI-TOF) technique, on an Applied Biosystems Voyager STR-DE spectrometer using either anthracene or 2,5-dihydroxibenzoic acid (DHB) as adjuvant matrices, or by the electrospray ionization - ion trap (ESI-IT) technique on a Finnigan Surveyor LCQ DECA XP MAX quadrupole mass spectrometer.
General procedure for the synthesis of imidazoquines 5a–h
The synthetic route followed is depicted on Scheme 2. Synthesis detailed procedures, percentage yields and analytical/spectroscopic data for all test compounds, with the exception of 5e and its Boc-protected precursor Boc-5e, have been described elsewhere.25,26,29
Briefly, PQ (1) was coupled to the relevant Boc-protected amino acid to produce Boc-3, which was then deprotected by acidolysis to give the trifluoroacetic acid salt of 3 that was converted to the free base (3) by addition of 30% aq. Na2CO3 until pH 10 was reached, followed by extraction with chloroform (Scheme 2 and respective legend).25,26 Each compound 3 (2 equiv) was then mixed with an excess (4 equiv) of propanone in dry methanol (10 mL) containing triethylamine (TEA, 2 equiv), and the mixture was refluxed for 3 days in the presence of 4 Å molecular sieves (1 g).25,26 The reaction was monitored by TLC and propanone was re-added (2 equiv) once per day. The molecular sieves were removed by decantation and the solution was evaporated to dryness, producing an oily residue that was submitted to column chromatography on silica gel. The product was isolated as yellow-orange oil and correctly identified as the target imidazolidin-4-one 4 (Scheme 2).25,26
Compounds 4 (1 equiv) were dissolved in DMF (20 mL) containing TEA (3 equiv) and the mixture was stirred at −10 °C for 20 min, under inert atmosphere. The appropriate Boc-protected amino acid (5 equiv) was added together with DIC (5 equiv) and HOBt (5 equiv), and the suspension was kept at −10 °C for further 4 h, under stirring. The temperature was then increased to 10 °C and thus maintained till the end of reaction (24 h by TLC). The solid was removed by vacuum filtration and the filtrate was evaporated to dryness in vacuum at 90 °C, yielding a residue that was dissolved in 40 mL of DCM. This solution was washed three times with 15 mL portions of 10% aq. NaHCO3 and the organic layer dried over anhydrous MgSO4 and evaporated to dryness. The residue was submitted to column chromatography on silica using DCM/acetone, to give the Boc-protected imidazoquines, Boc-5, as yellow-orange oils presenting correct spectral data.26,29 Each compound Boc-5 was then dissolved in TFA at 30% in DCM and the reaction allowed proceeding for 2 h at room temperature. 30% aq. Na2CO3 was added to the resulting trifluoroacetic acid salt of 5 until pH 10, and the supernatant oily layer formed was extracted six times with 10 mL portions of chloroform. The organic layers were pooled, dried over anhydrous MgSO4 and evaporated to dryness, yielding a chromatographically homogeneous yellow-orange oils that were identified as the target structures 5.26,29
1-(N-tert-butoxycarbonyl)isoleucyl-3-(4-(6-methoxyquinolin-8-ylamino)pentyl)-2,2-dimethylimidazolidin-4-one (Boc-5e)
1H NMR (400 MHz, CDCl3) δ 0.86-0.83 (m, 6H), 1.24 (d, 3H, J = 6.4 Hz), 1.37 (s, 9H), 1.54-1.53 (s+s, 6H), 1.82-1.58 (m, 7H), 3.27-3.10 (m, 2H), 3.61-3.55 (m, 1H), 3.82 (s, 2H), 3.97-3.91 (m, 1H), 4.01 (d, 1H, J = 15.0 Hz), 4.36 (d, 1H, J = 14.9 Hz), 4.97 (d, 1H, J = 9.0 Hz), 6.22 (d, 1H, J = 2.5 Hz), 6.26 (d, 1H, J = 2.5 Hz), 7.23 (dd, 1H, J = 4.2, 8.3 Hz), 7.85 (dd, 1H, J = 1.6, 8.3 Hz), 8.45 (dd, 1H, J = 1.6, 4.1 Hz). 13C NMR (100 MHz, CDCl3) δ 9.58, 13.93, 19.22, 22.91, 23.57, 24.31+24.36, 26.00, 26.82, 32.62+32.65, 35.94, 36.61, 38.39, 46.39, 47.14, 50.50, 53.76, 55.52, 78.36, 79.34, 90.32, 95.35, 120.41, 128.45, 133.35, 133.85, 142.86, 143.47, 154.42, 157.95, 164.35, 169.08. m/z [M+H+] = 570.8003 (calcd, 570.3655); [M+Na+] = 592.7314 (calcd, 592.3475).
1-isoleucyl-3-(4-(6-methoxyquinolin-8-ylamino)pentyl)-2,2-dimethylimidazolidin-4-one (5e)
1H NMR (400 MHz, CDCl3) δ 0.87-0.74 (m, 3H), 1.12-0.96 (m, 3H), 1.25 (d, 3H, J = 6.4 Hz), 1.57-1.54 (m, 2H), 1.55-1.54 (s+s, 3H), 1.57-1.56 (s+s, 3H), 1.82-1.60 (m, 7H), 3.06 (d, 1H, J = 6.2 Hz), 3.23-3.12 (m, 2H), 3.62-3.54 (m, 1H), 3.82 (s, 3H), 4.03-4.01 (s+s, 2H), 5.94 (d, 1H, J = 8.4 Hz), 6.22 (d, 1H, J = 2.5 Hz), 6.27 (d, 1H, J = 2.5 Hz), 7.24 (dd, 1H, J = 4.2, 8.2 Hz), 7.85 (dd, 1H, J = 1.6, 8.3 Hz), 8.45 (dd, 1H, J = 1.6, 4.2 Hz). 13C NMR (100 MHz, CDCl3) δ 10.31, 15.09, 19.69, 22.60, 23.37, 24.78, 28.68, 33.09, 37.94, 38.84, 47.36, 54.21, 57.38, 79.80, 90.74, 92.63, 95.76, 120.86, 128.88, 133.77, 134.32, 143.32, 143.92, 158.38, 164.75, 171.91. m/z [M+H+] = 470.0712 (calcd, 470.3131).
Biology
In vivo transmission-blocking activity assays
Balb/C mice were infected by intraperitoneal inoculations of 107 erythrocytes parasitized with P. berghei ANKA. After 4 days, when the presence of gametocytes was observed by microscopic observation of Giemsa stained blood films, mice were randomly separated into eight different groups of six animals. Each group was treated by intraperitoneal administration with one single dose of each compound 5 and PQ (10 and 50 µmol/kg in inoculation volumes of 0.1–0.2 mL; controls consisted of mice given a PBS solution). Two hours after administration, mice were anesthetized and placed on top of individual cages containing ca. 50 glucose-starved Anopheles stephensi female mosquitoes, which were allowed to feed for 2 h. After the blood meal, unfed females mosquitoes were removed from each cage. Ten days after the blood meal, 10 mosquitoes of each cage were randomly collected and dissected for microscopic detection of oocysts in midguts.
In vitro blood-schizontocidal activity assays
These assays have been conducted as reported elsewhere.28,32,49–52 Briefly, synchronized ring-stage W2 strain P. falciparum parasites were cultured with multiple concentrations of test compounds (added from 1,000× stocks in DMSO) in RPMI 1640 medium with 10% human serum. After a 48 h incubation, when control cultures contained new rings, parasites were fixed with 1% formaldehyde in PBS, pH 7.4, for 48 h at room temperature and then labeled with YOYO-1 (1 nM; Molecular Probes) in 0.1% Triton X- 100 in PBS. Parasitemias were determined from dot plots (forward scatter vs. fluorescence) acquired on a FACSort flow cytometer using CELLQUEST software (Becton Dickinson). IC50s for growth inhibition were determined with GraphPad Prism software from plots of percentages of the level of parasitemia of the control relative to inhibitor concentration. In each case, goodness of curve fit was documented by R2 values of > 0.95.
In vitro Plasmodium liver stage infection assays
Cells and parasites
Huh-7 cells, a human hepatoma cell line, were cultured in 1640 RPMI medium supplemented with 10% v/v fetal calf serum (FCS), 1% v/v non-essential amino acids, 1% v/v penicillin/streptomycin, 1% v/v glutamine and 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulphonic acid (HEPES), pH 7, and maintained at 37 °C with 5% CO2. For infection assays, Huh-7 cells (7 × 104 per well) were seeded in 24-well plates the day before drug treatment and infection. Medium in the cells was replaced by medium containing the appropriate concentration od each compound immediately prior to infection by addition of 30000 sporozoites, followed by centrifugation at 1700 g for 5 min at 37 °C. GFP-expressing P. berghei (parasite line 259 cL2) sporozoites were obtained by disruption of the salivary glands of freshly dissected infected female Anopheles stephensi mosquitoes.
Parasite development assays
Intracellular parasite development was determined by measuring GFP intensity in cells infected with GFP-expressing P. berghei parasites by fluorescence-activated cell sorting (FACS), as previously described.33 Briefly, cell samples for FACS analysis were washed with 1 ml of phosphate buffered saline (PBS), incubated with 100 µL of trypsin for 5 min at 37 °C and collected in 400 µl of 10% v/v FCS in PBS at the selected time points post sporozoite addition. Cells were then centrifuged at 0.1 g for 5 min at 4 °C and resuspended in 150 µL of 2% v/v FCS in PBS. Cells were analysed on a Becton Dickinson FACScalibur with the appropriate settings for the fluorophore used. Data acquisition and analysis were carried out using the CELLQuest (version 3.2.1fl1, Becton Dickinson) and FlowJo (version 6.3.4, FlowJo) software packages respectively.
In vitro anti-P. carinii activity assays
P. carinii organisms
Organisms for the ATP assays were obtained from chronically immunosuppressed Long Evans and Brown Norway rats housed under barrier conditions at the Cincinnati VA Medical Center (VAMC) and inoculated intratracheally with P. carinii. These were extracted and purified from the lungs of rats after 8–12 weeks of immunosuppression, enumerated, cryopreserved, and stored in liquid nitrogen. Typically, infected rat lungs yield up to 2×1010 organism nuclei with the vast majority (about 95%) of the life cycle forms present as trophic forms with the remainder (about 5%) being composed of cysts. P. carinii preparations were evaluated for microbial contamination, ATP content, karyotype, and host cell content prior to use in the ATP assay.
ATP assay
Isolated organisms used for ATP analyses were suspended in a supplemented RPMI 1640 medium containing 20% calf serum and other additives, pH 7.5 to 8.0, 380 mOsm, as previously described.40 Drugs were added to the culture medium in DMSO (the final concentration of DMSO was <0.2%, vol/vol), and 108 organisms (as total nuclei) per ml were added to 1 to 2 ml of the culture medium in multiwell plates. For every assay, each drug concentration was assayed in triplicate using different organism isolation batches. The final ATP content was expressed as the average relative light units of nine values (three readings per well). To assess the effects of extended exposure to PQ and imidazoquines, the ATP contents of cultures sampled after 24, 48, and 72 h of incubation at 35°C in a 10% CO2 humidified atmosphere were measured. The media of all wells were changed on a daily basis after centrifugation of the multiwell plates at 2,400×g and removal of the previous medium. The ATP content was determined by the luciferin-luciferase assay as described previously and was expressed as relative light units.40 The effects of the compounds on the P. carinii ATP content were compared with the ATP contents of P. carinii populations that did not receive experimental compounds and expressed as percentages of these control values. In addition, other controls for each assay included quench controls to evaluate the effects of the highest drug concentrations used on the luciferase-luciferin reaction; vehicle controls to evaluate the effects of any solvent on the same reaction and on the organism ATP content.
Cytotoxicity assays
Preparations with less than 85% viability were not used in the cytoxicity assay. After enumeration, organisms were added to 24-well plates containing 1 to 2 mL of RPMI supplemented with 20% FBS, vitamins, minerals, and other additives at a standard density of 108/mL with or without a test compound. At least three concentrations of each compound were evaluated (10, 1.0, and 0.1 µg/mL) by incorporation into the medium. Triplicate wells were used for each concentration of test compound evaluated and for control groups.40
Compound stability in human plasma
HPLC analysis
HPLC measurements were carried out using a Waters assembly equipped with a model 600 controlled pump and a model 991 photodiode-array detector. A Rheodyne 7725 injection valve equipped with 20-µl sample loop was used. Acquisition and treatment of data were made by means of NEC for MS-DOS, version 3.30 software. The separation was performed on a Purospher, 250 × 4.0-mm i.d. 5 µm (Merck, Germany) analytical column. A LiChrospher 100 RP-8 5 µm (Merck, Germany) was employed as pre-column. The solvent system used was a gradient of sodium acetate buffer (pH 4.75; 0.05 M) (A) and acetonitrile (B); 10−3 M triethylamine was added to the aqueous mobile phase in order to improve peak shape. The gradient was as follows: 0 min, 50% B; 4.5 min, 50% B; 5.0 min, 10% B; 20 min, 10% B. Elution was performed at a solvent flow-rate of 1 mL/min and a 15 mL/min nitrogen sparging was applied to remove dissolved gases. Chromatographic separation was monitored by UV detection at 265 nm. All analyses were performed at room temperature.
Hydrolysis in human plasma
Compounds 5 were incubated at 37°C in human plasma (from heparinised blood of healthy donors) diluted to 80% (v/v) with pH 7.4 isotonic phosphate buffer. At appropriate intervals, aliquots were added to acetonitrile to quench the reaction and precipitate plasma proteins. These samples were centrifuged and the supernatant analysed by the HPLC method described above for the presence of substrate and products.
Supplementary Material
Acknowledgments
This work was mainly supported by Fundação para a Ciência e a Tecnologia (FCT, Portugal) through projects PTDC/QUI/65142/2006 to PG and PTDC-BIA-BCM-71920-2006 to MP. NV thanks FCT for post-doctoral grant SFRH/BPD/4834572008. PG and RM thank FCT for financial support to CIQUP and CECF research units, respectively. MP is a holder of a Ciência 2007 position of the Portuguese Ministry of Science. MMM is a Howard Hughes Medical Institute International Scholar. PJR was supported by grants from the National Institutes of Health and Medicines for Malaria Venture and is a Distinguished Clinical Scientist of the Doris Duke Charitable Foundation.
Abbreviations
- Ac2O
acetic anhydride
- ATP
adenosine triphosphate
- 8-AQ
8-aminoquinoline
- BocAAOH
Nα-tert-butoxycarbonyl-protected amino acid
- DCM
dichloromethane
- DIC
N,N’-diisopropylcarbodiimide
- DMF
N,N-dimethylformamide
- EEF
exoerythrocytic form
- FACS
fluorescence activated cell sorting
- 6-GPD
glucose-6-phosphate dehydrogenase
- GFP
green fluorescent protein
- HIV
human immunodeficiency virus
- HOBt
1-hydroxybenzotriazole
- IC50
inhibitory concentration at 50%
- PQ
primaquine
- SAR
structure-activity relationship
- SMX
sulfamethoxazole
- TEA
triethylamine
- TFA
trifluoroacetic acid
- TMP
trimethoprim
Footnotes
The authors wish to share their joyfulness for the 100th anniversary of the Medicinal Chemistry Division of the American Chemical Society.
Supporting Information Available: HPLC traces for Imidazoquines 5, Supporting Figure 1 (flow cytometry-based Plasmodium liver stage infection assay of dose-dependencies of percentage of infected cells) and Table S1 (preliminary ADME data for Imidazoquines 5). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Tekwani BL, Walker LA. 8-Aminoquinolines: future role as antiprotozoal drugs. Curr. Opin. Infect. Dis. 2006;19:623–631. doi: 10.1097/QCO.0b013e328010b848. [DOI] [PubMed] [Google Scholar]
- 2.Baird KJ, Hoffman SL. Primaquine therapy for malaria. Clin. Infect. Dis. 2004;39:1336–1345. doi: 10.1086/424663. [DOI] [PubMed] [Google Scholar]
- 3.Chotivanich K, Sattabongkot J, Udomsangpetch R, Looareesuwan S, Day NPJ, Coleman RE, White NJ. Transmission-Blocking Activities of Quinine, Primaquine, and Artesunate. Antimicrob. Agents Chemother. 2006;50:1927–1930. doi: 10.1128/AAC.01472-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hill DR, Baird JK, Parise ME, Lewis LS, Ryan ET, Magill AJ. Primaquine: report from CDC expert meeting on malaria chemoprophylaxis I. Am. J. Trop. Med. Hyg. 2006;75:402–415. [PubMed] [Google Scholar]
- 5.Vale N, Moreira R, Gomes P. Primaquine revisited six decades after its discovery. Eur. J. Med. Chem. 2009;44:937–953. doi: 10.1016/j.ejmech.2008.08.011. [DOI] [PubMed] [Google Scholar]
- 6.Redhead SA, Cushion MT, Frenkel JK, Stringer JR. Pneumocystis and Trypanosoma cruzi: nomenclature and typifications. J. Eukaryot. Microbiol. 2006;53:2–11. doi: 10.1111/j.1550-7408.2005.00072.x. [DOI] [PubMed] [Google Scholar]
- 7.Hawksworth DL. Responsibility in naming pathogens: the case of Pneumocystis jirovecii, the causal agent of pneumocystis pneumonia. Lancet Infect. Dis. 2007;7:3–5. doi: 10.1016/S1473-3099(06)70663-6. [DOI] [PubMed] [Google Scholar]
- 8.Seage GR, Losina E, Goldie SJ, Paltiel AD, Kimmel AD, Freedberg KA. The Relationship of Preventable Opportunistic Infections, HIV-1 RNA, and CD4 Cell Counts to Chronic Mortality. J. Acquired Immune Defic. Syndr. 2002;30:421–428. doi: 10.1097/00042560-200208010-00008. [DOI] [PubMed] [Google Scholar]
- 9.Morris A, Lundgren JD, Masur H, Walzer PD, Hanson DL, Frederick T, Huang L, Beard CB, Kaplan JE. Current epidemiology of Pneumocystis pneumonia. Emerg. Infect. Dis. 2004;10:1713–1720. doi: 10.3201/eid1010.030985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thomas CF, Jr, Limper AH. Pneumocystis Pneumonia. New Engl. J. Med. 2004;350:2487–2498. doi: 10.1056/NEJMra032588. [DOI] [PubMed] [Google Scholar]
- 11.Peterson JC, Cushion MT. Pneumocystis: not just pneumonia. Curr. Opin. Microbiol. 2005;8:393–398. doi: 10.1016/j.mib.2005.06.010. [DOI] [PubMed] [Google Scholar]
- 12.Fishman JA. Prevention of infection due to Pneumocystis carinii. Antimicrob. Agents Chemother. 1998;42:995–1004. doi: 10.1128/aac.42.5.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mihaly GW, Ward SA, Edwards G, L’E Orme M, Breckenridge AM. Pharmacokinetics of primaquine in man: identification of the carboxylic acid derivative as a major plasma metabolite. Br. J. Clin. Pharmacol. 1984;17:441–446. doi: 10.1111/j.1365-2125.1984.tb02369.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Constantino L, Paixão P, Moreira R, Portela MJ, Rosário VE, Iley J. Metabolism of primaquine by liver homogenate fractions. Evidence for monoamine oxidase and cytochrome P450 involvement in the oxidative deamination of primaquine to carboxyprimaquine. Exp. Toxic. Pathol. 1999;51:299–303. doi: 10.1016/S0940-2993(99)80010-4. [DOI] [PubMed] [Google Scholar]
- 15.Srivastava P, Singh S, Jain GK, Puri SK, Pandey VC. A simple and rapid evaluation of methemoglobin toxicity of 8-aminoquinolines and related compounds. Ecotoxicol. Environ. Saf. 2000;45:236–239. doi: 10.1006/eesa.1999.1868. [DOI] [PubMed] [Google Scholar]
- 16.Haymond S, Cariappa R, Eby CS, Scott MG. Laboratory assessment of oxygenation in methemoglobinemia. Clin. Chem. 2005;51:434–444. doi: 10.1373/clinchem.2004.035154. [DOI] [PubMed] [Google Scholar]
- 17.Umbreit J. Methemoglobin-it's not just blue: a concise review. Am. J. Hematol. 2007;82:134–144. doi: 10.1002/ajh.20738. [DOI] [PubMed] [Google Scholar]
- 18.Portela MJ, Moreira R, Valente E, Constantino L, Iley J, Pinto J, Rosa R, Cravo P, Rosário VE. Dipeptide derivatives of primaquine as transmission-blocking antimalarials: effect of aliphatic side-chain acylation on the gametocytocidal activity and on the formation of carboxyprimaquine in rat liver homogenates. Pharm. Res. 1999;16:949–955. doi: 10.1023/a:1018922425551. [DOI] [PubMed] [Google Scholar]
- 19.Klixbüll U, Bundgaard H. Prodrugs as drug delivery systems. XXX. 4-Imidazolidinones as potential bioreversible derivatives for the α-aminoamide moiety in peptides. Int. J. Pharm. 1984;20:273–284. [Google Scholar]
- 20.Klixbüll U, Bundgaard H. Kinetics of reversible reactions of ampicillin with various aldehydes and ketones with formation of 4-imidazolidinones. Int. J. Pharm. 1985;23:163–173. [Google Scholar]
- 21.Rasmussen GJ, Bundgaard H. Prodrugs of peptides. 10. Protection of di- and tripeptides against aminopeptidase by formation of bioreversible 4-imidazolidinone derivatives. Int. J. Pharm. 1991;71:45–53. [Google Scholar]
- 22.Rasmussen GJ, Bundgaard H. Prodrugs of peptides. 15. 4-Imidazolidinone prodrug derivatives of enkephalins to prevent aminopeptidase-catalyzed metabolism in plasma and absorptive mucosae. Int. J. Pharm. 1991;76:113–122. [Google Scholar]
- 23.Bak A, Fich M, Larsen BD, Frokjaer S, Früs GJ. N-terminal 4-imidazolidinone prodrugs of Leu-enkephalin: synthesis, chemical and enzymatic stability studies. Eur. J. Pharm. Sci. 1999;7:317–323. doi: 10.1016/s0928-0987(98)00044-x. [DOI] [PubMed] [Google Scholar]
- 24.Rinnová M, Nefzi A, Houghten RA. An efficient approach for solid-phase synthesis of peptidomimetics based on 4-imidazolidinones. Tetrahedron Lett. 2002;43:2343–2346. [Google Scholar]
- 25.Gomes P, Araújo MJ, Rodrigues M, Vale N, Azevedo Z, Iley J, Chambel P, Morais J, Moreira R. Synthesis of imidazolidin-4-one and 1H-imidazo[2,1-a]isoindole-2,5(3H,9bH)-dione derivatives of primaquine: scope and limitations. Tetrahedron. 2004;60:5551–5562. [Google Scholar]
- 26.Araújo MJ, Bom J, Capela R, Casimiro C, Chambel P, Gomes P, Iley J, Lopes F, Morais J, Moreira R, Oliveira E, Rosário V, Vale N. Imidazolidin-4-ones of primaquine as novel transmission-blocking antimalarials. J. Med. Chem. 2005;48:888–892. doi: 10.1021/jm0494624. [DOI] [PubMed] [Google Scholar]
- 27.Chambel P, Capela R, Lopes F, Iley J, Morais J, Gouveia L, Gomes JRB, Gomes P, Moreira R. Reactivity of imidazolidin-4-one derivatives of primaquine: implications for prodrug design. Tetrahedron. 2006;62:9883–9891. [Google Scholar]
- 28.Vale N, Collins MS, Gut J, Ferraz R, Rosenthal PJ, Cushion MT, Moreira R, Gomes P. Anti-Pneumocystis carinii and antiplasmodial activities of primaquine-derived imidazolidin-4-ones. Bioorg. Med. Chem. Lett. 2008;18:485–488. doi: 10.1016/j.bmcl.2007.11.105. [DOI] [PubMed] [Google Scholar]
- 29.Vale N, Matos J, Moreira R, Gomes P. Amino acids as selective acylating agents: regiosselective N1-acylation of imidazolidin-4-one derivatives of the antimalarial drug primaquine. Tetrahedron. 2008;64:11144–11149. [Google Scholar]
- 30.Vale N, Matos J, Gut J, Nogueira F, do Rosário V, Rosenthal PJ, Moreira R, Gomes P. Imidazolidin-4-one peptidomimetic derivatives of primaquine: synthesis and antimalarial activity. Bioorg. Med. Chem. Lett. 2008;18:4150–4153. doi: 10.1016/j.bmcl.2008.05.076. [DOI] [PubMed] [Google Scholar]
- 31.Schimdt LH. Relationships between chemical structures of 8-aminoquinolines and their capacities for radical cure of infections with Plasmodium cynomolgi in rhesus monkeys. Antimicrob. Agents Chemother. 1983;24:615–652. doi: 10.1128/aac.24.5.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nodiff EA, Chatterjee S, Musallam HA. Antimalarial activity of 8-aminoquinolines. Prog. Med. Chem. 1991;28:1–40. doi: 10.1016/s0079-6468(08)70362-x. [DOI] [PubMed] [Google Scholar]
- 33.Prudêncio M, Rodrigues CD, Ataíde R, Mota MM. Dissecting in vitro host cell infection by Plasmodium sporozoites using flow cytometry. Cell. Microbiol. 2008;10:218–224. doi: 10.1111/j.1462-5822.2007.01032.x. [DOI] [PubMed] [Google Scholar]
- 34.Queener SF. New drug developments for opportunistic infections in immunosuppressed patients: Pneumocystis carinii. J. Med. Chem. 1995;38:4739–4759. doi: 10.1021/jm00024a001. [DOI] [PubMed] [Google Scholar]
- 35.Fishman JA. Treatment of infection due to Pneumocystis carinii. Antimicrob. Agents Chemother. 1998;42:1309–1314. doi: 10.1128/aac.42.6.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chan C, Montaner J, Lefebvre EA, Morey G, Dohn M, McIvor RA, Scott J, Marina R, Caldwell P. Atovaquone suspension compared with aerosolized pentamidine for prevention of Pneumocystis carinii pneumonia in human immunodeficiency virus-infected subjects intolerant of trimethoprim or sulfonamides. J. Infect. Dis. 1999;180:369–376. doi: 10.1086/314893. [DOI] [PubMed] [Google Scholar]
- 37.Bartlett SR, Queener SF, Tidwell RR, Milhouse WK, Berman JD, Ellis WY, Smith JW. 8-Aminoquinolines from Walter Reed Army Institute for Research for treatment and prophylaxis of Pneumocystis pneumonia in rat models. Antimicrob. Agents Chemother. 1991;35:277–282. doi: 10.1128/aac.35.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Queener SF, Bartlett MS, Nasr M, Smith JW. 8-aminoquinolines effective against Pneumocystis carinii in vitro and in vivo. Antimicrob. Agents Chemother. 1993;37:2166–2172. doi: 10.1128/aac.37.10.2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goodwin TE, Boylan CJ, Current WL, Byrd JC, Fuller DA, Green JL, Larocca CD, Raney KD, Ross AS, Tucker WA. Enhanced pneumocystis carinii activity of new primaquine analogues. Bioorg. Med. Chem. Lett. 2000;10:2205–2208. doi: 10.1016/s0960-894x(00)00436-4. [DOI] [PubMed] [Google Scholar]
- 40.Cushion MT, Chen F, Kloepfer N. A cytotoxicity assay for evaluation of candidate anti-Pneumocystis carinii agents. Antimicrob. Agents Chemother. 1997;41:379–384. doi: 10.1128/aac.41.2.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cushion MT, Walzer PD, Collins MS, Rebholz S, Vanden Eynde JJ, Mayence A, Huang TL. Highly active anti-Pneumocystis carinii compounds in a library of novel piperazine-linked bisbenzamidines and related compounds. Antimicrob Agents Chemother. 2004;48:4209–4216. doi: 10.1128/AAC.48.11.4209-4216.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Van den Eynde JJ, Mayence A, Huang TL, Collins MS, Rebholz S, Walzer PD, Cushion MT. Novel bisbenzamidines as potential drug candidates for the treatment of Pneumocystis carinii pneumonia. Bioorg. Med. Chem. Lett. 2004;14:4545–4548. doi: 10.1016/j.bmcl.2004.06.034. [DOI] [PubMed] [Google Scholar]
- 43.Flipo M, Beghyn T, Leroux V, Florent I, Deprez BP, Deprez-Poulain RF. Novel selective inhibitors of the zinc Plasmodial aminopeptidase PfA-M1 as potential antimalarial agents. J. Med. Chem. 2007;50:1322–1334. doi: 10.1021/jm061169b. [DOI] [PubMed] [Google Scholar]
- 44.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 45. http://www.vcclab.org/lab/alogps/
- 46. http://www.organic-chemistry.org/prog/peo/
- 47.Vale N, Moreira R, Gomes P. Characterization of primaquine imidazolidin-4-ones with antimalarial activity by electrospray ionization-ion trap mass spectrometry. Int. J. Mass Spectrom. 2008;270:81–93. [Google Scholar]
- 48.Vale N, Matos J, Moreira R, Gomes P. Electrospray ionization-ion trap mass spectrometry study of PQAAPro and PQProAA mimetic derivatives of the antimalarial primaquine. J. Am. Soc. Mass Spectrom. 2008;19:1476–1490. doi: 10.1016/j.jasms.2008.06.019. [DOI] [PubMed] [Google Scholar]
- 49.Rosenthal PJ, Olson JE, Lee GK, Palmer JT, Klaus JL, Rasnick D. Antimalarial effects of vinyl sulfone cysteine proteinase inhibitors. Antimicrob. Agents Chemother. 1996;40:1600–1603. doi: 10.1128/aac.40.7.1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shenai BR, Lee BJ, Alvarez-Hernandez A, Chong PY, Emal CD, Neitz RJ, Roush WR, Rosenthal PJ. Structure-activity relationships for inhibition of cysteine protease activity and development of Plasmodium falciparum by peptidyl vinyl sulfones. Antimicrob. Agents Chemother. 2003;47:154–160. doi: 10.1128/AAC.47.1.154-160.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sijwali PS, Rosenthal PJ. Gene disruption confirms a critical role for the cysteine protease falcipain-2 in haemoglobin hydrolysis by Plasmodium falciparum. Proc. Natl. Acad. Sci. USA. 2004;101:4384–4389. doi: 10.1073/pnas.0307720101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sijwali PS, Kato K, Seydel KB, Gut J, Lehman J, Klemba M, Goldberg DE, Miller LH, Rosenthal PJ. Plasmodium falciparum cysteine protease falcipain-1 is not essential in erythrocytic stage malaria parasites. Proc. Natl. Acad. Sci. USA. 2004;101:8721–8726. doi: 10.1073/pnas.0402738101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.