

Abstract
GAPDH (glyceraldehyde 3-phosphate dehydrogenase) is a glycolytic enzyme that displays several non-glycolytic activities, including the maintenance and/or protection of telomeres. In this study, we determined the molecular mechanism and biological role of the interaction between GAPDH and human telomeric DNA. Using gel shift assays, we show that recombinant GAPDH binds directly with high affinity (Kd = 45 nM) to a single-stranded oligonucleotide comprising three telomeric DNA repeats and that nucleotides T1, G5 and G6 of the TTAGGG repeat are essential for binding. The stoichiometry of the interaction is 2:1 (DNA: GAPDH), and GAPDH appears to form a high-molecular weight complex when bound to the oligonucleotide. Mutation of Asp32 and Cys149, which are localized to the NAD-binding site and the active site center of GAPDH, respectively, produced mutants that almost completely lost their telomere-binding functions both in vitro and in situ (in A549 human lung cancer cells). Treatment of A549 cells with the chemotherapeutic agents gemcitabine and doxorubicin resulted in increased nuclear localization of expressed wild-type GAPDH, where it protected telomeres against rapid degradation, concomitant with increased resistance to the growth inhibitory effects of these drugs. The non-DNA-binding mutants of GAPDH also localized to the nucleus when expressed in A549 cells, but did not confer any significant protection of telomeres against chemotherapy-induced degradation or growth inhibition, and this occurred without the involvement of caspase activation or apoptosis regulation. Overall, these data demonstrate that GAPDH binds telomeric DNA directly in vitro and may have a biological role in the protection of telomeres against rapid degradation in response to chemotherapeutic agents in A549 human lung cancer cells.
Keywords: protection of telomeres, glycolytic enzyme, A549 lung carcinoma cells, chemotherapeutics & protein-DNA interactions
Introduction
Glyceraldehyde 3-phosphate dehydrogenase (GAPDH, E.C. 1.2.1.12) is a well-known and ubiquitous enzyme that functions in the glycolytic and gluconeogenic pathways of sugar metabolism, but has been linked with a diverse array of non-glycolytic activities. These include several transactions with nucleic acids (tRNA transport, DNA repair, transcription and binding with viral RNAs), membrane fusion, vesicle transport and apoptosis (for reviews see 1–4). Its binding to the β-amyloid precursor protein 5 and the pathogenic protein huntington 6,7 implicates GAPDH in the neurodegenerative disorders Alzheimer’s and Huntington’s diseases, respectively. GAPDH also plays a role in cancer pathogenesis 8–10 and translocates to the nucleus in response to several anticancer agents 11–13. Additionally, GAPDH exhibits pro-apoptotic activity 14–17, which involves binding to the E3 ubiquitin ligase, Siah1 18, and translocation of the enzyme to the nucleus, mostly in non-cancerous neuronal cells 12,19,20.
In a recent study, Sundararaj et al found that GAPDH binds telomeric DNA in vivo and its overexpression in A549 lung adenocarcinoma cells prevents shortening of telomeres following treatment with the anticancer agents gemicitabine (GMZ) and doxorubicin (DOX), suggesting a possible role for GAPDH in chemotherapeutic resistance 21. Telomeres are DNA-protein assemblies that protect the ends of chromosomes from being recognized by the DNA repair machinery as double-stranded breaks. Protection of the chromosome ends is essential for cell viability, while their shortening is associated with cell senescence (for review see 22). The finding that GAPDH may function to protect telomeres was intriguing, but raised many questions, including whether GAPDH can bind directly to telomeric DNA or requires other factors, whether it binds single-stranded or double-stranded regions of telomeric DNA, whether it exhibits the high affinity and specificity expected of a telomere-binding protein, whether the DNA-binding site overlaps with the enzyme’s catalytic site or is elsewhere and, finally, whether the interaction serves to protect telomeres against rapid degradation in response to treatment with chemotherapeutic agents in human cancer cells and, if so, whether this protection occurs in a caspase-dependent or independent manner.
Here, we present data showing that recombinant GAPDH binds a single stranded (ss) telomeric DNA with high affinity, in the absence of any other factors, as a large molecular weight complex comprising at least two tetramers. We have also identified three bases of the hexameric telomere repeat that are essential for binding to GAPDH. NAD+-competition experiments, coupled with site-directed mutagenesis, show that the telomeric DNA-binding site comprises the NAD-binding and the active sites of the enzyme. It is also demonstrated here that the binding of GAPDH to telomeres is not unique to A549 cells because GAPDH also co-localizes with telomeres in non-cancerous murine-lung epithelial (MLE-15) cells. In addition, we find that over-expressed wild-type (wt) GAPDH binds to telomeres in A549 cells and prevents rapid degradation of telomeres in response to GMZ and DOX, whereas similar expression of mutants of GAPDH that do not bind telomeric DNA does not confer such protection. Importantly, we also show that ectopic expression of wt-GAPDH, but not of the non-DNA-binding mutants, was concomitant with the prevention of the anti-proliferative effects of GMZ and DOX in these cells in a caspase-independent manner. Overall, the data reveal a specific and direct interaction between GAPDH and telomeric DNA that is associated in vivo with the prevention of chemotherapy-induced rapid degradation of telomeres and growth inhibition of cells.
Results
GAPDH binds directly to telomeric DNA
Previous in vivo studies suggested that GAPDH binds to telomeric DNA and protects the ends of telomeres from degradative enzymes 21. In vitro binding between GAPDH and telomeric DNA, however, was only observed in a gel-binding assay that used UV radiation to cross link the complexes 21. Hence, it was not known whether GAPDH could bind directly to telomeric DNA or whether other proteins were required in vivo. To address this, we developed an electrophoretic mobility shift assay (EMSA) and used this to demonstrate direct binding between GAPDH and an 18 nucleotide single-stranded DNA molecule containing three repeats of human telomeric DNA ([32P]-ss-5′-(TTAGGG)3-3′). In this assay, 10 pM of the radiolabeled oligonucleotide was titrated with increasing concentrations of GAPDH. A clear shift was observed with maximal binding at approximately 500 nM (Fig. 1A). The dissociation constant was then calculated by quantifying the disappearance of the band corresponding to free DNA (from three independent experiments), resulting in an apparent Kd of 70 ± 5 nM (Fig. 1B).
Fig. 1.

GAPDH binds directly to single-stranded telomeric DNA. (A). An EMSA of the complex formed by titration of 10 pM of [32P]-ss-(TTAGGG)3 into increasing concentrations of GAPDH. (B). Binding isotherm of the data from (A) and two other representative assays, plotted on a log scale. The apparent dissociation constant (Kd) is 70 ± 5 nM. (C). 25 nM [32P]-ss-(TTAGGG)3 and 25 nM [32P]-ds-(TTAGGG)3 were both titrated into increasing concentrations of GAPDH and analyzed by EMSA, revealing a stronger affinity of GAPDH for ss-telomeric DNA.
Telomeres contain both single-stranded (ss) and double-stranded (ds) regions of DNA (see 23 for review). To establish whether GAPDH exhibits a higher affinity for either of these, an EMSA was performed in which increasing concentrations of GAPDH were added to a mixture of 25 nM [32P]-ss-(TTAGGG)3-3′ and 25 nM [32P]-ds-(TTAGGG)3. A shifted band was observed whose intensity correlated with the decrease in free ss-(TTAGGG)3, whereas that for double-stranded DNA was unchanged (Fig. 1C), indicating that GAPDH has a higher affinity for ss- over ds-telomeric DNA. At least 500 nM of GAPDH is required before an observable shift occurs with ds-DNA, corresponding to an approximately 10-fold higher affinity for ss-telomeric DNA over ds-telomeric DNA (data not shown).
GAPDH specifically recognizes the GGT sequence of human ss-telomeric DNA
To determine which bases of the telomeric DNA repeat are recognized by GAPDH, a series of single-stranded oligonucleotides that contained single base changes in the telomere repeat were constructed as follows: ((ATAGGG)3, (TAAGGG)3, (TTTGGG)3, (TTACGG)3, (TTAGCG)3 and (TTAGGC)3. GAPDH was then incubated in the presence of 50 nM [32P]-ss-(TTAGGG)3 and increasing concentrations of each unlabeled mutated competitor DNA. Increasing concentrations of oligonucleotides (TAAGGG)3, (TTTGGG)3, (TTACGG)3) competed with the probe, indicating that efficient binding to GAPDH is retained by these oligonucleotides (Fig. 2A). Of these, (TTTGGG)3-3′ competed the most strongly, whereas (T AAGGG)3 and (TTACGG)3 showed lower levels of competition. In contrast, (ATAGGG)3, (TTAGCG)3 and (TTAGGC)3 oligonucleotides did not compete, suggesting that the bases replaced in these are required for binding to GAPDH (Fig. 2A). Hence, GAPDH appears to recognize specifically bases T1, G5 and G6 of the human telomeric DNA sequence, which form a contiguous sequence when considered in the context of DNA containing several telomeric repeats (Fig. 2B).
Fig. 2.

The base specificity of GAPDH for telomeric DNA. (A). 50, 250 or 1000 nM unlabeled oligonucleotide competitors with a single base change (shown in italics) at each nucleotide of the telomere repeat were added to 50 nM GAPDH and 50 nM [32P]-ss-(TTAGGG)3, and analyzed by EMSA. A failure to compete with the labeled probe shows that the mutated base in that oligonucleotide is required for binding to GAPDH. (B). Three telomeric repeats showing the GGT recognition sequence (underlined) as a contiguous sequence.
Since one of the GGT recognition sequences was split in the 18-mer oligonucleotide used previously, a new oligonucleotide was designed that contained three intact recognition motifs 5′-AGGGTTAGGGTTAGGGTT-3′ and used for a new measurement of the dissociation constant using EMSA (Fig. 3A). This indicated an apparent Kd of 45 ± 5 nM (n=3) (Fig. 3B). The increase in affinity, compared to the original oligonucleotide used, suggests that all three GGT motifs in the 18mer oligonucleotide are involved in binding to GAPDH.
Fig 3.

The binding affinity of GAPDH for a redesigned oligonucleotide of telomeric DNA. (A). An EMSA of the complex formed by titration of 10 pM of [32P]-ss-(AGGGTT)3 into increasing concentrations of GAPDH. (B). Binding isotherm of the data from (A) plotted on a log scale (n=3). The apparent dissociation constant (Kd) is 45 ±5 nM. (C). Increasing concentrations of unlabeled ss-(AGGGTT)3 or control 18mer oligonucleotide (as shown) were incubated with 50 nM GAPDH and 100 nM [32P]-ss-(AGGGTT)3 and analyzed by EMSA as follows: lane 1, free [32P]-(AGGGTT)3; lane 2, [32P]-(AGGGTT)3 and GAPDH; lanes 3 and 4, addition of (unlabeled) ss-(AGGGTT)3 at a concentration of 500 nM (lane 3) and 2 μM (lane 4); lanes 5 & 6, addition of unlabeled control oligonucleotide, also at 500 nM and 2 μM, respectively.
The specificity of binding to the modified higher affinity oligonucleotide was examined using a control oligonucleotide that contained randomly chosen bases in each of the GGT regions. (5′-AGATGTAGCAATAGTAGT-3′). Addition of 500 nM or 2 μM ss-(AGGGTT)3 competed with the radiolabeled probe for binding to GAPDH, whereas the same amounts of the control oligonucleotide did not (Fig. 3C).
Stoichiometry and molecular mass of the GAPDH-ssDNA complex
When considering the various non-glycolytic activities of GAPDH, a key issue is the oligomeric state of the protein, as this could provide one mechanism for distinguishing the different functional forms of the protein. Some evidence suggests that GAPDH is monomeric or dimeric when bound to nucleic acid or protein partners 24,25. Additionally, various compounds such as the anti-apoptotic drug (−)-deprenyl convert the GAPDH tetramer to its dimeric form, which blocks the pro-apoptotic activity of GAPDH 26. In the case of binding to tubulin, though, GAPDH forms a complex with a stoichiometry of three tubulin dimers per GAPDH tetramer 27.
The stoichiometry of the interaction of GAPDH with ss-telomeric DNA was elucidated by performing an EMSA at a concentration of DNA (100 nM) above the apparent Kd (45 nM). In this experiment, 100 nM of [32P]-ss-(AGGGTT)3 was titrated into increasing concentrations of GAPDH tetramer (from 0 to 200 nM). As shown in Fig. 4A, half of the DNA probe was bound at 25 nM GAPDH and nearly all was bound at 50 nM GAPDH. These data suggest a complex with a molar ratio of 2:1 and would be consistent with one oligonucleotide per GAPDH dimer or two oligonucleotides per GAPDH tetramer.
Fig. 4.

The stoichiometry and size of the complex of GAPDH with telomeric DNA. (A). An EMSA of GAPDH and [32P]-ss-(AGGGTT)3 performed under stoichiometric conditions. (B). Blue Native-PAGE, where the lane assignments are as follows: lane 1, protein markers with molecular masses (in kDa): 480, apoferritin; 242, β-phycoerythrin; 146, lactate dehydrogenase (LDH); and 66, bovine serum albumin; lane 2, LDH protein standard; lane 3, 1 μg GAPDH; lane 4, 1 μg GAPDH and 2 μg ss-(AGGGTT)3; lane 5, 1 μg GAPDH and 4 μg ss-(AGGGTT)3.
Blue Native-PAGE (BN-PAGE) was then performed to investigate whether GAPDH dissociates into monomers or dimers when bound to telomeric DNA. This electrophoretic technique integrates negative charges into proteins using the anionic dye Coomassie G250, but is non-denaturing. Rather than causing dissociation of GAPDH, addition of ss-(AGGGTT)3 generated a complex of high molecular mass that migrated between markers corresponding to 242 and 480 kDa (Fig. 4B). In this range, the complex might comprise two or three tetramers of GAPDH bound with DNA. Surprisingly, GAPDH alone appeared to migrate as a monomer, which suggests that Coomassie G250 causes dissociation of the tetramer. Together, the results indicate that GAPDH forms a high molecular weight complex when bound to the ss-telomeric DNA oligonucleotide, and that binding of the telomeric DNA blocks the dissociation of the tetramer in the presence of Coomassie G250.
The binding sites for telomeric DNA and NAD+ overlap
NAD+ competes with tRNA and viral RNA for GAPDH binding in an EMSA 28,29 and with telomeric DNA in a UV cross-linked gel assay 21. To examine whether NAD+ can also compete with the telomere-binding activity of GAPDH under the more stringent conditions of an EMSA, increasing concentrations of NAD+ were titrated into 50 nM GAPDH and 100 nM of [32P]-ss-(AGGGTT)3 under stoichiometric conditions (Fig. 5A). The GAPDH-DNA complex was abolished at high concentrations of NAD+, consistent with there being overlapping binding sites on GAPDH for telomeric DNA and NAD+. The approximate concentration of NAD+ that reduced 50% binding of DNA was 50–75 nM, which indicates that NAD+ has a similar or slightly lower affinity for GAPDH compared to the 18-mer oligonucleotide.
Fig. 5.

Telomeric DNA binds at the NAD+-binding and active site of GAPDH. (A). In this competition EMSA, increasing concentrations of NAD+ (0 to 500 nM) were titrated into a fixed (50 nM) concentration of GAPDH and 100 nM of [32P]-ss-(AGGGTT)3. (B). Increasing concentrations of wild-type (WT) and the D32A mutant of GAPDH were titrated with 10 pM of [32P]-ss-(AGGGTT)3 and analyzed by EMSA, showing the failure of the mutant to shift telomeric DNA (C). The same experiment and result performed with the C149A mutant of GAPDH.
Although the competition between NAD+ and telomeric DNA for binding to GAPDH is most easily explained by overlapping binding sites, it does not rule out a mechanism where the binding of NAD+ causes a conformational change that lowers the affinity for DNA at a separate site. Hence, we mutated a residue within the NAD+-binding site and determined whether the mutated protein bound telomeric DNA. In a high-resolution crystal structure of human GAPDH in complex with NAD+, Asp32 forms several hydrogen bonds with O3 and O2 hydroxyl groups of the ribose sugar of NAD+30. Asp32 was mutated to Ala and the mutant GAPDH was cloned and expressed in E. coli. Proper folding of the protein was confirmed using circular dichroism (CD) spectroscopy (data not shown). Isothermal titration calorimetry demonstrated that the D32A mutant was unable to bind NAD+ and an activity assay showed it to be enzymatically inactive (Supplemental Figs. 1 & 2). To examine whether this mutation affects the binding of GAPDH to telomeric DNA, 10 pM [32P]-ss-(AGGGTT)3 was titrated into increasing concentrations of D32A-GAPDH (0 to 200 nM) and analyzed by EMSA. Compared to wild-type GAPDH, the D32A mutant failed to shift the oligonucleotide (Fig. 5B), which shows Asp32 is required for DNA binding and argues in favor of a direct competition mechanism between NAD+ and DNA rather than one involving conformational changes.
We also determined whether Cys149, which is the nucleophilic residue in the enzymatic reaction catalyzed by GAPDH, is required for the binding of telomeric DNA. In a similar manner as for D32A, a C149A mutant of GAPDH was prepared and purified, and was found to be enzymatically inactive (Supplemental Fig. 2). This mutant also failed to shift the labeled oligonucleotide (Fig. 5C), showing that Cys149 is also required for binding telomeric DNA. Hence, the binding site for DNA in GAPDH appears to include both the active site center and the NAD+-binding site.
Modeling the GAPDH-telomeric DNA complex
In order to visualize how GAPDH interacts with telomeric DNA at the molecular level, information gained from the stoichiometry and mutagenesis experiments was used to construct a model of the complex. Isoelectric-surface representations of the crystal structure of human GAPDH 30 shows two regions of positive charge that form a distinct groove on each side of the tetramer (Fig. 6A and B). This groove contains the active site region, including the NAD-binding site. In accordance with the observed stoichiometry of 2 (AGGGTT)3 oligonucleotides per GAPDH tetramer, each groove could accommodate one such oligonucleotide. The plausibility of this model was assessed by positioning (AGGGTT)3 into one of the grooves (Fig. 6C and D). Within the oligonucleotide, each GGT recognition motif is approximately 30 A apart and this corresponds very closely to the distance between adjacent NAD-binding sites in the tetramer, consistent with these bases being required for binding to telomeric DNA, as well as the NAD+-competition data. Given the presence of two or three tetramers of GAPDH in the complex with an 18 nucleotide molecule of telomeric DNA, the third GGT motif could contact the same groove in an adjacent tetramer (but is not long enough to reach an NAD-binding site). Although speculative, this model does highlight a plausible DNA-binding region of GAPDH that includes the active site and also suggests how extended regions of single-stranded DNA, such as those found in telomeres, could be accommodated by a complex of several tetramers of GAPDH in which the basic grooves on the surface of the protein line up in a contiguous fashion.
Fig. 6.

Model of the complex between GAPDH and telomeric DNA. (A). Isoelectric-surface representation of a tetramer of GAPDH tetramer, where red is negatively charged, blue is positive and white is uncharged. The active sites are indicated (A.S.). (B). The same representation as (A), but rotated 90°. (C). A model of an 18 nucleotide molecule of ss-telomeric DNA, in CPK format, in which the GGT recognition sequence is colored orange and where nucleotides containing bases that are not required for binding to GAPDH are green. The approximate distance between adjacent DNA-recognition motifs is marked (in Å), which also corresponds to the distance between active sites in the tetramer shown in (A). (D). The same oligonucleotide docked onto the surface of a GAPDH tetramer (similar orientation as (A)) showing the overlap of GGT sequence and active sites.
GAPDH co-localizes with telomeres in a non-cancerous cell line
Previously, GAPDH was demonstrated to localize to telomeres in human A549 lung adenocarcinoma cells in a ChIP assay 21. To investigate whether this observation is unique to A549 cells, we used a ChiP assay to determine whether endogenously expressed GAPDH co-localizes with telomeres in MLE-15 cells, which is a non-cancerous mouse distal lung epithelial cell line. The same experiment was also performed with A549 cells. In both cases, GAPDH-DNA complexes were immunoprecipitated with anti-GAPDH monoclonal antibody and the telomeric probe was visualized by anti-digoxigenin antibody. GAPDH co-localized with telomeric DNA in both cell types (Fig. 7), suggesting that the association of GAPDH with telomeres is not cell specific. The apparently stronger band observed for A549 cells, however, may suggest stronger binding of GAPDH in A549 human lung cancer cells, compared to MLE-15 non-cancerous cell lines.
Fig. 7.
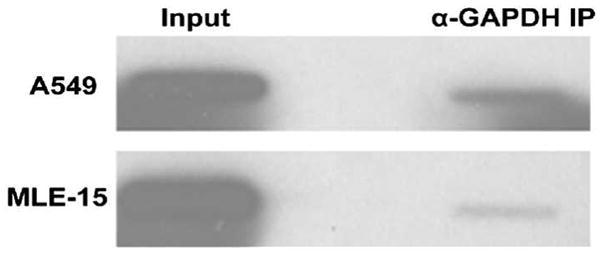
GAPDH localizes to the telomeres of A549 cells and MLE-15 cells. The binding of endogenously expressed GAPDH to telomeres of A549 cells and MLE-15 cells was observed in a ChIP assay in which GAPDH-DNA complexes were immunoprecipitated with anti-GAPDH monoclonal antibody and telomeric DNA was visualized by digoxigenin-labeled telomeric probe and anti-digoxigenin antibody.
Non-DNA-binding mutants of GAPDH no longer protect A549 cells against chemotherapeutic agents
Having determined that GAPDH binds telomeric DNA in vitro, and that in vivo binding occurs in more than one cell type, it was then important to examine the role this interaction plays in telomere maintenance. FLAG-tagged constructs of wt-, D32A- and C149A- variants of GAPDH were transiently transfected in A549 cells and the in vivo binding to telomeres of each was determined using ChIP assays, as described in Materials & Methods. FLAG-tagged constructs were used in order to distinguish overexpressed from endogenous GAPDH. There was no detectable binding of D32A-GAPDH-FLAG to telomeres in vivo, compared to wt-GAPDH-FLAG, and binding was almost completely abolished for C149A-GAPDH-FLAG (Fig. 8A). A Western blot showed similar expression levels of the three proteins (Fig. 8B & C). Thus, Asp32 and Cys149 appear to be important for binding to telomeric DNA both in vitro and in vivo.
Fig. 8.

Non-DNA-binding mutants of GAPDH do not co-localize with telomeres in vivo. (A). The binding of GAPDH-FLAG (WT), D32A-FLAG (D32A) or C149A-FLAG (C149A) to telomeres of A549 cells in a ChIP assay in which GAPDH-DNA complexes were immunoprecipitated with an anti-FLAG-M2 monoclonal antibody and telomeric DNA was visualized by a digoxigenin-labeled telomeric probe and anti-digoxigenin antibody. (B). To ensure the transfection efficiency of exogenously expressed proteins in A549 cells was equivalent, the expression levels of FLAG-tagged wt-, D32A-, or C149A- forms of GAPDH were determined by Western blotting using the anti-FLAG-M2 antibody, as described in Materials & Methods (lanes 2–4, respectively). Lysates obtained from cells transfected with the vector only were used as controls (lane 1). (C) The β-actin levels of cell extracts as loading controls. (D). The same A549 cells were treated with 250 nM GMZ and 150 nM DOX, and GAPDH was visualized using fluorescence confocal microscopy by a rhodamine-conjugated secondary antibody (colored red) directed against the primary anti-FLAG-M2 monoclonal antibody, showing that the nuclear localization of GAPDH in response to DOX/GMX is not affected by either of the D32A or C149A mutations.
To exclude the possibility that the lack of telomere binding of the D32A-FLAG and C149A-FLAG mutants of GAPDH was due to their failure to localize to the nucleus, we examined their sub-cellular localization in the presence and absence of GMZ/DOX treatment in A549 cells using immunofluorescence and confocal microscopy. As shown in Fig. 8D, treatment with GMZ/DOX triggers the nuclear localization of wt-GAPDH-FLAG and, importantly, this translocation also occurs for both mutants of GAPDH.
Telomere length in the presence or absence of GMZ/DOX, which results in the degradation of telomeres 21, was then measured in the transfected A549 cells (Fig. 9A). For vector-transfected control cells, there was a characteristic smearing of the DNA band toward lower molecular mass species after treatment with GMZ/DOX, indicative of telomere degradation. The average telomere restriction fragment (TRF) length for untreated cells was 5900 ± 300 bp, whereas that for vector-control treated was 4900 ± 200, a decrease of approximately 1000 bp. Overexpression of wt-GAPDH-FLAG protected A549 cells from telomere shortening in response to GMZ and DOX (TRF=5600 ± 200 bp), in agreement with previous findings 21. Expression of the D32A and C149A non-DNA-binding mutants of GAPDH, however, did not confer any significant protection of telomeres (TRF=4600 ± 200 and 4700 ± 300 bp, respectively). Thus, neither of the two non-telomeric DNA-binding mutants of GAPDH co-immunoprecipitates with telomeres in vivo, and neither protect telomeres from shortening in response to GMZ or DOX.
Fig 9.

A549 cells transfected with D32A or C149A mutants of GAPDH are no longer protected against treatment by GMZ and DOX. (A). A549 cells were transfected with vector control (VC), WT, D32A or C149A constructs and telomeric restriction fragment lengths were analyzed after 48 hours treatment with 250 nM GMZ and 150 nM DOX. M= markers; H= high molecular weight markers; L= low molecular weight markers. (B). Cell viability was measured using an MTT assay in which untransfected cells (control), or cells transfected with constructs expressing WT, D32A or C149A GAPDH, were treated with 250 nM GMZ and 150 nM DOX. Percent viability was calculated from the differences in absorbance of treated vs. untreated cells. Data were analyzed using 2-way ANOVA (p<0.001) (n=3). (C). Telomere length of A549 cells treated with GMZ/DOX in the presence or absence of Z-VAD (20 μM) was analyzed as described in (A), and the results were compared to untreated controls. (D). Effects of GMZ/DOX on cell survival or death were determined using trypan blue exclusion assay. Error bars represent standard deviations.
To investigate whether the protection of telomeres by GAPDH is linked to the protection of cells from anti-proliferative effects of GMZ/DOX, we measured the growth of A549 cells expressing wt-, D32A and C149A-FLAG-tagged constructs of GAPDH, compared to vector-transfected controls in the presence or absence of GMZ/DOX (250 nM GMZ and 150 nM DOX for 48 hours) using an MTT assay. Treatment of vector controls with GMZ/DOX decreased the growth of A549 cells by about 40% compared to untreated controls, whereas overexpression of wt-GAPDH-FLAG almost completely prevented the growth inhibition in these cells (Fig. 9B). On the other hand, expression of the mutant GAPDH proteins did not result in any significant protection against the inhibition of growth in response to GMZ/DOX treatment in these cells. In addition, prevention of caspase-induced apoptosis using Z-VAD did not have any preventive effect on GMZ/DOX-induced rapid telomere reduction, or growth inhibition in these cells (Figs. 9C and D, respectively). These data suggest that rapid degradation of telomeres in response to GMZ/DOX is not due to general apoptosis-induced DNA damage in these cells. Collectively, these data suggest that wt-GAPDH-FLAG, but not its mutants that lack the ability to bind telomeres, confers protection against GMZ/DOX-mediated growth inhibition, concomitant with the protection of telomeres in A549 cells.
Discussion
In the present study, we have determined that GAPDH interacts with single-stranded telomeric DNA with high affinity and specificity, and that binding requires Asp32 and Cys149, which are located in the active site region of the enzyme. We also show that the binding of GAPDH to telomeres in vivo results in the protection of telomere length, which correlates with the protection of A549 cells from the growth inhibitory effects of the chemotherapeutic agents GMZ and DOX.
GAPDH-nucleic acid interactions
The affinity of GAPDH for the ss-5′-(AGGGTT)3-3′ telomeric DNA used in this study (Kd = 45 nM) is generally lower than for GAPDH binding to other nucleic acids. For instance, GAPDH binds to E. coli tRNATyr and tRNASer with a Kd of 18 nM 28 and to hepatitis A virus mRNA with a Kd of 0.2 nM 31. Most likely, GAPDH exhibits higher affinity for these targets because they are much larger than the 18 base oligonucleotides used here. In addition, GAPDH may bind RNA more tightly than DNA 32. The specificity of GAPDH for ss- over the ds-telomeric DNA is consistent with previous studies that calculated a 300-fold preference of GAPDH for ss- over ds-DNA 33 and suggests that GAPDH preferentially recognizes single-stranded regions of telomeres.
The binding affinity of GAPDH for the telomeric DNA oligonucleotide is comparable to the affinities of members of the shelterin family of telomere-binding proteins. The DNA-binding (myb-like) domain of TRF1 binds a two-repeat of telomeric DNA with a Kd of ~3 nM, as determined via an EMSA 34, (although a Kd of 200 nM was determined with the same oligonucleotide using surface plasmon resonance 35). TRF2 exhibits a similar affinity as TRF1 35,36. Like GAPDH, POT1 (Protection of Telomeres 1) preferentially binds ssDNA, but at higher affinity (e.g. Kd = 9.5 nM for [GGTTAG]2 37). Hence, GAPDH is most comparable to POT1 in terms of its preference for ss-DNA and its binding affinity. Interestingly, the sites on telomeric DNA recognized by GAPDH and POT1 overlap by only one nucleotide (AGGGTT vs. AGGGTT 37, respectively), making it possible that the proteins bind adjacent to one another on single-stranded regions of telomeres. Hence, it is probable that, at least at some stage in the cell cycle, GAPDH is a de facto telomere-binding protein. Given that the nuclear localization of GAPDH is at its maximum during S phase of the cell cycle 21, its presence at telomeres may coincide with DNA synthesis.
Mechanism of competition with NAD
The observed competition between NAD+ and ss-telomeric DNA for binding GAPDH supports previous data obtained using a UV-cross-linking binding assay that suggested that telomeric DNA binds to the NAD-binding site of GAPDH 21. Similar observations have also been made for the binding of tRNA 28 and mRNA 29. These previous data, though, do not exclude an indirect mechanism of competition, where binding of NAD+ lowers the affinity for GAPDH via conformational changes that alter a separate DNA-binding site. Indeed, E. coli GAPDH undergoes conformational changes upon binding NAD+38. The mutation of a key residue at the heart of the NAD-binding site, Asp32, to alanine, which produced a mutant GAPDH that no longer binds telomeric DNA, shows that the NAD- and DNA-binding sites do overlap. Likewise, the failure of the C149A mutant to bind DNA shows that the DNA-binding site also extends to include the catalytic center of the enzyme. A crystal structure of the C149A mutant of GAPDH shows no significant structural difference from the wild-type structure (data not shown), thus ruling out a conformational mechanism of NAD+ competition.
Based on our competition assay, the concentration of NAD+ required to compete half of the bound oligonucleotide was approximately 75 nM, indicating that the relative affinity of GAPDH for NAD+ is slightly lower than the affinity of GAPDH for (AGGGTT)3 (see Fig. 5A). This is consistent with the measured binding constants of GAPDH for NAD+, which is approximately 100 nM for the enzyme from E. coli 39. The physiological relevance of this for the putative nuclear functions of GAPDH, however, is not clear. The concentration of free NAD+ in the nucleus (of Cos7 cells) is estimated to be ~70 μM 40, which suggests that any GAPDH in the nucleus would be fully saturated by NAD+ and unavailable to bind telomeric DNA. There are a number of possibilities, though, that might explain this apparent discrepancy. The nuclear form of GAPDH might be modified in some way that increases its affinity for DNA and lowers its affinity for NAD+. Indeed, distinct nuclear and cytoplasmic isoforms of GAPDH are detectable by isoelectric focusing 21 (although the molecular basis for this difference is unknown). In addition, because telomeres are large and extended, the effective concentration of DNA may be high enough for GAPDH to bind in the presence of NAD+. Our model, in which tetramers of GAPDH bind adjacent to one another along linear telomeric DNA, supports this idea. Finally, although GAPDH binds telomeric DNA directly in vitro, other proteins may also be involved in binding in vivo that increases the affinity of GAPDH. Two such candidates are the transcription factor Oct-1 41 and the DNA repair enzyme APE1 42.
The nuclear roles of GAPDH in the protection of telomeres and prevention of growth inhibition in A549 cells
GAPDH translocates to the nucleus after treatment by several anticancer agents, including saframycins 11, thiopurine and mercaptopurine 43, dexamethasone 17, and DOX/GMZ (this study). GAPDH also translocates to the nucleus in response to S-nitrosylation of the active site cysteine (Cys149) 18 in a mechanism that involves binding to the E3 ubiquitin ligase Siah1. Increased nuclear localization of GAPDH is associated with apoptosis in non-malignant cells 18,44 and some anti-apoptotic agents appear to work by preventing binding to Siah1 with subsequent nuclear translocation of the GAPDH-Siah1 complex 26,45.
Treatment of A549 cells with GMZ/DOX also results in the translocation of GAPDH to the nucleus (see Fig. 7C), but in this case appears to be a pro-survival activity because it both protects telomere length against degradation and prevents inhibition of growth, which is caspase independent, in response to treatment (see Figs. 8A and B). These data are in agreement with recently published results that demonstrated a role for GAPDH in the protection of human cancer cells against caspase-independent cell death 46. The underlying mechanism governing the nuclear localization of GAPDH in response to chemotherapy-induced stress remains unclear, but one possible regulator involved is the signaling molecule ceramide, which is associated with induction of growth arrest, but not apoptosis, in A549 cells 47. Another possible mechanism might involve the cell cycle, because treatment with GMZ blocks cancer cells in S phase 48 and nuclear localization of GAPDH seems to be dependent on S-phase in A549 cells 21. The increased nuclear localization of GAPDH during S phase may be associated with its various postulated functions in the nucleus, specifically those involved with DNA repair, such as activity as a uracil DNA glycosylase 24 and its binding to the DNA repair protein APE1 42. However, the molecular details of how GAPDH is localized to the nucleus during cellular stress, how it protects telomeres against chemotherapy-induced rapid degradation, and whether this protection counteracts the growth inhibitory effects of chemotherapeutic agents in human cancer cells remain unknown questions that warrant further investigation.
Materials and Methods
Oligonucleotides
Oligonucleotides used in this study are presented in Table 1. The oligonucleotides were purchased from Integrated DNA Technologies, Inc. (Coralville, IA) and gel-purified by denaturing polyacrylamide gel electrophoresis. For electromobility shift assays (EMSA), 2-4 pmol of ss-5′-(TTAGGG)3-3′ or ss-5′-(AGGGTT)3-3′ oligonucleotides were labeled with 10 μCi [γ-32P]ATP and polynucleotide kinase. Double-stranded-oligonucleotides were constructed by annealing the complementary ss-telomeric DNA strand (5′-CCCTAA)3-3′) to [32P]-ss-5′-(TTAGGG)3-3′.
Table 1.
Oligonucleotides used in this study:
| [32P]-5′-(TTAGGG)3-3′ |
| 5′-(ATAGGG)3-3′ |
| 5′-(TTAGGG)3-3′ |
| 5′-(TTTGGG)3-3′ |
| 5′-(TTACGG)3-3′ |
| 5′-(TTAGCG)3-3′ |
| 5′-(TTAGGC)3-3′ |
| 5′-(CCCTAA)3-3′ |
| [32P]-5′-(AGGGTT)3-3′ |
| 5′-(AGATGTAGCAATAGTAGT)-3′ |
Cloning, expression and purification of GAPDH: The cDNA of human liver GAPDH (kindly provided by Dr. S. Lemon, University of Texas) was cloned into the pT7-HTb vector (kindly provided by Dr. R. Nicholas, UNC-Chapel Hill), which expresses the protein with an N- terminal His6 tag together with an intervening sequence that contains a cleavage site for TEV protease. BL21 (DE3) E. coli transformed with the vector were grown in LB medium at 37°C to an OD600 of 0.5 and induced with 0.5 mM isopropyl-β-D-thiogalactopyranoside (IPTG) for four hours. After centrifugation, the cell pellet was resuspended in Buffer A (20mM Tris pH 8, 500 mM NaCl, 1mM DTT, 0.1 mM benzamidine, 0.1 mM phenylmethylsulfonyl fluoride (PMSF), 1 mM EDTA, 10% glycerol) and frozen at −80°C. After thawing, the cells were lysed by sonication, centrifuged, and the resulting supernatant was precipitated with 30% w/v ammonium sulphate. The pellet was redissolved in Buffer A and loaded onto a HiTrap Ni2+-affinity column (GE Healthcare). GAPDH was eluted with a linear gradient 0–1.0 M imidazole in Buffer A. The purification tag was cleaved by adding 0.1 mg/mL of TEV protease (purified from a recombinant source) per 5 mg of GAPDH, followed by incubation at 30°C for 4 hours. The digestion reaction was then loaded onto a HiPrep 26/60 Sephacryl S-200 gel filtration column (GE Healthcare) equilibrated with Buffer A. Purified GAPDH eluted at ~120 mL.
Mutagenesis
Mutants of C149A and D32A were constructed in both pT7Htb and pcDNA3.1-Flag vectors using the QuikChangeR Site-Directed Mutagenesis Kit (Stratagene). Following confirmation of the mutation via DNA sequencing, plasmids were transformed into BL21 (DE3) E. coli or A549 lung adenocarcinoma cells.
Electromobility shift assay (EMSA)
EMSAs were performed in 10 μL reactions in binding buffer (20 mM Tris pH 8, 100 mM NaCl, 10% glycerol, 25 ng/μL poly (dIdC) and 25 ng/μL BSA). Each reaction contained unlabeled ss-telomeric oligonucleotide, 10,000 cpm [γ-32P]-ss-telomeric oligonucleotide and GAPDH. For EMSA competition assays, NAD+ or unlabeled oligonucleotide was titrated into the reaction. The reactions were then loaded onto 5% polyacrylamide gels and run in 0.25X TBE (Tris, borate & EDTA) buffer. The gels were dried onto NyTran blotting membrane (Schleicher & Schuell Bioscience, Inc.), exposed to a PhosphorImager (Amersham Biosciences) and quantified using ImageQuant software (GE Healthcare). The binding isotherms were constructed using GRAPHPAD PRISM (GraphPad Software, Inc.)
Blue native PAGE (BN-PAGE)
BN-PAGE was performed in 20 μL reactions of binding buffer (20mM Tris pH 8, 100mM NaCl and 10% glycerol), ss-5′-(TTAGGG)3-3′ and GAPDH. Two binding reactions were set up, where each consisted of 1 μg GAPDH and either 2 μg or 4 μg, of unlabeled ss-5′-(AGGGTT)3-3′. GAPDH without ssDNA was used as a control and the gel also included lanes for lactate dehydrogenase (2 μg) and 2.5 μL NativeMark Unstained Protein Standard (Invitrogen). The samples were resolved on a NativePage Novex 4–16% Bis-Tris Gel (Invitrogen). Cathode buffer (50 mM tricine, 15 mM bis-Tris pH 7.0) containing 0.02% of the anionic dye Coomassie G250 and anode buffer (50mM Bis-Tris pH 7.0) were both chilled at 4°C before samples were loaded. After approximately 1.5 hours of electrophoresis (120V at 25°C), cathode buffer was replaced with the same buffer containing 0.005 % Coomassie G250 and electrophoresis then continued until the dye front reached the bottom of the gel. The complexes were visualized by silver staining.
Cell culture and transfection
A549 cells (ATCC) were cultured at 37°C in Dulbecco’s Modification of Eagle’s Medium (DMEM) (supplemented with 10 % fetal bovine serum). MLE-15 (ATCC) cells were cultured at 37°C in RPMI 1640 media (supplemented with insulin, sodium selenite, hydrocortisone, β-estradiol, HEPES, L-glutamine, penicillin/streptomycin and 2% fetal bovine serum). Both cell lines were grown at atmospheric conditions plus 5% carbon dioxide (CO2). Cells were transfected with the pcDNA3.1 mammalian expression vector containing the cDNA of wild type wt-GAPDH-FLAG (kindly provided by Dr. Y. Hannun, MUSC), D32A-FLAG or C149A-FLAG GAPDH mutants, using the GenJet DNA In Vitro Transfection Reagent (SignaGen Laboratories) following the manufacturer’s instructions.
ChIP assay
ChIP assays were performed using a ChIP assay kit (Upstate Biotechnology, Inc.) using MLE-15 cells or A549 cells that were either transfected with plasmid DNA or untransfected. After growth, the cells were treated with formaldehyde to a final concentration of 1% and incubated for 20 mins. at room temperature. Cells were washed with ice-cold PBS, containing protease inhibitors, and after scraping, were pelleted and lysed. The GAPDH-DNA complexes were then immunoprecipitated with a primary monoclonal anti-GAPDH monoclonal antibody or anti-Flag M2 antibody (Sigma) and pulled down with salmon sperm DNA-protein A-agarose beads. The beads were washed in buffers supplied by the manufacturer and protein was eluted from the beads in solution of 1% SDS and 0.1% NaHCO3. The protein-DNA crosslinks were reversed by incubation at 65°C in 5 M NaCl for 4 hours and protein was separated from the DNA by phenol:chloroform extraction. DNA samples were heated at 98 °C and loaded onto a nylon membrane under vacuum pressure using a slot-blot vacuum manifold (Schleicher and Schuell Bioscience, Inc.). Telomeric DNA was detected with the TeloTAGG Telomere Length Assay kit (Roche Applied Science) using the digoxigenin-labeled telomeric probe and anti-digoxigenin antibody.
Confocal microscopy
Transfected A549 cells were grown on coverslips in a 6-well plate and then treated with 250 nM GMZ and 150 nM DOX. These are below the μM concentrations used clinically for these compounds, where general cell toxicity would occur, but are within range of their IC50 values (see Fig. 9B). After 48 hours incubation at 37°C, the cells were washed using fresh media and fixed after an additional four hours by 3.7% paraformaldahyde (PFA) at 25° for 15 minutes. The cells were then permeabilized by incubation in 3.7% PFA in 0.1% Triton X-100 for 15 minutes followed by 1 hour incubation in a 5% BSA blocking solution. WT-GAPDH-Flag, D32A-Flag and C149A-Flag were visualized after incubation with the monoclonal anti-Flag M2 antibody (Sigma) (1:200) at 25°C for 1 hour, and rhodamine-labeled secondary antibody (1:1000). Confocal imaging was conducted using a Leica TSC SP2 AOBS Laser Scanning Confocal Microscope.
Telomere-length assay
Transfected A549 cells were treated with 250nM GMZ and 150nM DOX. Following 48 hours incubation at 37°C, A549 cells were scraped and chromosomal DNA was extracted using the DNeasy® Tissue Kit (Qiagen) following the manufacturer’s protocol. 1 μg of chromosomal DNA was digested with restriction enzymes Rsa1 and Hinf1 (Roche Applied Science) and the telomeric restriction fragments were then resolved on a 0.8% agarose gel. The gel was transferred to a positively charged nylon membrane (Roche Applied Science) and telomeric DNA was visualized via Southern blot following the manufacturer’s protocol.
Telomere Restriction Fragment (TRF) length was calculated using the manufacturer’s protocol by measuring the RF values for each length of DNA within the DNA ladder (Roche Applied Science). A standard curve for the DNA ladder was constructed by plotting the quotient of 1/(log10(RF)) against DNA length (Microsoft Office Excel). Linear regression analysis calculated the equation for the DNA ladder (y = 17.525x - 17.958). The quotient of 1/(log10(RF)) for each experimental sample was then substituted for “x”. Standard deviations were calculated using GRAPHPAD PRISM (GraphPad Software, Inc.).
MTT assay
This is a colorimetric assay used to measure cellular growth and to determine the cytotoxicity of agents. A549 cells were transfected with WT-GAPDH-Flag, D32A-Flag or C149A-Flag and plated in a 96-well plate (5×103 cells/well). After treatment with a mixture of 250 nM GMZ and 150 nM DOX for 48 hours, MTT (3-(4, 5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) was then added to each well, followed by incubation at 37°. After 5 hours, the cells were solubilized in lysis buffer (20% sodium dodecyl sulfate (w/v), 50% N,N-dimethyl formamide (v/v), 0.8% acetic acid (v/v) pH 4.6) overnight at room temperature. The percent viability of cells was determined by measuring the absorbance at OD595 with a 96-well plate reader (Thermo Scientific).
Trypan blue exclusion assay
Living and dead A549 cells in response to GMZ/DOX treatment in the absence or presence of the pan-caspase inhibitor Z-VAD were examined using a trypan blue exclusion assay, as described previously 49.
Supplementary Material
Acknowledgments
The authors wish to thank Dr. S. Lemon (University of Texas) for providing cDNA of human liver GAPDH and Dr. R. Nicholas (UNC-Chapel Hill) for the pT7-HTb vector, both of which were used in this study. We also thank the following for technical advice: Adam Smolka, Sivakumar Ramalingam and Can Emre Senkal. This work was supported by the National Institutes of Health grant CA88932 (to BO).
Abbreviations
- BN-PAGE
blue native-polyacrylamide gel electrophoresis
- ChIP
chromatin immunoprecipitation
- DOX
doxorubicin
- EMSA
electrophoretic mobility shift assay
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GMZ
gemcitibine
- NAD
nicotinamide adenine dinucleotide
- TRF
telomere-restriction fragment
References
- 1.Sirover MA. Role of the glycolytic protein, glyceraldehyde-3-phosphate dehydrogenase, in normal cell function and in cell pathology. J Cell Biochem. 1997;66:133–40. [PubMed] [Google Scholar]
- 2.Sirover MA. New insights into an old protein: the functional diversity of mammalian glyceraldehyde-3-phosphate dehydrogenase. Biochim Biophys Acta. 1999;1432:159–184. doi: 10.1016/s0167-4838(99)00119-3. [DOI] [PubMed] [Google Scholar]
- 3.Berry MD, Boulton AA. Glyceraldehyde-3-phosphate dehydrogenase and apoptosis. J Neurosci Res. 2000;60:150–154. doi: 10.1002/(SICI)1097-4547(20000415)60:2<150::AID-JNR3>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 4.Sirover MA. New nuclear functions of the glycolytic protein, glyceraldehyde-3-phosphate dehydrogenase, in mammalian cells. J Cell Biochem. 2005;95:45–52. doi: 10.1002/jcb.20399. [DOI] [PubMed] [Google Scholar]
- 5.Schulze H, Schuler A, Stuber D, Dobeli H, Langen H, Huber G. Rat brain glyceraldehyde-3-phosphate dehydrogenase interacts with the recombinant cytoplasmic domain of Alzheimer’s beta-amyloid precursor protein. J Neurochem. 1993;60:1915–1922. doi: 10.1111/j.1471-4159.1993.tb13420.x. [DOI] [PubMed] [Google Scholar]
- 6.Burke JR, Enghild JJ, Martin ME, Jou YS, Myers RM, Roses AD, Vance JM, Strittmatter WJ. Huntingtin and DRPLA proteins selectively interact with the enzyme GAPDH. Nat Med. 1996;2:347–350. doi: 10.1038/nm0396-347. [DOI] [PubMed] [Google Scholar]
- 7.Bae BI, Hara MR, Cascio MB, Wellington CL, Hayden MR, Ross CA, Ha HC, Li XJ, Snyder SH, Sawa A. Mutant huntingtin: nuclear translocation and cytotoxicity mediated by GAPDH. Proc Natl Acad Sci U S A. 2006;103:3405–3409. doi: 10.1073/pnas.0511316103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Du ZX, Wang HQ, Zhang HY, Gao DX. Involvement of glyceraldehyde-3-phosphate dehydrogenase in tumor necrosis factor-related apoptosis-inducing ligand-mediated death of thyroid cancer cells. Endocrinology. 2007;148:4352–4361. doi: 10.1210/en.2006-1511. [DOI] [PubMed] [Google Scholar]
- 9.Kim S, Lee J, Kim J. Regulation of oncogenic transcription factor hTAF(II)68-TEC activity by human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) Biochem J. 2007;404:197–206. doi: 10.1042/BJ20061297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harada N, Yasunaga R, Higashimura Y, Yamaji R, Fujimoto K, Moss J, Inui H, Nakano Y. Glyceraldehyde-3-phosphate dehydrogenase enhances transcriptional activity of androgen receptor in prostate cancer cells. J Biol Chem. 2007;282:22651–22661. doi: 10.1074/jbc.M610724200. [DOI] [PubMed] [Google Scholar]
- 11.Xing C, LaPorte JR, Barbay JK, Myers AG. Identification of GAPDH as a protein target of the saframycin antiproliferative agents. Proc Natl Acad Sci U S A. 2004;101:5862–5866. doi: 10.1073/pnas.0307476101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ishitani R, Tanaka M, Sunaga K, Katsube N, Chuang DM. Nuclear localization of overexpressed glyceraldehyde-3-phosphate dehydrogenase in cultured cerebellar neurons undergoing apoptosis. Mol Pharmacol. 1998;53:701–707. doi: 10.1124/mol.53.4.701. [DOI] [PubMed] [Google Scholar]
- 13.Krynetski EY, Krynetskaia NF, Gallo AE, Murti KG, Evans WE. A novel protein complex distinct from mismatch repair binds thioguanylated DNA. Mol Pharmacol. 2001;59:367–374. doi: 10.1124/mol.59.2.367. [DOI] [PubMed] [Google Scholar]
- 14.Sunaga K, Takahashi H, Chuang DM, Ishitani R. Glyceraldehyde-3-phosphate dehydrogenase is over-expressed during apoptotic death of neuronal cultures and is recognized by a monoclonal antibody against amyloid plaques from Alzheimer’s brain. Neurosci Lett. 1995;200:133–136. doi: 10.1016/0304-3940(95)12098-o. [DOI] [PubMed] [Google Scholar]
- 15.Ishitani R, Sunaga K, Hirano A, Saunders P, Katsube N, Chuang DM. Evidence that glyceraldehyde-3-phosphate dehydrogenase is involved in age-induced apoptosis in mature cerebellar neurons in culture. J Neurochem. 1996;66:928–935. doi: 10.1046/j.1471-4159.1996.66030928.x. [DOI] [PubMed] [Google Scholar]
- 16.Ishitani R, Kimura M, Sunaga K, Katsube N, Tanaka M, Chuang DM. An antisense oligodeoxynucleotide to glyceraldehyde-3-phosphate dehydrogenase blocks age-induced apoptosis of mature cerebrocortical neurons in culture. J Pharmacol Exp Ther. 1996;278:447–454. [PubMed] [Google Scholar]
- 17.Sawa A, Khan AA, Hester LD, Snyder SH. Glyceraldehyde-3-phosphate dehydrogenase: nuclear translocation participates in neuronal and nonneuronal cell death. Proc Natl Acad Sci U S A. 1997;94:11669–11674. doi: 10.1073/pnas.94.21.11669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hara MR, Agrawal N, Kim SF, Cascio MB, Fujimuro M, Ozeki Y, Takahashi M, Cheah JH, Tankou SK, Hester LD, Ferris CD, Hayward SD, Snyder SH, Sawa A. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat Cell Biol. 2005;7:665–674. doi: 10.1038/ncb1268. [DOI] [PubMed] [Google Scholar]
- 19.Saunders PA, Chalecka-Franaszek E, Chuang DM. Subcellular distribution of glyceraldehyde-3-phosphate dehydrogenase in cerebellar granule cells undergoing cytosine arabinoside-induced apoptosis. J Neurochem. 1997;69:1820–1828. doi: 10.1046/j.1471-4159.1997.69051820.x. [DOI] [PubMed] [Google Scholar]
- 20.Saunders PA, Chen RW, Chuang DM. Nuclear translocation of glyceraldehyde-3-phosphate dehydrogenase isoforms during neuronal apoptosis. J Neurochem. 1999;72:925–932. doi: 10.1046/j.1471-4159.1999.0720925.x. [DOI] [PubMed] [Google Scholar]
- 21.Sundararaj KP, Wood RE, Ponnusamy S, Salas AM, Szulc Z, Bielawska A, Obeid LM, Hannun YA, Ogretmen B. Rapid shortening of telomere length in response to ceramide involves the inhibition of telomere binding activity of nuclear glyceraldehyde-3-phosphate dehydrogenase. J Biol Chem. 2004;279:6152–6162. doi: 10.1074/jbc.M310549200. [DOI] [PubMed] [Google Scholar]
- 22.Blackburn EH. Telomere states and cell fates. Nature. 2000;408:53–56. doi: 10.1038/35040500. [DOI] [PubMed] [Google Scholar]
- 23.Rhodes D, Fairall L, Simonsson T, Court R, Chapman L. Telomere architecture. EMBO Rep. 2002;3:1139–1145. doi: 10.1093/embo-reports/kvf246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meyer-Siegler K, Mauro DJ, Seal G, Wurzer J, deRiel JK, Sirover MA. A human nuclear uracil DNA glycosylase is the 37-kDa subunit of glyceraldehyde-3-phosphate dehydrogenase. Proc Natl Acad Sci U S A. 1991;88:8460–8464. doi: 10.1073/pnas.88.19.8460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Engel M, Seifert M, Theisinger B, Seyfert U, Welter C. Glyceraldehyde-3-phosphate dehydrogenase and Nm23-H1/nucleoside diphosphate kinase A. Two old enzymes combine for the novel Nm23 protein phosphotransferase function. J Biol Chem. 1998;273:20058–20065. doi: 10.1074/jbc.273.32.20058. [DOI] [PubMed] [Google Scholar]
- 26.Carlile GW, Chalmers-Redman RM, Tatton NA, Pong A, Borden KE, Tatton WG. Reduced apoptosis after nerve growth factor and serum withdrawal: conversion of tetrameric glyceraldehyde-3-phosphate dehydrogenase to a dimer. Mol Pharmacol. 2000;57:2–12. [PubMed] [Google Scholar]
- 27.Muronetz VI, Wang ZX, Keith TJ, Knull HR, Srivastava DK. Binding constants and stoichiometries of glyceraldehyde 3-phosphate dehydrogenase- tubulin complexes. Arch Biochem Biophys. 1994;313:253–260. doi: 10.1006/abbi.1994.1385. [DOI] [PubMed] [Google Scholar]
- 28.Singh R, Green MR. Sequence-specific binding of transfer RNA by glyceraldehyde-3-phosphate dehydrogenase. Science. 1993;259:365–368. doi: 10.1126/science.8420004. [DOI] [PubMed] [Google Scholar]
- 29.Schultz DE, Hardin CC, Lemon SM. Specific interaction of glyceraldehyde 3-phosphate dehydrogenase with the 5′-nontranslated RNA of hepatitis A virus. J Biol Chem. 1996;271:14134–14142. doi: 10.1074/jbc.271.24.14134. [DOI] [PubMed] [Google Scholar]
- 30.Jenkins JL, Tanner JJ. High-resolution structure of human D-glyceraldehyde-3-phosphate dehydrogenase. Acta Crystallogr D Biol Crystallogr. 2006;62:290–301. doi: 10.1107/S0907444905042289. [DOI] [PubMed] [Google Scholar]
- 31.Yi M, Schultz DE, Lemon SM. Functional significance of the interaction of hepatitis A virus RNA with glyceraldehyde 3-phosphate dehydrogenase (GAPDH): opposing effects of GAPDH and polypyrimidine tract binding protein on internal ribosome entry site function. J Virol. 2000;74:6459–6468. doi: 10.1128/jvi.74.14.6459-6468.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arutyunova EI, Danshina PV, Domnina LV, Pleten AP, Muronetz VI. Oxidation of glyceraldehyde-3-phosphate dehydrogenase enhances its binding to nucleic acids. Biochem Biophys Res Commun. 2003;307:547–552. doi: 10.1016/s0006-291x(03)01222-1. [DOI] [PubMed] [Google Scholar]
- 33.Grosse F, Nasheuer HP, Scholtissek S, Schomburg U. Lactate dehydrogenase and glyceraldehyde-phosphate dehydrogenase are single-stranded DNA-binding proteins that affect the DNA-polymerase-alpha-primase complex. Eur J Biochem. 1986;160:459–467. doi: 10.1111/j.1432-1033.1986.tb10062.x. [DOI] [PubMed] [Google Scholar]
- 34.Konig P, Fairall L, Rhodes D. Sequence-specific DNA recognition by the myb-like domain of the human telomere binding protein TRF1: a model for the protein-DNA complex. Nucleic Acids Res. 1998;26:1731–1740. doi: 10.1093/nar/26.7.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hanaoka S, Nagadoi A, Nishimura Y. Comparison between TRF2 and TRF1 of their telomeric DNA-bound structures and DNA-binding activities. Protein Sci. 2005;14:119–130. doi: 10.1110/ps.04983705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sperry JB, Shi X, Rempel DL, Nishimura Y, Akashi S, Gross ML. A mass spectrometric approach to the study of DNA-binding proteins: interaction of human TRF2 with telomeric DNA. Biochemistry. 2008;47:1797–1807. doi: 10.1021/bi702037p. [DOI] [PubMed] [Google Scholar]
- 37.Lei M, Podell ER, Cech TR. Structure of human POT1 bound to telomeric single-stranded DNA provides a model for chromosome end-protection. Nat Struct Mol Biol. 2004;11:1223–1229. doi: 10.1038/nsmb867. [DOI] [PubMed] [Google Scholar]
- 38.Yun M, Park CG, Kim JY, Park HW. Structural analysis of glyceraldehyde 3-phosphate dehydrogenase from Escherichia coli: direct evidence of substrate binding and cofactor-induced conformational changes. Biochemistry. 2000;39:10702–10710. doi: 10.1021/bi9927080. [DOI] [PubMed] [Google Scholar]
- 39.Corbier C, Mougin A, Mely Y, Adolph HW, Zeppezauer M, Gerard D, Wonacott A, Branlant G. The nicotinamide subsite of glyceraldehyde-3-phosphate dehydrogenase studied by site-directed mutagenesis. Biochimie. 1990;72:545–554. doi: 10.1016/0300-9084(90)90119-2. [DOI] [PubMed] [Google Scholar]
- 40.Zhang Q, Piston DW, Goodman RH. Regulation of corepressor function by nuclear NADH. Science. 2002;295:1895–1897. doi: 10.1126/science.1069300. [DOI] [PubMed] [Google Scholar]
- 41.Zheng L, Roeder RG, Luo Y. S phase activation of the histone H2B promoter by OCA-S, a coactivator complex that contains GAPDH as a key component. Cell. 2003;114:255–266. doi: 10.1016/s0092-8674(03)00552-x. [DOI] [PubMed] [Google Scholar]
- 42.Azam S, Jouvet N, Jilani A, Vongsamphanh R, Yang X, Yang S, Ramotar D. Human glyceraldehyde-3-phosphate dehydrogenase plays a direct role in reactivating oxidized forms of the DNA repair enzyme APE1. J Biol Chem. 2008;283:30632–30641. doi: 10.1074/jbc.M801401200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brown VM, Krynetski EY, Krynetskaia NF, Grieger D, Mukatira ST, Murti KG, Slaughter CA, Park HW, Evans WE. A novel CRM1-mediated nuclear export signal governs nuclear accumulation of glyceraldehyde-3-phosphate dehydrogenase following genotoxic stress. J Biol Chem. 2004;279:5984–5992. doi: 10.1074/jbc.M307071200. [DOI] [PubMed] [Google Scholar]
- 44.Maruyama W, Oya-Ito T, Shamoto-Nagai M, Osawa T, Naoi M. Glyceraldehyde-3-phospate dehydrogenase is translocated into nuclei through Golgi apparatus during apoptosis induced by 6-hydroxydopamine in human dopaminergic SH-SY5Y cells. Neurosci Lett. 2002;321:29–32. doi: 10.1016/s0304-3940(01)02490-9. [DOI] [PubMed] [Google Scholar]
- 45.Hara MR, Thomas B, Cascio MB, Bae BI, Hester LD, Dawson VL, Dawson TM, Sawa A, Snyder SH. Neuroprotection by pharmacologic blockade of the GAPDH death cascade. Proc Natl Acad Sci U S A. 2006;103:3887–3889. doi: 10.1073/pnas.0511321103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Colell A, Ricci JE, Tait S, Milasta S, Maurer U, Bouchier-Hayes L, Fitzgerald P, Guio-Carrion A, Waterhouse NJ, Li CW, Mari B, Barbry P, Newmeyer DD, Beere HM, Green DR. GAPDH and autophagy preserve survival after apoptotic cytochrome c release in the absence of caspase activation. Cell. 2007;129:983–97. doi: 10.1016/j.cell.2007.03.045. [DOI] [PubMed] [Google Scholar]
- 47.Ogretmen B, Hannun YA. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat Rev Cancer. 2004;4:604–616. doi: 10.1038/nrc1411. [DOI] [PubMed] [Google Scholar]
- 48.Mose S, Class R, Weber HW, Rahn A, Brady LW, Bottcher HD. Radiation enhancement by gemcitabine-mediated cell cycle modulations. Am J Clin Oncol. 2003;26:60–69. doi: 10.1097/00000421-200302000-00013. [DOI] [PubMed] [Google Scholar]
- 49.Ogretmen B, Schady D, Usta J, Wood R, Kraveka JM, Luberto C, Birbes H, Hannun YA, Obeid LM. Role of ceramide in mediating the inhibition of telomerase activity in A549 human lung adenocarcinoma cells. J Biol Chem. 2001;276:24901–24910. doi: 10.1074/jbc.M100314200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.