

Abstract
Objectives:
Studies of autosomal dominant Alzheimer disease (AD) have shown structural and cognitive changes in mutation carriers decades prior to clinical disease. Whether such changes are detectable in offspring of persons with sporadic dementia remains unknown. We related prospectively verified parental dementia to brain MRI and cognitive testing in the offspring, within a 2-generational community-based cohort.
Methods:
A total of 717 Framingham offspring (mean age: 59 ± 8 years) were studied. In multivariate analyses, we compared offspring with and without verified parental dementia (and AD) for 1) performance on tests of memory, abstract reasoning, and cognitive flexibility, and 2) volumetric brain MRI measures of total cerebral brain volume (TCBV), hippocampal volume (HV), and white matter hyperintensity volume (WMHV), assessed cross-sectionally and longitudinally.
Results:
When testing the association of parental dementia and AD with baseline cognitive performance, we observed an interaction of parental dementia and AD with APOE ε4 status (p < 0.002). In APOE ε4 carriers only (n = 165), parental dementia was associated with poorer scores on tests of verbal memory (beta = −1.81 ± 0.53, p < 0.001) and visuospatial memory (beta = −1.73 ± 0.47, p < 0.001). These associations were stronger for parental AD (beta = −1.97 ± 0.52, p < 0.001, beta = −1.95 ± 0.48, p < 0.001), equivalent to 14–16 years of brain aging. Among APOE ε4 carriers, offspring of participants with dementia were also more likely to show an annual decline in TCBV in the top quartile (odds ratio = 4.67 [1.26–17.30], p = 0.02). Regardless of APOE ε4 status, participants with parental dementia were more likely to be in the highest quartile of decline in executive function test scores (odds ratio = 1.61 [1.02–2.53], p = 0.04).
Conclusions:
Among middle-aged carriers of the APOE ε4 allele, parental dementia and Alzheimer disease were associated with poorer verbal and visuospatial memory and a higher rate of global brain atrophy.
GLOSSARY
- AD
= Alzheimer disease;
- DSM-IV
= Diagnostic and Statistical Manual of Mental Disorders, 4th edition;
- FSRP
= Framingham Stroke Risk Profile;
- HV
= hippocampal volume;
- LM-d
= Logical Memory test;
- NP
= neuropsychological test battery;
- PAR-d
= Paired Associate Learning test;
- SIM
= Similarities test;
- TCBV
= total cerebral brain volume;
- THV
= temporal horn volume;
- TrB-TrA
= Trail Making Test B minus A;
- VR-d
= Visual Reproductions test;
- WMHV
= white matter hyperintensity volume.
Family studies have shown that first-degree relatives of patients with sporadic, late-onset dementia and Alzheimer disease (AD) have a higher risk of developing dementia.1–3 The only well-established genetic susceptibility factor is the APOE ε4 allele,4 which is neither necessary nor sufficient to cause disease.
Interestingly, in autosomal dominant familial AD, studies have shown structural and cognitive changes in mutation carriers several years prior to onset of clinical disease.5,6 Whether such changes are detectable in offspring of persons with sporadic dementia remains largely unknown. Determining whether cognitive and structural changes can be detected early in asymptomatic offspring of patients with dementia is important, as these changes might help identify target populations for preventive interventions and serve as quantitative endophenotypes to broaden the search for genetic risk factors underlying sporadic dementia.
Our aim was to examine the association of prospectively verified parental dementia with tests of memory, abstract reasoning, and cognitive flexibility, and with MRI measures of total brain volume, hippocampal volume, and white matter hyperintensity volume in middle-aged community subjects, both cross-sectionally and longitudinally.
METHODS
Study population.
The Framingham Offspring Cohort has undergone 8 periodic physical and medical examinations since 1971.7 A key criterion for enrollment was that at least one of the participant's biologic parents or their spouse's parents was a member of the Framingham Original Cohort. As part of an ancillary study, Offspring participants who survived until the seventh examination (1998–2001) and attended at least one of examinations between the fifth and the seventh, or had moved away from Framingham but continued to be followed up offsite (n = 3,623), were invited to undergo a neuropsychological test battery (NP) and volumetric brain MRI (1999–2005). The acceptance rate was 72%: 2,607 subjects underwent NP, of whom 2,262 also had an MRI. After exclusion of subjects with a neurologic condition that might confound the assessment of cognitive function and measurement of brain volumes and 1,855 subjects who were not informative for both maternal and paternal dementia, the sample size for the present analysis was 717 persons with NP and 629 with MRI (see flow diagram, figure e-1 on the Neurology® Web site at www.neurology.org). Participants informative for parental dementia were younger, more educated, less likely to have hypertension, and more likely to be smokers than noninformative participants; they had greater brain volume, less white matter hyperintensities, and scored higher on neuropsychological tests except for the difference between Trail Making Test B minus A (TrB-TrA) (table e-1).
Since 2005, all participants have been invited to undergo a second NP and MRI. Of the 717 participants selected above, 485 with a second NP and 408 with a second MRI performed between 2005 and 2007 could be included in the longitudinal analysis (data from 2008 to 2009 are still being processed at this time).
Diagnosis of dementia and AD.
The Framingham Original cohort (parental generation) was screened for prevalent dementia in 1974–1976. Participants free of dementia at inception have been monitored for incident dementia using previously described surveillance techniques (appendix e-1: Methods). The diagnosis was made by a committee of neurologists and neuropsychologists according to the criteria of the DSM-IV for dementia8 and of the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer Disease and Related Disorders Association for definite, probable, or possible AD.9
Definition of parental dementia and AD.
Offspring participants with parental dementia were subjects whose mother, father, or both had verified dementia at the time of the offspring's NP/MRI (including participants whose mother or father had died with dementia). Participants without parental dementia were subjects whose mother and father were both known to be alive and free of dementia, or had died free of dementia. For parental AD similar definitions applied (participants with parental dementia other than AD were included in the group of participants without parental AD). All analyses were performed both for parental dementia and AD, as familial aggregation may differ according to the type of dementia.
Neuropsychological tests.
We selected a subset of tests from the NP battery that were representative for measures of memory, abstract reasoning, and cognitive flexibility (table e-2).10 The delayed recall component of the Logical Memory test (LM-d) provides a savings measure for long-term verbal memory. The delayed recall component of the Visual Reproductions test (VR-d) assesses long-term visuospatial memory. The delayed recall component of the Paired Associate Learning test (PAR-d) measures the ability to learn new information. The Similarities test (SIM) measures abstract reasoning skills. TrB-TrA is a marker of executive function. We transformed TrB-TrA so that higher scores reflected better performance.
MRI scans.
MRI techniques used in the Framingham Offspring Study have been described previously.11–13 Briefly, participants were evaluated with a 1 or 1.5-Tesla Siemens Magnetom. T2-weighted double spin-echo coronal sequences were acquired in 4-mm contiguous slices. All images were read centrally blind to parental dementia status. We computed total cerebral brain volume (TCBV) as the ratio of total brain parenchymal volume to total cranial volume, to correct for differences in head size. Hippocampal volume (HV) was estimated using operator-defined, manually traced boundaries, a previously validated method.14 HV at the second MRI examination was available only in a small subset of participants at this time, therefore change in hippocampal size was estimated using change in temporal horn volume of the lateral ventricles (THV).15 White matter hyperintensity volume (WMHV) was determined according to previously published methods.12 HV, THV, and WMHV were computed as ratios to total cranial volume.
Definition of covariates.
Educational achievement was studied as a 3-class variable (no college; some college; college degree). Vascular risk factors (systolic blood pressure, smoking, diabetes mellitus, history of cardiovascular disease, and atrial fibrillation) were defined as in the Framingham Stroke Risk Profile (FSRP).16 Offspring participants were categorized according to the presence or absence of at least one APOE ε4 allele.
Statistical analysis.
For cross-sectional analyses, TrB-TrA and WMHV were log-transformed to normalize their distribution. Annual change in neuropsychological test scores and brain volume measures was calculated as the difference between the last and first measurement, divided by the time interval between the 2 examinations. Change in neuropsychological test scores and in brain volume measures was analyzed using quartiles, comparing the top quartile of change to the rest. The top quartile represents the greatest increase for THV, WMHV, and TrB-TrA and the greatest decrease for other measures.
To examine the association of parental dementia and AD with neuropsychological test scores and brain volume measures (cross-sectionally and longitudinally), we used a multivariable generalized estimation equation, adjusted for sex, age at examination, and sibship among offspring. For cognitive outcomes, we also adjusted for education. Persons with missing covariate data (<5%) were excluded from these analyses. We systematically investigated interactions with APOE ε4 carrier status.
The following secondary analyses were performed: 1) additional adjustment for vascular risk factors; 2) exclusion of participants with stroke or dementia at baseline; 3) testing for an association with paternal and maternal dementia or AD; 4) investigating interactions with age and sex with analyses stratified on these parameters (for age we categorized participants as younger than 55 years vs 55 years or older); and 5) restriction to parental dementia by age 85 (participants without parental dementia being individuals whose parents remained free of dementia to age 85), as dementia occurring at a very old age is less likely to have a strong genetic component.
Finally, to estimate the equivalency between the mean change in LM-d and VR-d associated with chronological aging and that associated with parental dementia, we divided the regression coefficient for parental dementia by the regression coefficient for age.
All analyses were performed using Statistical Analyses System® software (SAS Institute, Cary, NC).
RESULTS
Of 717 participants, 285 had at least 1 parent with dementia: 192 had a mother with dementia (mean age at dementia: 86 ± 7 years), 106 had a father with dementia (mean age at dementia: 82 ± 6 years), 13 had both parents with dementia. At the baseline NP/MRI evaluation, the offspring were 21 ± 7 years younger than the mean age at diagnosis of dementia in their affected parent; 1 participant had prevalent dementia and 11 participants had prevalent stroke.
For the 485 participants with data on longitudinal change, the mean duration of follow-up was 6.15 ± 1.25 years. During this period, no participant developed dementia and 3 participants developed a stroke.
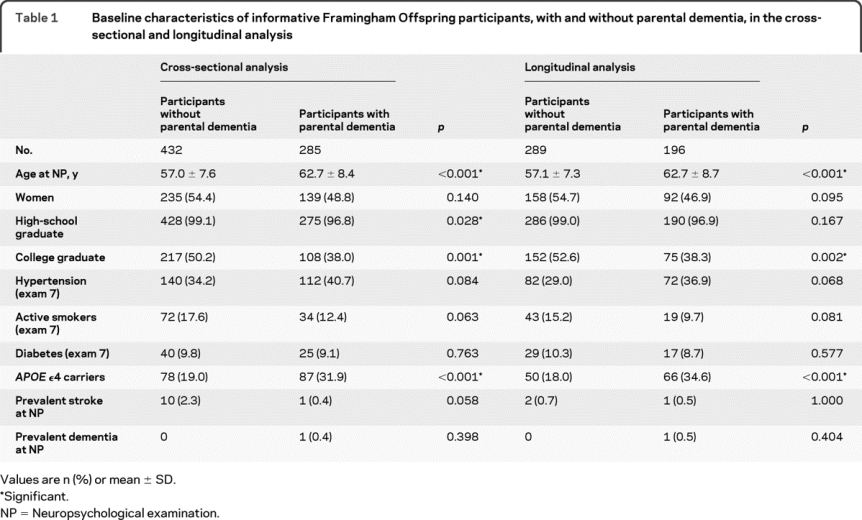
Participants with parental dementia or AD were older, were less educated, and more often carried the APOE ε4 allele compared to participants without parental dementia or AD (table 1, table e-3).
Table 1 Baseline characteristics of informative Framingham Offspring participants, with and without parental dementia, in the cross-sectional and longitudinal analysis
Association of parental dementia with baseline cognitive performance and brain structure.
Baseline cognitive performance.
Parental dementia and AD were not associated with poorer NP test performance overall (table 2). However, we found a significant interaction of parental dementia (and AD) with APOE ε4 carrier status in the offspring, for the association with LM-d and VR-d. We therefore conducted analyses stratified on APOE ε4 carrier status (table 3).
Table 2 Association of parental dementia and parental Alzheimer disease (AD) with baseline cognitive and brain MRI measures
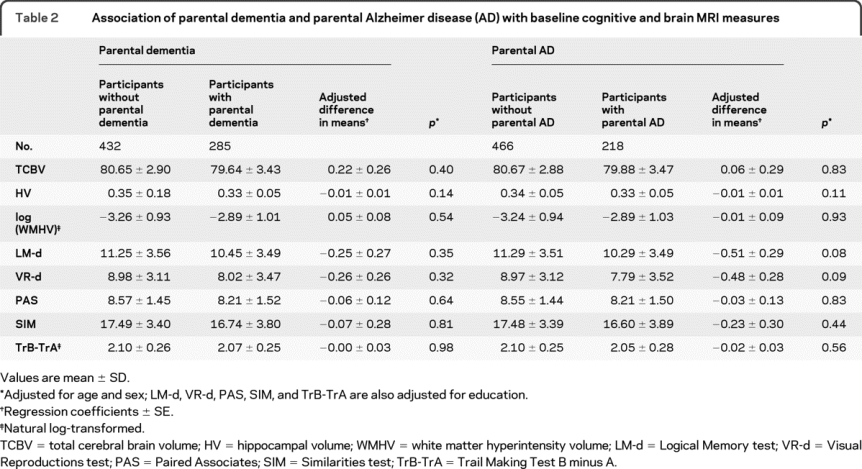
Table 3 Association of parental dementia and parental Alzheimer disease (AD) with baseline cognitive and brain MRI measures, stratified on APOE ε4 status
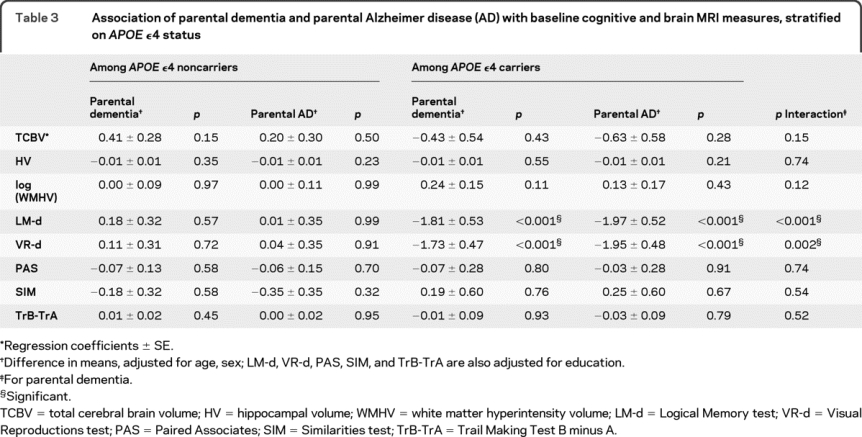
In APOE ε4 carriers only, parental dementia was associated with significantly lower LM-d and VR-d scores (table 3). For parental AD these associations were similar, or even stronger (table 3). The magnitude of effect of parental AD on performances in LM-d and VR-d among APOE ε4 carriers was equivalent to >16 years of brain aging for LM-d and approximately 14 years for VR-d.
Among APOE ε4 carriers, results were unchanged when stratifying on age (data not shown); conversely, we found an interaction with gender for LM-d (p = 0.04), an association with parental dementia being observed in women only (β = −2.89 ± 0.66, p < 0.001).
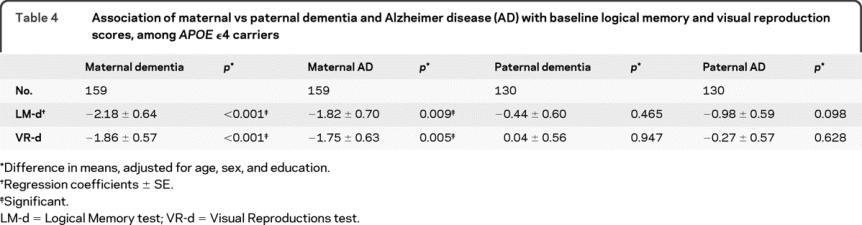
When looking separately at maternal and paternal dementia or AD in APOE ε4 carriers, we found that only maternal dementia or AD was significantly associated with LM-d and VR-d (table 4), even after adjusting for age at death of the parent, to account for differences in survival between mothers and fathers (data not shown).
Table 4 Association of maternal vs paternal dementia and Alzheimer disease (AD) with baseline logical memory and visual reproduction scores, among APOE ε4 carriers
Of note, when assessing the relationship of APOE ε4 carrier status with memory performance in the offspring overall, there was no association (β = −0.07 ± 0.32, p = 0.83 for LM-d and β = 0.15 ± 0.29, p = 0.60 for VR-d). When stratifying on parental dementia status, APOE ε4 carriers had reduced performances in LM-d only if they also had a parent with dementia (β = −1.09 ± 0.48, p = 0.02). A similar trend was observed for VR-d (β = −0.71 ± 0.43, p = 0.10). Conversely, APOE ε4 was associated with better memory performance in offspring without parental dementia (β = 1.04 ± 0.39, p = 0.007 for LM-d and β = 1.00 ± 0.33, p = 0.003 for VR-d).
Baseline brain MRI characteristics.
We found no association of parental dementia or AD with TCBV, HV, and WMHV overall and after stratifying on APOE ε4 status (tables 2 and 3).
Association of parental dementia with change in cognitive performance and brain structure.
Change in cognitive performance.
We observed a significant association of parental dementia and AD with worsening performance in TrB-TrA (table 5). There was no interaction with APOE ε4 (data not shown).
Table 5 Association of parental dementia and parental Alzheimer disease (AD) with quartiles of change in cognitive and brain MRI measures
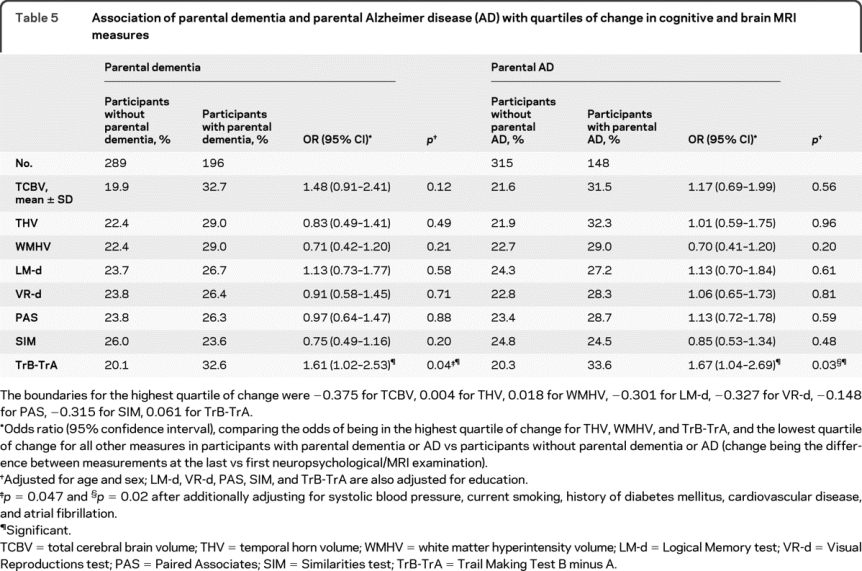
Change in brain MRI characteristics.
We did not observe any association of parental dementia or AD with change in brain volume measures overall (table 5). There was however an interaction of parental dementia with APOE ε4 status when looking at change in TCBV (p for interaction = 0.04), an association of parental dementia with the highest quartile of decrease in TCBV being observed in APOE ε4 carriers only (OR = 4.67 [1.26–17.30], p = 0.02) (table e-4).
The results described above were similar after adjusting for vascular risk factors, when restricting the analysis to parental dementia (or AD) by age 85, and after excluding the few individuals with prevalent stroke and prevalent dementia at baseline (data not shown).
DISCUSSION
In a middle-aged, community-based sample of 717 Framingham Offspring participants, parental dementia was associated with poorer performance in verbal and visuospatial memory tasks, among APOE ε4 carriers only. Similar associations were found for parental AD and these associations were driven mainly by maternal dementia. Among APOE ε4 carriers, participants with parental dementia also had greater brain atrophy rates over 6 years of follow-up. Finally, parental dementia and AD were associated with worsening performance in executive function, regardless of APOE ε4 status.
To our knowledge, the finding that significant changes in verbal and visuospatial memory can be detected in asymptomatic middle-aged offspring of individuals with sporadic dementia, 2 decades before the age at onset of dementia in the affected parent, is new. The magnitude of the effect of parental AD on memory performance, corresponding to approximately 15 years of brain aging, is particularly striking. Preliminary data from previous publications lend support to our findings. Indeed, an altered memorization pattern on the Rey Auditory Verbal Learning Test in middle-aged persons with a family history of AD has been shown recently, suggesting greater reliance on immediate working memory as opposed to consolidated episodic memory, as seen in early AD.17 In another study, first-degree relatives of patients with AD more often showed evidence of decline in memory and intelligence measures compared to controls, but the sample was relatively small and included both siblings and children.18 Moreover, functional imaging data suggest modified activation patterns in asymptomatic individuals with a positive parental history of AD compared to controls, more than a decade before their parent's onset age.19
Importantly, neither the APOE ε4 allele nor parental dementia alone was associated with impaired memory performances, while the combination of the 2 was associated with significantly worse performances in verbal and visuospatial memory. The fact that the association of parental dementia and AD with poorer performance in verbal and visuospatial memory was seen only in APOE ε4 carriers is consistent with previous work suggesting that genetic risk factors besides APOE ε4 contribute to the familial component of dementia and verbal memory performance,20,21 and that APOE ε4 interacts with these genes to determine risk.22,23 The observation that parental dementia was associated with poorer memory performance only among APOE ε4 carriers could imply either that parental dementia impacts memory only if the APOE ε4 allele is present (via an interaction of the latter with other genetic risk factors and shared environment) or that APOE ε4 accelerates the clinical expression of memory impairment in predisposed persons.4,24
Our finding that the association of memory performance with parental dementia was largely attributable to maternal dementia is intriguing. Previous studies have reported that maternal transmission of AD is significantly more frequent than paternal transmission,25 and that having an AD-affected mother confers a greater risk than having an AD-affected father.26 Moreover, in a recent analysis of PET scans from cognitively intact individuals, persons with affected mothers showed a modified pattern of glucose consumption, while findings were unremarkable in subjects with an AD-affected father.27 Potential explanations for a predominantly maternal inheritance include chromosome X variants, genetic imprinting, involvement of mitochondrial DNA, or intrauterine exposure to risk factors.28–30 Alternatively, the stronger association with maternal dementia could reflect the fact that women live longer, up to an age when they are more likely to develop dementia, which reduces the probability for mothers to be misclassified as having no dementia due to early death by competing causes. This is unlikely to be the only explanation, as our results were unchanged when restricting the analysis to dementia by age 85, or adjusting for age at the parent's death.
In previous publications on the Framingham Original cohort and the PAQUID study, poor performance in verbal and visuospatial memory heralded dementia approximately 10 years before it was diagnosed.31,32 However, whether the cognitive changes we observed here in middle-aged offspring of individuals with dementia are associated with subsequent dementia in these persons, or just reflect a less favorable but not necessarily ominous cognitive profile, remains to be determined.
Among APOE ε4 carriers, individuals with parental dementia exhibited a significantly higher rate of global brain atrophy compared to individuals without parental dementia, suggesting that the poorer memory performance we observed in the same subgroup of participants could be related to an underlying degenerative process. Higher rates of global brain atrophy have previously been related to an increased risk of incident dementia.33 The absence of association between parental dementia and change in THV or baseline TCBV and HV does not necessarily mean that these associations do not exist. In a study on familial autosomal dominant AD, differences between mutation carriers and controls in hippocampal and total brain volume and atrophy rates became evident less than 5 years before diagnosis of AD.5 Given the young age of our population, we had little power to detect such differences. Besides, early changes from AD pathology involve the entorhinal cortex, which may be missed when looking at a global measure of hippocampal size. The absence of a significant association with annual increase in THV ought to be interpreted with particular caution, as THV is only a surrogate marker of HV.
The significant association of parental dementia and AD with a steeper decline in executive function but not memory-related tests could be explained by the young age of our sample and the relatively short duration of follow-up. Previous studies suggest that in persons who subsequently develop dementia the magnitude of preclinical cognitive deficits remains relatively stable until a few years before clinical diagnosis,34 due perhaps to compensatory mechanisms.35 Besides, executive functions, especially cognitive speed, decline earlier with age than other cognitive domains such as memory.36
The strengths of this study are its population-based setting and the careful prospective surveillance and validation of dementia and AD. Although the acceptance rate was high, persons included in this study are not perfectly representative of the general population, as they are more educated with fewer risk factors and less disease than persons excluded. This limitation is common to all population-based studies involving time-consuming examinations and follow-up. Finally, our sample was largely Caucasian, reflecting the racial composition of Framingham in 1948 when the Original cohort was enrolled.
If the present findings are confirmed, their public health implications are substantial. First, they reinforce prior data suggesting a prolonged subclinical phase before the onset of dementia. Second, they suggest that in addition to age and APOE ε4, parental dementia and its interaction with APOE ε4 are important to consider when designing a risk score for dementia or cognitive impairment. Third, they indicate that heritable factors other than APOE ε4 are important in determining the familial aggregation of parental dementia with memory performance, and that at least some of them interact with APOE ε4. Fourth, our results underscore that measures of verbal and visuospatial memory are valid endophenotypes that can be used to broaden the search for genetic risk factors of sporadic dementia, as in addition to being heritable,21,37,38 and predicting an increased risk of dementia,31,32 they are also associated with parental dementia.
AUTHOR CONTRIBUTIONS
Statistical analysis was conducted by Dr. Alexa S. Beiser.
DISCLOSURE
Dr. Debette reports no disclosures. Dr. Wolf receives royalties from publishing Stroke: Pathophysiology, Diagnosis, and Management, 4th edition (Elsevier, 2004); and receives research support from the NIH [NHLBI N01-HC-25195 (PI), NINDS 2 R01 NS 17950 (PI), NIA/NINDS 1-R01-AG033040 (PI), NIA 5-R01-AG-08122 (PI), NIA 1-R01-AG16495 (PI), NHLB R01HL093029-01A1 (Co-I), NIA R01-AG033193 (Co-I), NIA R01-AG029451 (Co-I), NHLBI 1-R01-HL083197 (Co-I), and NIA P-30-AG13846 (Co-I)]. Dr. Beiser receives royalties from publishing Introductory Applied Statistics (Brooks Cole, 2005); and receives research support (as biostatistician) from the NIH [NINDS 2 R01 NS017950-28, NIA 5 R01 AG08122, NIA 2 R01 AG16495, NIA/NINDS 1 R01 AG033040-01, and NHLBI R01 HL093029-01A1]. Dr. Au receives research support from the NIH [NIA 5 R01 AG08122 (Investigator), NIA 2 R01 AG16495 (Investigator), NINDS R01 NS017950 (Neuropsychologist), NIA 1 R01 AG033040 (Investigator), and NIA R01-AG029451 (Neuropsychologist)] and from the Wing Tat Lee Fund. J.J. Himali and Dr. Pikula report no disclosures. Dr. Auerbach served on a scientific advisory board for Forest Laboratories, Inc., and serves/has served on speakers' bureaus for Forest Laboratories, Inc., Takeda Pharmaceutical Company Limited, and GlaxoSmithKline. Dr. DeCarli serves as Editor-in-Chief of Alzheimer Disease and Associated Disorders; has received honoraria for lectures or educational activities not funded by industry; serves/has served as a consultant to Eisai Inc. and Merck Serono; and receives research support from Eisai Inc., Merck Serono, the Hillblom Foundation, and Network for Cognitive Neuroscience of Diabetes and Aging, and the NIH [P30 AG10129 (PI), DHS 98–14970 (Co-I), P01 AG12435 (Co-I), P01 AG0027232 (Co-I), R01 AG111101 (Co-I), R01 AG08122 (Co-I), R01 AG16495 (Co-I), U01 AG024904 (Co-I), 1UL1RR024922-01 (Co-I), R01 AG033040 (Co-I), R01 AG010220 (Co-I), R01 AG031252 (Co-I), R01 AG 031563 (Co-I), and R01 AG012975 (Co-I)]. Dr. Seshadri receives research support from the NIH [NIA R01 AG08122 (Investigator), NINDS R01 NS017950-28 (Investigator), NIA R01 AG16495 (Investigator), NIA 1 R01 AG033040-01 (Investigator), NIA R01 AG033193 (PI), NIA R01 AG031287 (PI), NHLBI R01HL093029, and NINDS R01 NS062877 (PI of subcontracts)].
Supplementary Material
Address correspondence and reprint requests to Dr. Sudha Seshadri, Department of Neurology, Boston University School of Medicine, B602, 72 East Concord Street, Boston, MA 02118 suseshad@bu.edu
Editorial, page 2054
Supplemental data at www.neurology.org
e-Pub ahead of print on December 9, 2009, at www.neurology.org.
Supported by the Framingham Heart Study's National Heart, Lung, and Blood Institute contract (N01-HC-25195) and by grants from the National Institute of Neurological Disorders and Stroke (R01 NS17950; PI: P.A.W.) and from the National Institute on Aging (R01 AG16495 and AG08122; PI: P.A.W.; R01 AG033193; PI: S.S.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Neurological Disorders and Stroke, the National Heart Lung and Blood Institute, the National Institute of Aging or the NIH.
Dr. Debette is supported by a Fulbright grant and received an award from the Bettencourt-Schueller Foundation.
Disclosure: Author disclosures are provided at the end of the article.
Received June 10, 2009. Accepted in final form August 25, 2009.
REFERENCES
- 1.Green RC, Cupples LA, Go R, et al. Risk of dementia among white and African American relatives of patients with Alzheimer disease. JAMA 2002;287:329–336. [DOI] [PubMed] [Google Scholar]
- 2.van Duijn CM, Farrer LA, Cupples LA, Hofman A. Genetic transmission of Alzheimer's disease among families in a Dutch population based study. J Med Genet 1993;30:640–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Silverman JM, Li G, Zaccario ML, et al. Patterns of risk in first-degree relatives of patients with Alzheimer's disease. Arch Gen Psychiatry 1994;51:577–586. [DOI] [PubMed] [Google Scholar]
- 4.Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 1993;261:921–923. [DOI] [PubMed] [Google Scholar]
- 5.Ridha BH, Barnes J, Bartlett JW, et al. Tracking atrophy progression in familial Alzheimer's disease: a serial MRI study. Lancet Neurol 2006;5:828–834. [DOI] [PubMed] [Google Scholar]
- 6.Fox NC, Warrington EK, Seiffer AL, Agnew SK, Rossor MN. Presymptomatic cognitive deficits in individuals at risk of familial Alzheimer's disease: a longitudinal prospective study. Brain 1998;121:1631–1639. [DOI] [PubMed] [Google Scholar]
- 7.Garrison RJ, Kannel WB, Stokes J 3rd, Castelli WP. Incidence and precursors of hypertension in young adults: the Framingham Offspring Study. Prev Med 1987;16:235–251. [DOI] [PubMed] [Google Scholar]
- 8.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders: DSM-IV. Washington, DC: American Psychiatric Association; 1994. [Google Scholar]
- 9.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 1984;34:939–944. [DOI] [PubMed] [Google Scholar]
- 10.Au R, Seshadri S, Wolf PA, et al. New norms for a new generation: cognitive performance in the Framingham Offspring cohort. Exp Aging Res 2004;30:333–358. [DOI] [PubMed] [Google Scholar]
- 11.DeCarli C, Massaro J, Harvey D, et al. Measures of brain morphology and infarction in the Framingham Heart Study: establishing what is normal. Neurobiol Aging 2005;26:491–510. [DOI] [PubMed] [Google Scholar]
- 12.Jeerakathil T, Wolf PA, Beiser A, et al. Stroke risk profile predicts white matter hyperintensity volume: the Framingham Study. Stroke 2004;35:1857–1861. [DOI] [PubMed] [Google Scholar]
- 13.Seshadri S, Wolf PA, Beiser AS, et al. Association of plasma total homocysteine levels with subclinical brain injury: cerebral volumes, white matter hyperintensity, and silent brain infarcts at volumetric magnetic resonance imaging in the Framingham Offspring Study. Arch Neurol 2008;65:642–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeCarli C, Reed BR, Jagust W, Martinez O, Ortega M, Mungas D. Brain behavior relationships among African Americans, whites, and Hispanics. Alzheimer Dis Assoc Disord 2008;22:382–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davis PC, Gearing M, Gray L, et al. The CERAD experience, part VIII: neuroimaging-neuropathology correlates of temporal lobe changes in Alzheimer's disease. Neurology 1995;45:178–179. [DOI] [PubMed] [Google Scholar]
- 16.Wolf PA, D'Agostino RB, Belanger AJ, Kannel WB. Probability of stroke: a risk profile from the Framingham Study. Stroke 1991;22:312–318. [DOI] [PubMed] [Google Scholar]
- 17.La Rue A, Hermann B, Jones JE, Johnson S, Asthana S, Sager MA. Effect of parental family history of Alzheimer's disease on serial position profiles. Alzheimers Dement 2008;4:285–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.La Rue A, O'Hara R, Matsuyama SS, Jarvik LF. Cognitive changes in young-old adults: effect of family history of dementia. J Clin Exp Neuropsychol 1995;17:65–70. [DOI] [PubMed] [Google Scholar]
- 19.Bassett SS, Yousem DM, Cristinzio C, et al. Familial risk for Alzheimer's disease alters fMRI activation patterns. Brain 2006;129:1229–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martinez M, Campion D, Brice A, et al. Apolipoprotein E epsilon4 allele and familial aggregation of Alzheimer disease. Arch Neurol 1998;55:810–816. [DOI] [PubMed] [Google Scholar]
- 21.Lee JH, Flaquer A, Stern Y, Tycko B, Mayeux R. Genetic influences on memory performance in familial Alzheimer disease. Neurology 2004;62:414–421. [DOI] [PubMed] [Google Scholar]
- 22.Reiman EM, Webster JA, Myers AJ, et al. GAB2 alleles modify Alzheimer's risk in APOE epsilon4 carriers. Neuron 2007;54:713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang W, Qiu C, von Strauss E, Winblad B, Fratiglioni L. APOE genotype, family history of dementia, and Alzheimer disease risk: a 6-year follow-up study. Arch Neurol 2004;61:1930–1934. [DOI] [PubMed] [Google Scholar]
- 24.Locke PA, Conneally PM, Tanzi RE, Gusella JF, Haines JL. Apolipoprotein E4 allele and Alzheimer disease: examination of allelic association and effect on age at onset in both early- and late-onset cases. Genet Epidemiol 1995;12:83–92. [DOI] [PubMed] [Google Scholar]
- 25.Gomez-Tortosa E, Barquero MS, Baron M, et al. Variability of age at onset in siblings with familial Alzheimer disease. Arch Neurol 2007;64:1743–1748. [DOI] [PubMed] [Google Scholar]
- 26.Edland SD, Silverman JM, Peskind ER, Tsuang D, Wijsman E, Morris JC. Increased risk of dementia in mothers of Alzheimer's disease cases: evidence for maternal inheritance. Neurology 1996;47:254–256. [DOI] [PubMed] [Google Scholar]
- 27.Mosconi L, Brys M, Switalski R, et al. Maternal family history of Alzheimer's disease predisposes to reduced brain glucose metabolism. Proc Natl Acad Sci USA 2007;104:19067–19072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carrasquillo MM, Zou F, Pankratz VS, et al. Genetic variation in PCDH11X is associated with susceptibility to late-onset Alzheimer's disease. Nat Genet 2009;41:192–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Swerdlow RH. PET sheds light on Alzheimer's disease genetic risk. Proc Natl Acad Sci USA 2007;104:18881–18882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Touze E, Rothwell PM. Sex differences in heritability of ischemic stroke: a systematic review and meta-analysis. Stroke 2008;39:16–23. [DOI] [PubMed] [Google Scholar]
- 31.Elias MF, Beiser A, Wolf PA, Au R, White RF, D'Agostino RB. The preclinical phase of Alzheimer disease: a 22-year prospective study of the Framingham Cohort. Arch Neurol 2000;57:808–813. [DOI] [PubMed] [Google Scholar]
- 32.Amieva H, Jacqmin-Gadda H, Orgogozo JM, et al. The 9 year cognitive decline before dementia of the Alzheimer type: a prospective population-based study. Brain 2005;128:1093–1101. [DOI] [PubMed] [Google Scholar]
- 33.Jack CR Jr., Shiung MM, Weigand SD, et al. Brain atrophy rates predict subsequent clinical conversion in normal elderly and amnestic MCI. Neurology 2005;65:1227–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Small BJ, Mobly JL, Laukka EJ, Jones S, Backman L. Cognitive deficits in preclinical Alzheimer's disease. Acta Neurol Scand Suppl 2003;179:29–33. [DOI] [PubMed] [Google Scholar]
- 35.Armstrong RJ, Barker RA. Neurodegeneration: a failure of neuroregeneration? Lancet 2001;358:1174–1176. [DOI] [PubMed] [Google Scholar]
- 36.van den Heuvel DM, ten Dam VH, de Craen AJ, et al. Increase in periventricular white matter hyperintensities parallels decline in mental processing speed in a non-demented elderly population. J Neurol Neurosurg Psychiatry 2006;77:149–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carmelli D, Swan GE, DeCarli C, Reed T. Quantitative genetic modeling of regional brain volumes and cognitive performance in older male twins. Biol Psychol 2002;61:139–155. [DOI] [PubMed] [Google Scholar]
- 38.McClearn GE, Johansson B, Berg S, et al. Substantial genetic influence on cognitive abilities in twins 80 or more years old. Science 1997;276:1560–1563. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.