

Abstract
Reactive oxygen and nitrogen species (ROS and RNS) are produced by metabolism of normal cells. However, in liver diseases, redox is increased thereby damaging the hepatic tissue; the capability of ethanol to increase both ROS/RNS and peroxidation of lipids, DNA, and proteins was demonstrated in a variety of systems, cells, and species, including humans. ROS/RNS can activate hepatic stellate cells, which are characterized by the enhanced production of extracellular matrix and accelerated proliferation. Cross-talk between parenchymal and nonparenchymal cells is one of the most important events in liver injury and fibrogenesis; ROS play an important role in fibrogenesis throughout increasing platelet-derived growth factor. Most hepatocellular carcinomas occur in cirrhotic livers, and the common mechanism for hepatocarcinogenesis is chronic inflammation associated with severe oxidative stress; other risk factors are dietary aflatoxin B1 consumption, cigarette smoking, and heavy drinking. Ischemia–reperfusion injury affects directly on hepatocyte viability, particularly during transplantation and hepatic surgery; ischemia activates Kupffer cells which are the main source of ROS during the reperfusion period. The toxic action mechanism of paracetamol is focused on metabolic activation of the drug, depletion of glutathione, and covalent binding of the reactive metabolite N-acetyl-p-benzoquinone imine to cellular proteins as the main cause of hepatic cell death; intracellular steps critical for cell death include mitochondrial dysfunction and, importantly, the formation of ROS and peroxynitrite. Infection with hepatitis C is associated with increased levels of ROS/RNS and decreased antioxidant levels. As a consequence, antioxidants have been proposed as an adjunct therapy for various liver diseases.
Keywords: Oxidative stress, Liver damage, Liver injury, ROS, RNS, Cancer, Fibrosis, Paracetamol, HCV
Introduction
Oxygen toxicity
Oxygen is lethal to mammals within a few days when dioxygen is inhaled at 1 atm, whereas survival time at 5 atm is approximately 1 h. Oxygen toxicity is associated with the capacity of this molecule to oxidize organic molecules and to produce free radical species according to the general reactions:
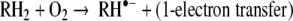 |
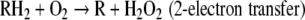 |
For these reactions to occur at significant rates, transition metal catalysts are required.
Properties of free radicals
All molecules have electrons as their outermost components. The behavior of these electrons determines the properties of the molecule. Modern quantum-mechanical theory describes electrons as having an intrinsic tendency to spin, thereby generating an electromagnetic field, the effect of which can be canceled by a similar charge spinning in the opposite direction. Thus, the most stable configuration of electrons is a paired one in which each member has opposite spins. Given this requirement for pairing, any situation in which species are generated with an unpaired electron will result in a potentially reactive entity known as a free radical. Therefore, a stable molecule contains an even number of electrons and a free radical is formed by gaining or losing one or more electrons. In order to have significant activity as a free radical, a molecule must have an unpaired electron and sufficient redox potential to be reactive. Free radicals can be generated in biological systems through a variety of processes. A major question in free radical biology is what they do after they have been formed.
Polyunsaturated lipids are essential to the entire supporting system of cells, including cell membranes, endoplasmic reticulum, and mitochondria. Disruption of their structural properties can, therefore, have dire consequences for cellular function. Peroxidation of lipids has traditionally been a major effect of free radicals. As a result of this, many of the assay methods to establish free radical-induced injury have measured by-products of the reaction of these molecules with lipids (Fig. 1).
Fig. 1.

Lipid peroxidation (LPO). X′ and X′• are free radicals, causing initiation and termination of the LPO sequence, respectively. L′•, lipid radical; LOO•, lipid peroxide; LOOH, lipid hydroperoxide
Reactive oxygen and nitrogen species (ROS and RNS, respectively; Fig. 2) are produced by normal cellular metabolism with beneficial effects such as cytotoxicity against bacteria and other pathogens. In fact, there are enzymes whose functions are to produce ROS/RNS, such as nicotinamide adenine dinucleotide phosphate (NAD(P)H) oxidases, nitric oxide synthases (NOS), and myeloperoxidases. Since these free radicals may also damage normal tissue, the balance between antioxidants and prooxidants is critical for normal function. An imbalance favoring prooxidants is defined as oxidative stress. Oxidative stress is proposed to be critical in various diseases including liver diseases.
Fig. 2.

a Main pathways for the formation of reactive oxygen species (ROS). b The three major mechanisms of reactive nitrogen species (RNS). CAT, catalase; GPX, glutathione peroxidase; NOS, nitric oxide synthase; NO•, nitric oxide; ONOO−, peroxinitrite anion; O2•−, superoxide anion
Alcoholic liver disease and free radicals
The World Health Organization has reported recently that alcohol-related diseases are the third cause of death and disability in most developed countries and are one of the leading causes in several of the developing countries of Central and South America, Eastern Europe, and East Asia [1]. It is interesting to note that the pharmacological treatment of alcohol liver disease is associated with free radicals.
Di Luzio [2] in 1966 was the first to observe lipid peroxidation after alcohol exposure; this was confirmed by other researchers [3]. The capacity of ethanol to increase both ROS/RNS and peroxidation of lipids, DNA, and proteins was demonstrated in a variety of systems, cells, species, including humans (Fig. 3). A lot was learned about alcohol metabolism, the various pathways and enzymes involved, and how alcohol directly via its solvent action affects cellular membranes or indirectly via its metabolism alters cell function. A major mechanism is lipid peroxidation and oxidative stress in alcohol toxicity. Several routes have been suggested to play a key role in the mechanism of alcohol-induced oxidative stress [4, 5]. It is likely that many systems contribute to the ability of alcohol to induce oxidative stress.
Fig. 3.

Effect of alcohol that exacerbates some of the toxic effects of acetaldehyde and generates a harmful condition called oxidative stress in the cells, characterized by excess levels of reactive oxygen species (ROS)
There are several studies that show that antioxidants administration or chelators of iron or reduced glutathione (GSH)-replenishing agents can ameliorate or prevent the toxic effects of ethanol. In the intragastric infusion of ethanol, liver damage was associated with enhanced lipid peroxidation, formation of 1-hydroxyethyl radical, decreased formation of protein carbonyl in GSH, and formation of lipid radicals [6–10]. Replacement of polyunsaturated lipids (necessary for peroxidation of lipids to occur) with medium-chain triglycerides or saturated lipids in the diets fed to rats intragastrically prevented or lowered the peroxidation of lipids and the alcohol-induced hepatic damage [9, 11]. Therefore, polyunsaturated lipids plus alcohol are needed for the production of liver damage. Iron is known both to produce •OH and to induce liver damage; importantly, addition of iron to these diets exacerbated hepatic damage [12]. Interestingly, administration of antioxidants, such as ebselen, vitamin E, superoxide dismutase (SOD), and precursors of GSH prevented alcohol-induced hepatic damage in rats [8]. Since ethanol-induced liver injury was linked to oxidative stress, Cederbaum and co-workers [13, 14] investigated the effect of a compromised antioxidant defense system, copper–zinc SOD1 knockout mice, in an alcohol-induced hepatic damage model. A moderate ethanol consumption induced oxidative stress and liver damage in these mice, indicating that compromised antioxidant defense exacerbates alcohol liver damage.
The previous in vivo studies were confirmed by in vitro studies with hepatocytes. Studies with isolated hepatocytes from long-term ethanol-fed rats and corresponding controls showed that ethanol metabolism via alcohol dehydrogenase leads to increased ROS production, hepatocyte damage, and apoptosis; these reactions were prevented by antioxidants [15, 16]. Studies with HepG2 cell lines expressing CYP2E1 indicated that addition of iron, polyunsaturated fatty acids, or ethanol or depletion of GSH resulted in hepatocytes toxicity, increased oxidative stress, and mitochondrial injury, events blocked by antioxidants [17]. CYP2E1 plays an important role in ethanol-induced oxidant stress—topic reviewed in depth recently by Lu and Cederbaum [10].
The role of free radicals in alcohol-induced hepatic injury and the capacity of ethanol to promote oxidative stress are important areas of research, in particular, because such information may possess very important therapeutic significance to prevent the hepatotoxic effects of ethanol by antioxidants, inhibitors of CYP2E1, iron chelators, or GSH replenishment among others.
Fibrosis/cirrhosis and free radicals
Liver fibrosis is the result of an exacerbated wound-healing process after chronic hepatic damage and is characterized by the activation of hepatic stellate cells (HSC) and excess production of extracellular matrix (ECM) components by these cells. The activation of HSC involves the transdifferentiation from a quiescent state into myofibroblast-like cells. The activated HSC are characterized by the enhanced production of ECM and accelerated proliferation.
Hepatic stellate cells
The embryologic origin of stellate cells has been elusive. Currently, the bulk of evidence supports their origin from either the endoderm or the septum transversum, as it forms from cardiac mesenchyme during invagination of the hepatic bud [18]. A separate issue pertains to whether stellate cells and sinusoidal endothelial cells derive from a common precursor cell, a likely possibility given their shared mesenchymal phenotype, close proximity in situ, and joint expression of some angiogenic factors, for example, vascular endothelial cell growth factor [19].
The source of activated stellate cells and myofibroblast in liver injury has provoked extensive study and debate [20], specially the notion that bone marrow contributes a substantial fraction of these cells.
ROS generated within cells or, more generally, in a tissue environment may easily turn into a source of cell and tissue injury. ROS and other oxidative stress-related intermediates contribute to death, the perpetuation of chronic inflammatory responses, fibrogenesis, with a major focus on hepatic chronic wound healing and liver fibrogenesis [21, 22].
Cross-talks between parenchymal and nonparenchymal cells are the most important event in liver injury and fibrogenesis. Soluble factors such as cytokines [23] and ROS are the most important factors in these cross-talks and are possible targets for therapeutic consideration.
ROS are involved in necrosis and apoptosis of hepatocytes and HSC activation [24, 25]. Several major classes of free radical scavengers, such as catalase, superoxide SOD, and glutathione peroxidase (GSH-P), were investigated in various types of liver damage, and they afforded effective protection against the oxidative insults to hepatic parenchyma [26].
High levels of ROS, from phagocytic cells, such as Kupffer cells (KC), protect the organism from external pathogens; however, lower amounts of ROS mainly from HSC actively participate in the regulation of intracellular signaling [25, 27]. Platelet-derived growth factor (PDGF) is the most potent mitogen of HSC and is, therefore, likely to be an important mediator during liver fibrogenesis [28]. Interestingly, NAD(P)H is expressed in HSC and produce ROS, which, in turn, induces the production of PDGF; again, this molecule increases mitosis of HSC [27]. These results strongly suggest that ROS play an important role in fibrogenesis increasing PDGF throughout.
Hepatocellular carcinoma and free radicals
Hepatocellular carcinoma (HCC) is one of the most malignant and frequent worldwide spreading diseases; it is the third most common cause of cancer deaths [29, 30]. This disease occurrence is increasing in developed Western countries such as the United States, with an incidence ratio of 2.8 and 6.2 (whiteand African American, respectively) per 100,000 habitants [31]; it is endemic in Korea, Taiwan, China, and sub-Saharan Africa [32]. The major risk factors for HCC are chronic hepatitis B and C viruses (HBV and HCV), accounting for 80% of HCC cases; other risk factors are dietary aflatoxin B1 consumption, cigarette smoking, and heavy drinking [33].
Most HCC occur in cirrhotic livers, and the common mechanism for hepatocarcinogenesis is chronic inflammation associated with severe oxidative stress [34]. There is a large body of evidence indicating that ROS play a pathogenetic role in carcinogenesis [35]. During the initiation phase of this process, ROS may interact directly with DNA, damaging specific genes that control cell growth and differentiation, among others [36]. They can also increase the activity of carcinogenic xenobiotics by facilitating their activation to reactive compounds [37]. During the progression phase of carcinogenesis, ROS can directly stimulate the growth of cancer cells [38]. The hydroxyl radical is, among all the ROS produced during the inflammation phase, the most damaging; it has been proved that it is responsible for a number of base modifications, including the formation of thymine and thymidine glycol, 8-hydroxydeoxyguanosine, and 5-hydroxylmethyluracil [35]. 8-Hydroxydeoxyguanosine is a guanine modification that induces a point mutation in the daughter DNA strand and is commonly used as an indicator of DNA damage [39].
Chronic alcohol exposure elicits hepatocyte hyperegeneration due to the activation of survival factors and interference with retinoid metabolism. Direct DNA damage results from acetaldehyde, which can bind to DNA, inhibit DNA repair systems, and lead to the formation of carcinogenic exocyclic DNA ethenoadducts. Long-term alcohol abuse also interferes with the methyl group transfer and may alter gene expression [40].
The network linking HCV infection, inflammation, free radical production, and carcinogenesis applies very well to HCV-mediated chronic liver damage, just as it applies to any chronic inflammatory condition [41]. Research into the role of structural and nonstructural proteins of HCV and the changes induced in cytokine expression oncogenes, antioncogenes, and intracellular kinases shows that HCV is by itself and not only through inflammation able to induce ROS, an effect specific to this virus [42]. This free radical production, accompanied by oxidative genomic injury, constitutes the first step of a cascade of genomic and postgenomic events that play an important role in HCC [43]. More information is necessary from recently introduced technologies for proteomics that will hopefully close the gap between hypothesis and understanding.
Ischemia/reperfusion liver injury and free radicals
Interruption of blood flow of an organ with its subsequent lack of nutrient and oxygen supply is an inherent effect during various surgical processes. In hepatic surgery, there are situations in which the ischemic periods can be very long; this is the case during the resection of large hepatic tumors, vascular reconstructions, management of hepatic trauma from various origins, and hepatic procedure for transplantation [44, 45]. When the flow of oxygen and blood is re-established, reperfusion increases the damage induced during the ischemic period, worsening the injury produced at the cellular level [44, 46]. This process called ischemia–reperfusion (IR) injury affects directly on hepatocyte viability, particularly during transplantation and hepatic surgery [45, 47]. In the ischemic period, various modifications occur at the cellular level, which promote cell injury. A decline in oxidative phosphorylation leads to ATP depletion and loss of calcium homeostasis [48].
The detrimental effects of ATP catabolism are reinforced by the production of various compounds, including cytokines vasoactive agents, and specially ROS. These effects are associated by a decline in cytoprotective compounds such as prostacycline, nitric oxide, and others [49]. Liver cell death occurs due to both apoptosis and necrosis [50].
Aerobic metabolism produces ROS that are normally inactivated through diverse antioxidant mechanisms. However, during oxidative stress conditions, the balance between ROS and antioxidants shifts toward the former, resulting in liver damage [51].
Some of the process that participate both directly and indirectly in IR injury by ROS include the formation of xantine oxidase from xantine dehydrogenase (an oxygen-dependent process that releases ROS, hydrogen peroxide, and superoxide and produces uric acid) [52], induction of NADPH oxidase by activated KC and neutrophils (ROS production is blocked when NADPH oxidase is inhibited), and NO formation and its conversion to peroxynitrite (both are RNS) [53]. The cytotoxic effects of ROS and RNS in the liver translate into tyrosine residues, lipid peroxidation, inactivation of the heme group, and nitrosylation of iron–sulfur group [44, 53].
Strong evidence indicates that KC (the resident macrophages of the liver) may cause hepatic injury in various disease processes, including cold [54] and warm [55] ischemic injury. Ischemia activates KC, which are the main source of ROS during the reperfusion period [53]. Various studies show that newly recruited nonocytes and leukocytes are partially responsible for the ischemic damage. They play an important role in the synthesis of ROS such as superoxide and hydrogen species [55]. In hepatocytes, proinflammatory cytokines, such as TNF-α, IL-1, or interferon-γ, can induce the production of ROS [56]. In addition, ischemic cell damage leads to intracellular oxidant stress during reoxygenation [57]. Mitochondria are recognized as the major intracellular source of ROS that are produced by cellular respiration [57].
Since antioxidants can inhibit ROS, various studies have aimed at modulating the severity of IR damage utilizing different mechanisms, including pharmacological allopurinol [58, 59], α-tocopherol [60], N-acethylcysteine [61], and enzymatic catalase [62, 63] and SOD [57]. Endogenous antioxidant concentrations decrease significantly during reperfusion [60, 64]; thus, administration of antioxidants, especially in the early stages of reperfusion, may significantly diminish IR injury in transplanted livers.
Genetic, pharmacological, and surgical approaches to decrease liver IR damage have been applied and are increasingly being used. Therapeutic approaches include ischemic preconditioning and the pharmacological treatment with N-acetylcysteine, prostaglandins, and prostacycline [44].
Paracetamol-induced liver damage and free radicals
Paracetamol (acetaminophen; N-acetyl-p-aminophenol [APAP]) is a safe and effective analgesic and antipyretic drug when used at therapeutic doses. However, an overdose can induce severe liver injury both in experimental animals and in humans [65]. In the past, researchers studying the toxic action mechanism of APAP focused on metabolic activation of the drug, depletion of glutathione, and covalent binding of the reactive metabolite N-acetyl-p-benzoquinone imine (NAPQI) to cellular proteins as the main cause of hepatic cell death [66]. More recently, it was discovered that covalent binding is not sufficient to kill liver cells but is a signal for the toxicity that requires amplification in the cell [67]. Intracellular steps critical for cell death include mitochondrial dysfunction and, importantly, the formation of ROS and peroxynitrite (Fig. 4). Oxidant stress of mitochondria triggers the mitochondrial membrane permeability transition pore, loss of the membrane potential of the mitochondria, depletion of ATP, and release of intermembrane proteins that are responsible for the typical nuclear DNA fragmentation of APAP-induced cell death [67]. We have found that antioxidants, such as silymarin [68], protect the liver of rats intoxicated with APAP [69]. Reduced glutathione can effectively protect the liver both by scavenging NAPQI and by detoxifying ROS and RNS, such as peroxynitrite. This mechanism is the basis for the rational clinical use of N-acetylcysteine, a GSH precursor, as antidote against APAP toxicity [70].
Fig. 4.

Schematic representation depicting the role of O2• and NO• in paracetamol toxicity
Viral hepatitis and free radicals
Infection with HCV is associated with increased levels of ROS/RNS and decreased antioxidant levels in patients [71–74]. Patients infected with HCV show increases in lipid peroxidation levels in liver samples, serum, and peripheral blood mononuclear cells [72, 75–80]. In addition, other indicators of oxidative stress such as 4-hydroxynonenal and 8-hydroxydeoxyguanosine were found to be increased in HCV [72, 74, 80–83]. The content of GSH decreased in the blood, liver, and lymphatic system, whereas that of GSSG increased, indicating a high glutathione turnover [83].
The presence of ROS and RNS is, interestingly, more pronounced with HCV than with HBV [75]. The mechanisms for more severe increases of oxidative and nitrosative stresses during HCV disease may include chronic inflammation (i.e., phagocytic NAD(P)H oxidase activation) and overload of iron, which is more specific to HCV [72, 75, 84]. Furthermore, the production of ROS in the hepatocytes may lead to the activation of KC [85]. These cells, when activated, produce and secrete cytokines; cytokines may be proinflammatory, such as TNF-α and IL-1, or profibrotic, such as TGF-β. These proteins can further increase ROS and play important roles in the mediation of hepatic injury [23, 85–87], such as fatty liver, by inhibiting lipase of lipoprotein and adiponectin and fibrosis as a result of HSC activation [88–90].
In addition, proteins of HCV can increase ROS and RNS in the infected cells; proteins of the HCV core have been shown to augment the oxidative and nitrosative stress, lipid peroxidation, oxidized thioredoxin, and antioxidant gene expression such as that of metallothionein family proteins and manganese superoxide dismutase (MnSOD) as well as to enhance sensitivity to toxins such as ethanol and CCl4 [81, 91–95]. HCV core gene expression diminishes the intracellular GSH levels and the mitochondrial NADPH content that are associated with increased uptake of calcium and oxidative stress generation at complex I in mitochondria, providing an action mechanism for HCV-induced ROS production [42, 91, 92, 96]. On the contrary, core protein modulates the production of cytokines and host enzymes, such as cyclooxygenase-2 and inducible nitric oxide synthase (iNOS), which can increase ROS and RNS [97–103].
Nonstructural proteins may also modulate the host redox status by HCV. Host antioxidant defenses, such as GSH, catalase, MnSOD, and heme oxygenase-1, are augmented, suggesting adaptation to ROS/RNS stress [92, 104, 105].
Stress produced by ROS/RNS has been implicated in HCV-induced hepatic cancer. HCV core-induced iNOS generates RNS, which may cause damage to the DNA, and augments mutations within the immunoglobulin and tumor suppressor genes [103, 106]. The genotoxic effects of ROS/RNS may contribute to the development of B-cell lymphoma or HCC during HCV infection. In fact, this association was documented in vivo in HCV core-transgenic mice [42, 107]. Other mechanisms by which core protein increases HCC include the modulation of tumor suppressor genes and proto-oncogenes as well as the inhibition of apoptosis [83]. In this regard, it should be noted that oxidative and nitrosative stress may possess diverse effects on cell growth and apoptosis [108]. As a consequence, antioxidants have been proposed as an adjunct therapy for chronic hepatitis C [109].
Nonalcoholic fatty liver and free radicals
Oxidative stress in nonalcoholic steatohepatitis (NASH) may be associated with potential etiologic mechanism. Three factors have been proposed: lipid peroxidation, hepatic iron, and hyperinsulinemia.
Lipid peroxidation
Increased lipid peroxidation was demonstrated in both animal models of fatty liver [110–112] and patients with nonalcoholic fatty liver diseases (NAFLD) [113–117]. NASH patients have increased levels of oxidative stress as compared with patients with steatosis alone [113, 115].
In these patients, free fatty acids (FFA) are the likely source of oxidative stress. Patients with NAFLD show increased lipolysis and augmented delivery of FFA to the liver [113, 118], the concentration of which are associated with more severe liver disease [119]. Elevated FFA in the liver [120] act as ligands for the transcription factor PPAR-α, which upregulates the oxidation of FFA inside the mitochondria, microsomes, and peroxisomes [121]. The FFA oxidation products (lipid peroxides and superoxide and hydrogen peroxide radicals) can generate oxidative stress and subsequent lipid peroxidation.
Insulin
Increased insulin is a frequently occurring finding in NAFLD; however, it is frequently overlooked in its pathophysiology. Insulin can damage the liver directly and indirectly [122, 123]. Patients with long-term ambulatory dialysis develop fatty liver, but only when insulin is added to the peritoneal fluid dialysate [124–126]. The steatosis is seen only at the surface of the liver and sometimes has the histological appearance of NASH [124]. This direct effect may be due to the ability of insulin to produce ROS [123]. In addition, insulin seems to posses direct profibrogenic effects by stimulating connective tissue growth factor, especially in the presence of hyperglycemia [124]. This may explain the observation that patients with NAFLD and type 2 diabetes mellitus have a very poor prognosis [127–129]. Evidence [130] indicates that insulin may be directly involved in causing endoplasmic reticulum stress along with the unfolding protein response and apoptosis. This may exacerbate insulin resistance [131].
Iron
Some evidence [132, 133] suggests that iron is important in inducing ROS and lipid peroxidation. However, most studies do not [134–138]. On the contrary, 30% of the patients with NAFLD have elevated ferritin levels [139–141], and there is an association between insulin resistance and liver iron [142, 143]. Therefore, it sounds rationale that iron causes oxidative stress because it is a well-known pro-oxidant and possesses negative effects upon the mitochondria [144, 145]. However, ongoing additional studies at present indicate that iron is likely to be important in only a minority of patients with NAFLD.
Conclusions
Reactive oxygen and nitrogen species are involved in liver damage induced by several conditions such as alcohol abuse, fibrosis/cirrhosis of various etiologies, HCC, IR liver injury, paracetamol overdose, and viral hepatitis. Oxygen and NO radicals may affect the energetic, respiratory, and regenerative pathways in hepatocytes. The imbalance of proinflammatory/anti-inflammatory cytokine in immune and inflammatory cells, the expression of collagen genes, and angiogenesis in endothelial and stellate cells aggravates the disease. On the basis of these facts, antioxidant therapy alone or in combination with other pharmacological strategies appears as the most reasonable treatment of a variety of liver diseases.
Acknowledgments
The author expresses his gratitude to biologist Mario G. Moreno in the preparation of the figures, and Liseth Rubí and Aldaba Muruato for their careful review of the manuscript.
References
- 1.Tsukamoto H. Conceptual importance of identifying alcoholic liver disease as a lifestyle disease. J Gastroenterol. 2007;42:603–609. doi: 10.1007/s00535-007-2075-3. [DOI] [PubMed] [Google Scholar]
- 2.Luzio NR. A mechanism of the acute ethanol-induced fatty liver and the modification of liver injury by antioxidants. Lab Invest. 1966;15:50–63. [PubMed] [Google Scholar]
- 3.Minicis S, Brenner DA. Oxidative stress in alcoholic liver disease: role of NADPH oxidase complex. J Gastroenterol Hepatol. 2008;23:S98–S103. doi: 10.1111/j.1440-1746.2007.05277.x. [DOI] [PubMed] [Google Scholar]
- 4.Nordmann R, Ribiere C, Rouach H. Implication of free radical mechanisms in ethanol-induced cellular injury. Free Radic Biol Med. 1992;12:219–240. doi: 10.1016/0891-5849(92)90030-k. [DOI] [PubMed] [Google Scholar]
- 5.Cederbaum AI. Microsomal generation of reactive oxygen species and their possible role in alcohol hepatotoxicity. Alcohol Alcohol. 1991;1:S291–S296. [PubMed] [Google Scholar]
- 6.Knecht KT, Adachi Y, Bradford BU, Iimuro Y, Kadiiska M, Xuang QH, et al. Free radical adducts in the bile of rats treated chronically with intragastric alcohol: inhibition by destruction of Kupffer cells. Mol Pharmacol. 1995;47:1028–1034. [PubMed] [Google Scholar]
- 7.Tsukamoto H, Lu SC. Current concepts in the pathogenesis of alcoholic liver injury. FASEB J. 2001;15:1335–1349. doi: 10.1096/fj.00-0650rev. [DOI] [PubMed] [Google Scholar]
- 8.Iimuro Y, Bradford BU, Yamashina S, Rusyn I, Nakagami M, Enomoto N, et al. The glutathione precursor l-2-oxothiazolidine-4-carboxilic acid protects against liver injury due to chronic enteral ethanol exposure in the rat. Hepatology. 2000;31:391–398. doi: 10.1002/hep.510310219. [DOI] [PubMed] [Google Scholar]
- 9.Morimoto M, Zern MA, Hagbjork AL, Ingelman-Sundberg M, French SW. Fish oil, alcohol, and liver pathology: role of cytochrome P450 2E1. Proc Soc Exp Biol Med. 1994;207:197–205. doi: 10.3181/00379727-207-43807. [DOI] [PubMed] [Google Scholar]
- 10.Lu Y, Cederbaum AI. CYP2E1 and oxidative liver injury by alcohol. Free Radic Biol Med. 2008;44:723–734. doi: 10.1016/j.freeradbiomed.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nanji AA, Zhao S, Sadrzadeh SM, Dannenberg AJ, Tahan SR, Waxman DJ. Markedly enhanced cytochrome P450E1 induction and lipid peroxidation is associated with severe liver injury in fish oil-ethanol-fed rats. Alcohol Clin Exp Res. 1994;18:1280–1285. doi: 10.1111/j.1530-0277.1994.tb00119.x. [DOI] [PubMed] [Google Scholar]
- 12.Tsukamoto H, Horne W, Kamimura S, Niemelä O, Parkkila S, Ylä-Herttuala S, et al. Experimental liver cirrhosis induced by alcohol and iron. J Clin Invest. 1995;96:620–630. doi: 10.1172/JCI118077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kessova IG, Ho YS, Thung S, Cederbaum AI. Alcohol-induced liver injury in mice lacking Cu, Zn-superoxide dismutase. Hepatology. 2003;38:1133–1145. doi: 10.1053/jhep.2003.50450. [DOI] [PubMed] [Google Scholar]
- 14.Kessova IG, Cederbaum AI. Mitochondrial alterations in livers of Sod1−/− mice fed alcohol. Free Radic Biol Med. 2007;42:1470–1480. doi: 10.1016/j.freeradbiomed.2007.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adachi M, Ishii H. Role of mitochondria in alcoholic liver injury. Free Radic Biol Med. 2002;32:487–491. doi: 10.1016/s0891-5849(02)00740-2. [DOI] [PubMed] [Google Scholar]
- 16.Bailey SM, Cunningham CC. Contribution of mitochondria to oxidative stress associated with alcoholic liver disease. Free Radic Biol Med. 2002;32:11–16. doi: 10.1016/s0891-5849(01)00769-9. [DOI] [PubMed] [Google Scholar]
- 17.Wu D, Cederbaum AI. Ethanol-induced apoptosis to stable HepG2 cell lines expressing human cytochrome P-4502E1. Alcohol Clin Exp Res. 1999;23:67–76. [PubMed] [Google Scholar]
- 18.Friedman SL. Hepatic stellate cells: protean multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88:125–172. doi: 10.1152/physrev.00013.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ankoma-Sey V, Matli M, Chang KB, Lalazar A, Donner DB, Wong L, et al. Coordinated induction of VEGF receptors in mesenchymal cell types during rat hepatic wound healing. Oncogene. 1988;17:115–121. doi: 10.1038/sj.onc.1201912. [DOI] [PubMed] [Google Scholar]
- 20.Shackel N, Rockey D. In pursuit of the “Holy Grail”-stem cells, hepatic injury, fibrogenesis and repair. Hepatology. 2005;41:16–18. doi: 10.1002/hep.20551. [DOI] [PubMed] [Google Scholar]
- 21.Novo E, Parola M. Redox mechanisms in hepatic chronic wound healing and fibrogenesis. Fibrog Tissue Repair. 2008;13:5–23. doi: 10.1186/1755-1536-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–1669. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muriel P. Cytokines in liver diseases. In: Sahu S, editor. Hepatotoxicity: From Genomics to In Vitro and In Vivo Models. West Sussex, UK: Wiley; 2007. pp. 371–389. [Google Scholar]
- 24.Wu J, Zern MA. Hepatic stellate cells: a target for the treatment of liver fibrosis. J Gastroenterol. 2000;35:665–672. doi: 10.1007/s005350070045. [DOI] [PubMed] [Google Scholar]
- 25.Bataller R, Schwabe RF, Choi YH, Yang L, Paik YH, Lindquist J, et al. NADPH oxidase signal transduces angiotensin II in hepatic stellate cells and is critical in hepatic fibrosis. J Clin Invest. 2003;112:1383–1394. doi: 10.1172/JCI18212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prosser CC, Yen RD, Wu J. Molecular therapy for hepatic injury and fibrosis: where are we? World J Gastroenterol. 2006;12:509–515. doi: 10.3748/wjg.v12.i4.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adachi T, Togashi H, Suzuki A, Kasai S, Ito J, Sugahara K, et al. NAD(P)H oxidase plays a crucial role in PDGF-induced proliferation of hepatic stellate cells. Hepatology. 2005;41:1272–1281. doi: 10.1002/hep.20719. [DOI] [PubMed] [Google Scholar]
- 28.Pinzani M, Gesualdo L, Sabbah GM, Abboud HE. Effects of platelet-derived growth factor polypeptide mitogens on DNA synthesis and growth of cultured rat liver fat-storing cells. J Clin Invest. 1989;84:1786–1793. doi: 10.1172/JCI114363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pisani P, Parkin DM, Bray F, Ferlay J. Estimates of the worldwide mortality from 25 cancers in 1990. Int J Cancer. 1999;83:18–29. doi: 10.1002/(sici)1097-0215(19990924)83:1<18::aid-ijc5>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 30.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 31.El-Serag HB, Mason AC. Rising incidence of hepatocellular carcinoma in the United States. N Engl J Med. 1999;340:745–750. doi: 10.1056/NEJM199903113401001. [DOI] [PubMed] [Google Scholar]
- 32.Dominguez-Malagon H, Gaytan-Graham S. Hepatocellular carcinoma: an update. Ultraestruct Pathol. 2001;25:497–516. doi: 10.1080/019131201753343539. [DOI] [PubMed] [Google Scholar]
- 33.Wang XW, Hussain SP, Huo TI, Wu CG, Forgues M, Hofseth LJ, et al. Molecular pathogenesis of human hepatocellular carcinoma. Toxicology. 2002;181–182:43–47. doi: 10.1016/s0300-483x(02)00253-6. [DOI] [PubMed] [Google Scholar]
- 34.Seitz HK, Stickel F. Risk factors and mechanisms of hepatocarcinogenesis with special emphasis on alcohol and oxidative stress. Biol Chem. 2006;387:349–360. doi: 10.1515/BC.2006.047. [DOI] [PubMed] [Google Scholar]
- 35.Marx J. Inflammation and cancer: the link grows stronger. Science. 2004;36:966–1008. doi: 10.1126/science.306.5698.966. [DOI] [PubMed] [Google Scholar]
- 36.Adelman R, Saul RL, Ames BN. Oxidative damage to DNA: relation to species metabolic rate and life span. Proc Natl Acad Sci USA. 1988;85:2706–2708. doi: 10.1073/pnas.85.8.2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ames BN. Mutagenesis and carcinogenesis: endogenous and exogenous factors. Environ Mol Mutagen. 1989;16:S66–S77. doi: 10.1002/em.2850140614. [DOI] [PubMed] [Google Scholar]
- 38.Troll W, Wiesner R. The role of oxygen radicals as a possible mechanism of tumor promotion. Annu Rev Pharmacol Toxicol. 1985;25:509–528. doi: 10.1146/annurev.pa.25.040185.002453. [DOI] [PubMed] [Google Scholar]
- 39.Kuchino Y, Mori F, Kasai InoueH, Iwai S, Miura K. et al. Misreading of DNA templates containing 8-hydroxydeoxyguanosine at the modified base and at adjacent residues. Nature. 1987;327:77–79. doi: 10.1038/327077a0. [DOI] [PubMed] [Google Scholar]
- 40.Seitz HK, Stickel F. Risk factors and mechanisms of hepatocarcinogenesis with special emphasis on alcohol and oxidative stress. Biol Chem. 2006;387:346–360. doi: 10.1515/BC.2006.047. [DOI] [PubMed] [Google Scholar]
- 41.Choi J, Ou JH. Mechanisms of liver injury. III. Oxidative stress in the pathogenesis of hepatitis C virus. Am J Physiol Gastrointest Liver Physiol. 2006;290:847–851. doi: 10.1152/ajpgi.00522.2005. [DOI] [PubMed] [Google Scholar]
- 42.Moriya K, Nakagawa K, Santa T, Shintani Y, Fujie H, Miyoshi H, et al. Oxidative stress in the absence of inflammation in a mouse model for hepatitis C virus-associated hepatocarcinogenesis. Cancer Res. 2001;61:4365–4370. [PubMed] [Google Scholar]
- 43.Farinati F, Cardin R, Bortolami M. Hepatitis C virus: from oxygen free radicals to hepatocellular carcinoma. J Viral Hepat. 2007;14:821–829. doi: 10.1111/j.1365-2893.2007.00878.x. [DOI] [PubMed] [Google Scholar]
- 44.Montalvo-Jave EE, Escalante-Tattersfield T, Ortega-Salgado JA, Piña E, Geller DA. Factors in the pathophysiology of the liver ischemia–reperfusion injury. J Surg Res. 2008;147:153–159. doi: 10.1016/j.jss.2007.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Powner DJ. Factors during donor care that may affect liver transplantation outcome. Prog Transplant. 2004;14:241–247. doi: 10.1177/152692480401400310. [DOI] [PubMed] [Google Scholar]
- 46.Serracino-Inglott F, Habib NA, Mathie RT. Hepatic ischemia–reperfusion injury. Am J Surg. 2001;181:160–166. doi: 10.1016/s0002-9610(00)00573-0. [DOI] [PubMed] [Google Scholar]
- 47.Henderson JM. Liver transplantation and rejection: an overview. Hepatogastroentelogy. 1999;46(Suppl 2):1482–1484. [PubMed] [Google Scholar]
- 48.Groot H, Rauen U. Ischemia–reperfusion injury: processes in pathogenetic networks: a review. Transplant Proc. 2007;39:481–484. doi: 10.1016/j.transproceed.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 49.Jaeschke H. Molecular mechanism of hepatic ischemia–reperfusion injury and preconditioning. Am J Physiol Gastrointest Liver Physiol. 2003;284:G15–G26. doi: 10.1152/ajpgi.00342.2002. [DOI] [PubMed] [Google Scholar]
- 50.Malhi H, Gores GJ, Lemasters JJ. Apoptosis and necrosis in the liver: a tale of two deads? Hepatology. 2006;43:S31–S44. doi: 10.1002/hep.21062. [DOI] [PubMed] [Google Scholar]
- 51.Anaya-Prado R, Toledo-Pereyra LH, Lentsch AB, Ward PA. Ischemia/reperfusion injury. J Surg Res. 2002;105:248–258. doi: 10.1006/jsre.2002.6385. [DOI] [PubMed] [Google Scholar]
- 52.McCord JM. Oxygen-derived radicals: a link between reperfusion injury and inflammation. Fed Proc. 1987;46:2402–2406. [PubMed] [Google Scholar]
- 53.Selzner N, Rudiger H, Graf R, Clavien PA. Protective strategies against ischemic injury of the liver. Gastroenterology. 2003;125:917–936. doi: 10.1016/s0016-5085(03)01048-5. [DOI] [PubMed] [Google Scholar]
- 54.Caldwell-Kenkel J, Currin R, Tanaka Y, Thurman R, Lemasters J. Kupffer cell activation and endothelial cell damage after storage of rat livers: effects of reperfusion. Hepatology. 1991;13:83–95. [PubMed] [Google Scholar]
- 55.Jaeschke H, Farhood A. Neutrophil and Kupffer cell-induced oxidant stress and ischemia–reperfusion injury in rat liver in vivo. Am J Physiol. 1991;260:G355–G362. doi: 10.1152/ajpgi.1991.260.3.G355. [DOI] [PubMed] [Google Scholar]
- 56.Adamson G, Billings R. Tumor necrosis factor induced oxidative stress in isolated mouse hepatocytes. Arch Biochem Biophys. 1992;294:223–229. doi: 10.1016/0003-9861(92)90161-o. [DOI] [PubMed] [Google Scholar]
- 57.Jaeschke H, Mitchell J. Mitochondria and xanthine oxidase both generate reactive oxygen species after hypoxic damage in isolated perfused rat liver. Biochem Biophys Res Commun. 1989;160:140–147. doi: 10.1016/0006-291x(89)91632-x. [DOI] [PubMed] [Google Scholar]
- 58.Nordstrom G, Seeman T, Hasselgren PO. Beneficial effect of allopurinol in liver ischemia. Surgery. 1985;97:679–984. [PubMed] [Google Scholar]
- 59.Kusumoto K, Morimoto T, Minor T, Uchino J, Isselhard W. Allopurinol effects in rat liver transplantation on recovery of energy metabolism and free radical-induced damage. Eur Surg Res. 1995;27:285–291. doi: 10.1159/000129411. [DOI] [PubMed] [Google Scholar]
- 60.Marubayashi S, Dohi K, Ochi K, Kawasaki T. Protective effects of free radical scavenger and antioxidant administration on ischemic liver cell injury. Transplant Proc. 1987;19:1327–1328. [PubMed] [Google Scholar]
- 61.Koeppel TA, Lehmann TG, Thies JC, Gehrcke R, Gebhard MM, Herfarth C, et al. Impact of N-acetylcysteine on the hepatic microcirculation after orthotopic liver transplantation. Transplantation. 1996;61:1397–1402. doi: 10.1097/00007890-199605150-00020. [DOI] [PubMed] [Google Scholar]
- 62.Mizoe A, Kondo S, Azuma T, Fujioka H, Tanaka K, Hashida M, et al. Preventive effects of superoxide dismutase derivatives modified with monosaccharides on reperfusion injury in rat liver transplantation. J Surg Res. 1997;73:160–165. doi: 10.1006/jsre.1997.5215. [DOI] [PubMed] [Google Scholar]
- 63.Younes M, Strubelt O. The involvement of reactive oxygen species in hypoxic injury to rat liver. Res Commun Chem Pathol Pharmacol. 1988;59:369–381. [PubMed] [Google Scholar]
- 64.Marubayashi S, Dohi K, Yamada K, Kawasaki T. Changes in the levels of endogenous coenzyme Q homologs, alpha-tocopherol, and glutathione in rat liver after hepatic ischemia and reperfusion, and the effect of pretreatment with coenzyme Q10. Biochim Biophys Acta. 1984;797:1–9. [PubMed] [Google Scholar]
- 65.Larson AM, Polson J, Fontana RJ, Davern TJ, Lalani E, Hynan LS, et al. Acute Liver Failure Study Group. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology. 2005;42:1364–1372. doi: 10.1002/hep.20948. [DOI] [PubMed] [Google Scholar]
- 66.Nelson SD. Molecular mechanisms of the hepatotoxicity caused by acetaminophen. Semin Liver Dis. 1990;10:267–278. doi: 10.1055/s-2008-1040482. [DOI] [PubMed] [Google Scholar]
- 67.Jaeschke H, Bajt ML. Intracellular signaling mechanism of acetaminophen-induced liver cell death. Toxicol Sci. 2006;89:31–41. doi: 10.1093/toxsci/kfi336. [DOI] [PubMed] [Google Scholar]
- 68.Pascual C, González R, Armesto J, Muriel P. Effect of silymarin and silybinin on oxygen radicals. Drug Dev Res. 1993;29:73–77. [Google Scholar]
- 69.Muriel P, Garciapiña T, Pérez-Alvarez V, Mourelle M. Silymarin protects against paracetamol-induced lipid peroxidation and liver damage. J Appl Toxicol. 1992;12:439–442. doi: 10.1002/jat.2550120613. [DOI] [PubMed] [Google Scholar]
- 70.Polson J, Lee WM. American Association for the Study of Liver Diseases. AASLD position paper: the management of acute liver failure. Hepatology. 2005;41:1179–1197. doi: 10.1002/hep.20703. [DOI] [PubMed] [Google Scholar]
- 71.Choi J, Ou JH. Mechanism of liver injury: III. Oxidative stress in the pathogenesis of hepatitis C virus. Am J Physiol Gastrointest Liver Physiol. 2006;290:G847–G851. doi: 10.1152/ajpgi.00522.2005. [DOI] [PubMed] [Google Scholar]
- 72.Kageyama F, Kobayashi Y, Kawasaki T, Toyokuni S, Uchida K, Nakamura H. Successful interferon therapy reverses enhanced hepatic iron accumulation and lipid peroxidation in chronic hepatitis C. Am J Gastroenterol. 2000;95:1041–1050. doi: 10.1111/j.1572-0241.2000.01979.x. [DOI] [PubMed] [Google Scholar]
- 73.Jain SK, Pemberton PW, Smith A, McMahon RF, Burrows PC, Aboutwerat A, et al. Oxidative stress in chronic hepatitis C: not just a feature of late stage disease. J Hepatol. 2002;36:805–811. doi: 10.1016/s0168-8278(02)00060-0. [DOI] [PubMed] [Google Scholar]
- 74.Mahmood S, Kawanaka M, Kamei A, Izumi A, Nakata K, Niiyama G, et al. Immunohistochemical evaluation of oxidative stress markers in chronic hepatitis C. Antioxid Redox Signal. 2004;6:19–24. doi: 10.1089/152308604771978318. [DOI] [PubMed] [Google Scholar]
- 75.Farinati F, Cardin R, Maria N, Della Libera G, Marafin C, Lecis E, et al. Iron storage, lipid peroxidation and glutathione turnover in chronic anti-HCV positive hepatitis. J Hepatol. 1995;22:449–456. doi: 10.1016/0168-8278(95)80108-1. [DOI] [PubMed] [Google Scholar]
- 76.Higueras V, Raya A, Rodrigo JM, Serra MA, Roma J, Romero FJ. Interferon decreases serum lipid peroxidation products of hepatitis C patients. Free Radic Biol Med. 1994;16:131–133. doi: 10.1016/0891-5849(94)90250-x. [DOI] [PubMed] [Google Scholar]
- 77.Mutlu-Turkoglu U, Ademoglu E, Turkoglu S, Badur S, Uysal M, Toker G. The effects of interferon-alpha on serum lipid peroxidation and total thiol content in patients with chronic active hepatitis-C. Res Commun Mol Pathol Pharmacol. 1997;96:357–361. [PubMed] [Google Scholar]
- 78.Swietek K, Juszczyk J. Reduced glutathione concentration in erythrocytes of patients with acute and chronic viral hepatitis. J Viral Hepat. 1997;4:139–141. doi: 10.1111/j.1365-2893.1997.tb00217.x. [DOI] [PubMed] [Google Scholar]
- 79.Konishi M, Iwasa M, Araki J. Increased lipid peroxidation in patients with non-alcoholic fatty liver disease and chronic hepatitis C as measured by the plasma level of 8-isoprostane. J Gastroenterol Hepatol. 2006;21:1821–1825. doi: 10.1111/j.1440-1746.2006.04420.x. [DOI] [PubMed] [Google Scholar]
- 80.Paradis V, Mathurin P, Kollinger M. In situ detection of lipid peroxidation in chronic hepatitis C: correlation with pathological features. J Clin Pathol. 1997;50:401–406. doi: 10.1136/jcp.50.5.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Farinati F, Cardin R, Degan P, Maria N, Floyd RA, Thiel DH, et al. Oxidative DNA damage in circulating leukocytes occurs as an early event in chronic HCV infection. Free Radic Biol Med. 1999;27:1284–1291. doi: 10.1016/s0891-5849(99)00161-6. [DOI] [PubMed] [Google Scholar]
- 82.Hara Y, Hino K, Okuda M, Furutani T, Hidaka I, Yamaguchi Y, et al. Hepatitis C virus core protein inhibits deoxycholic acid-mediated apoptosis despite generating mitochondrial reactive oxygen species. J Gastroenterol. 2006;41:257–268. doi: 10.1007/s00535-005-1738-1. [DOI] [PubMed] [Google Scholar]
- 83.Seronello S, Sheikh MY, Choi J. Redox regulation of hepatitis C in nonalcoholic and alcoholic liver. Free Radic Biol Med. 2007;43:869–882. doi: 10.1016/j.freeradbiomed.2007.05.036. [DOI] [PubMed] [Google Scholar]
- 84.Izumi N, Enomoto N, Uchihara M, Murakami T, Ono K, Noguchi O, et al. Hepatic iron contents and response to interferon-alpha in patients with chronic hepatitis C: relationship to genotypes of hepatitis C virus. Dig Dis Sci. 1996;41:989–994. doi: 10.1007/BF02091542. [DOI] [PubMed] [Google Scholar]
- 85.Poli G. Pathogenesis of liver fibrosis: role of oxidative stress. Mol Aspects Med. 2000;21:49–98. doi: 10.1016/s0098-2997(00)00004-2. [DOI] [PubMed] [Google Scholar]
- 86.Koike N, Takamura T, Kaneko S. Induction of reactive species from isolated rat glomeruli by protein kinase C activation and TNF-alpha stimulation, and effects of a phosphodiesterase inhibitor. Life Sci. 2007;80:1721–1728. doi: 10.1016/j.lfs.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 87.Garcia-Trevijano ER, Iraburu MJ, Fontana L, Domínguez-Rosales JA, Auster A, Covarrubias-Pinedo A, et al. Transforming growth factor beta 1 induces the expression of alpha(I) procollagen mRNA by a hydrogen peroxide-C/EBPbeta-dependent mechanism in rat hepatic stellate cells. Hepatology. 1999;29:910–960. doi: 10.1002/hep.510290346. [DOI] [PubMed] [Google Scholar]
- 88.Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to liver injury. J Biol Chem. 2000;275:2247–2250. doi: 10.1074/jbc.275.4.2247. [DOI] [PubMed] [Google Scholar]
- 89.Kota RS, Ramana CV, Tenorio FA, Enelow RI, Rutledge JC. Differential effects of lipoprotein lipase on tumor necrosis factor-alpha and interferon-gamma-mediated gene expression in human endothelial cells. J Biol Chem. 2005;280:31076–31084. doi: 10.1074/jbc.M412189200. [DOI] [PubMed] [Google Scholar]
- 90.Liu C, Gaca MD, Swenson ES, Vellucci VF, Reiss M, Wells RG. Smads 2 and 3 are differentially activated by transforming growth factor-beta (TGF-beta) in quiescent and activated hepatic stellate cells: constitutive nuclear localization of Smads in activated cells is TGF-beta-independent. J Biol Chem. 2003;278:11721–11728. doi: 10.1074/jbc.M207728200. [DOI] [PubMed] [Google Scholar]
- 91.Korenagua M, Wang T, Li Y, Showalter LA, Chang T, Sun J, et al. Hepatitis virus core protein inhibits mitochondrial electron transport and increases ROS production. J Biol Chem. 2005;280:37481–37488. doi: 10.1074/jbc.M506412200. [DOI] [PubMed] [Google Scholar]
- 92.Abdalla MY, Ahmad IM, Spitz DR, Schmidt WN, Britigan BE. Hepatitis C virus-core and non structural proteins lead to different effects on cellular antioxidant defenses. J Med Virol. 2005;76:489–497. doi: 10.1002/jmv.20388. [DOI] [PubMed] [Google Scholar]
- 93.Okuda M, Li K, Beard MR, Showalter LA, Scholle F, Lemon SM, et al. Mitochondrial injury, oxidative stress, and antioxidant gene expressions are induced by hepatitis C core proteins. Gastroenterology. 2002;122:366–375. doi: 10.1053/gast.2002.30983. [DOI] [PubMed] [Google Scholar]
- 94.Li K, Prow T, Lemon SM, Beard MR. Cellular response to conditional expression of hepatitis C virus core protein in Huh7 cultured human hepatoma cells. Hepatology. 2002;35:1237–1246. doi: 10.1053/jhep.2002.32968. [DOI] [PubMed] [Google Scholar]
- 95.Perlemuter G, Letteron P, Carnot F, Zavala F, Pessayre D, Nalpas B, et al. Alcohol and hepatitis C virus core protein additively increase lipid peroxidation and synergistically trigger hepatic cytokine expression in a transgenic mouse model. J Hepatol. 2003;39:1020–1027. doi: 10.1016/s0168-8278(03)00414-8. [DOI] [PubMed] [Google Scholar]
- 96.Li Y, Boehning DF, Qian T, Popov VL, Weinman SA. Hepatitis C virus core protein increases mitochondrial ROS production by stimulation of Ca2+ uniporter activity. FASEB J. 2007;21:2474–2485. doi: 10.1096/fj.06-7345com. [DOI] [PubMed] [Google Scholar]
- 97.Gochee PA, Jonsson JR, Clouston AD, Pandeya N, Purdie DM, Powell EE. Steatosis in chronic hepatitis C: association with increased messenger RNA expression of collagen I, tumor necrosis factor-alpha and cytochrome P450 2E1. J Gastroenterol Hepatol. 2003;18:386–392. doi: 10.1046/j.1440-1746.2003.02984.x. [DOI] [PubMed] [Google Scholar]
- 98.Lucas S, Bartolome J, Amaro MJ, Carreno V. Hepatitis C virus core protein transactivates the inducible nitric oxide synthase promoter via NF-kappaB activation. Antiviral Res. 2003;60:117–124. doi: 10.1016/j.antiviral.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 99.Garcia-Mediavilla MV, Sanchez-Campos S, Gonzalez-Perez P, Gómez-Gonzalo M, Majano PL, López-Cabrera M, et al. Differential contribution of hepatitis C virus NS5A and core proteins to the induction of oxidative and nitrosative stress in human hepatocyte-derived cells. J Hepatol. 2005;43:606–613. doi: 10.1016/j.jhep.2005.04.019. [DOI] [PubMed] [Google Scholar]
- 100.Waris G, Siddiqui A. Hepatitis C virus stimulates the expression of cyclooxigenase-2 via oxidative stress: role of prostaglandin E2 in RNA replication. J Virol. 2005;79:9725–9734. doi: 10.1128/JVI.79.15.9725-9734.2005. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 101.Nunez O, Fernandez-Martinez A, Majano PL, Apolinario A, Gómez-Gonzalo M, Benedicto I, et al. Increased intrahepatic cyclooxygenase 2, matrix metalloproteinase 2 and matrix metalloproteinase 9 expression is associated with progressive liver disease in chronic hepatitis C virus infection: role of viral core and NS5A proteins. Gut. 2004;53:1665–1672. doi: 10.1136/gut.2003.038364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Miller K, McArdle S, Gale MJ, Jr, Geller DA, Tenoever B, Hiscott J, et al. Effects of the hepatitis C virus core protein on innate cellular defense pathways. J Interferon Cytokine Res. 2004;24:391–402. doi: 10.1089/1079990041535647. [DOI] [PubMed] [Google Scholar]
- 103.Machida K, Cheng KT, Sung VM, Lee KJ, Levine AM, Lai MM. Hepatitis C virus infection activates the immunologic (type II) isoform of nitric oxide synthase and thereby enhances DNA damage and mutations of cellular genes. J Virol. 2004;78:8835–8843. doi: 10.1128/JVI.78.16.8835-8843.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gong G, Waris G, Tanveer R, Siddiqui A. Human hepatitis C virus NS5A protein alters intracellular calcium levels, induces oxidative stress, and activates STAT-3 and NF-kappa B. Proc Natl Acad Sci USA. 2001;98:9599–9604. doi: 10.1073/pnas.171311298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Qadri I, Iwahashi M, Capasso JM, Hopken MW, Flores S, Schaack J, et al. Induced oxidative stress and activated expression of manganese superoxide dismutase during hepatitis C virus replication: role of JNK, p38 MAPK and AP1. Biochem J. 2004;378:919–928. doi: 10.1042/BJ20031587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Machida K, Cheng KT, Sung VM. Hepatitis C virus induces a mutator phenotype: enhanced mutations of immunoglobulin and protooncogenes. Proc Natl Acad Sci USA. 2004;101:4262–4267. doi: 10.1073/pnas.0303971101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Moriya K, Fujie H, Shintani Y, Yotsuyanagi H, Tsutsumi T, Ishibashi K, et al. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat Med. 1998;4:1065–1067. doi: 10.1038/2053. [DOI] [PubMed] [Google Scholar]
- 108.Brune B. The intimate relation between nitric oxide and superoxide in apoptosis and cell survival. Antioxid Redox Signal. 2005;7:497–507. doi: 10.1089/ars.2005.7.497. [DOI] [PubMed] [Google Scholar]
- 109.Melhem A, Stern M, Shibolet O, Israeli E, Ackerman Z, Pappo O, et al. Treatment of chronic hepatitis C virus infection via antioxidants: results of a phase I clinical trial. J Clin Gastroenterol. 2005;39:737–742. doi: 10.1097/01.mcg.0000174023.73472.29. [DOI] [PubMed] [Google Scholar]
- 110.Lieber CS. CYP2E1 from ASH to NASH. Hepatol Res. 2004;28:1–11. doi: 10.1016/j.hepres.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 111.Leclercq IA, Farrell GC, Field J, Bell DR, Gonzalez FJ, Robertson GR. CY2P2E1 and CYP4A as microsomal catalysts of lipid peroxides in murine nonalcoholic steatohepatitis. J Clin Invest. 2000;105:1067–1075. doi: 10.1172/JCI8814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.George J, Pera N, Phung N, Leclercq I, Yun Hou J, Farrell G. Lipid peroxidation stellate cells activation and hepatic fibrogenesis in a rat model of chronic steatohepatitis. J Hepatol. 2003;39:756–764. doi: 10.1016/s0168-8278(03)00376-3. [DOI] [PubMed] [Google Scholar]
- 113.Sanyal AJ, Campbell-Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, et al. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology. 2001;120:1183–1192. doi: 10.1053/gast.2001.23256. [DOI] [PubMed] [Google Scholar]
- 114.Garcia-Monson C, Martin Perez E, Lojacono O. Characterization of pathogenic and prognostic factors of nonalcoholic steatohepatitis associated with obesity. J Hepatol. 2000;33:716–724. doi: 10.1016/s0168-8278(00)80301-3. [DOI] [PubMed] [Google Scholar]
- 115.Videla LA, Rodrigo R, Orelland M. Oxidation stress-related parameters in the liver of non-alcoholic fatty liver disease patients. Clin Sci. 2004;106:261–268. doi: 10.1042/CS20030285. [DOI] [PubMed] [Google Scholar]
- 116.Koruk M, Taysi S, Savas MC, Yilmaz O, Akcay F, Karakok M. Oxidative stress and enzymatic antioxidant status in patients with non alcoholic steatohepatitis. Ann Clin Lab Sci. 2004;34:57–62. [PubMed] [Google Scholar]
- 117.Loguercio C, Girolamo V, Sio I, Tuccillo C, Ascione A, Baldi F, et al. Non-alcoholic fatty liver disease in an area of southern Italy: main clinical histological, and pathophysiological aspects. J Hepatol. 2001;35:568–574. doi: 10.1016/s0168-8278(01)00192-1. [DOI] [PubMed] [Google Scholar]
- 118.Marchesini G, Brizi M, Bianchi G, Tomassetti S, Bugianesi E, Lenzi M, et al. Nonalcoholic fatty liver disease: a feature of the metabolic syndrome. Diabetes. 2001;50:1844–1850. doi: 10.2337/diabetes.50.8.1844. [DOI] [PubMed] [Google Scholar]
- 119.Nehra V, Angulo P, Buchman AL, Lindor KD. Nutritional and metabolic considerations in the etiology of non-alcoholic steatohepatitis. Dig Dis Sci. 2001;46:2347–2352. doi: 10.1023/a:1012338828418. [DOI] [PubMed] [Google Scholar]
- 120.Mavrelis PG, Ammon HV, Gleysteen JJ, Komorowski RA, Charaf UK. Hepatic free fatty acids in alcoholic liver disease and obesity. Hepatology. 1983;3:226–231. doi: 10.1002/hep.1840030215. [DOI] [PubMed] [Google Scholar]
- 121.Clarke SD. Nonalcoholic steatosis and steatohepatitis, part I: molecular mechanism for polyunsaturated fatty acid regulation of gene transcription. Am J Physiol Gastrointest Liver Physiol. 2001;281:G865–G869. doi: 10.1152/ajpgi.2001.281.4.G865. [DOI] [PubMed] [Google Scholar]
- 122.Parola M, Robino G. Oxidative stress-related molecules and liver fibrosis. J Hepatol. 2001;35:297–306. doi: 10.1016/s0168-8278(01)00142-8. [DOI] [PubMed] [Google Scholar]
- 123.Goldstein BJ, Kalyankar M, Wu X. Insulin action is facilitated by insulin-stimulated reactive oxygen species with multiple potential signaling targets. Diabetes. 2005;54:311–321. doi: 10.2337/diabetes.54.2.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wanless JR, Bargman JM, Oreopoullos DG. Subcapsular steatonecrosis in response to peritoneal insulin deliver: a clue to the pathogenesis of steatonecrosis in obesity. Mod Pathol. 1989;2:69–74. [PubMed] [Google Scholar]
- 125.Khalili K, Lan FP, Hanbidge AE, Muradali D, Oreopoulos DG, Wanless IR. Hepatic subcapsular steatosis in response to intraperitoneal insulin delivery: CT findings and prevalence. AJR Am J Roentgenol. 2003;180:1601–1604. doi: 10.2214/ajr.180.6.1801601. [DOI] [PubMed] [Google Scholar]
- 126.Paradis V, Perlemuter G, Benvoust F, Dargere D, Parfait B, Vidaud M, et al. High glucose and hyperinsulinemia stimulate connective growth factor expression: a potential mechanism involved in progression to fibrosis in nonalcoholic steatohepatitis. Hepatology. 2001;34:738–744. doi: 10.1053/jhep.2001.28055. [DOI] [PubMed] [Google Scholar]
- 127.Matteoni CA, Younossi ZM, Gramlich T, Boparai N, Liu YC, McCullough AJ. Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology. 1999;116:1413–1419. doi: 10.1016/s0016-5085(99)70506-8. [DOI] [PubMed] [Google Scholar]
- 128.Younossi ZM, Gramlich T, Matteoni CA, Boparai N, McCullough AJ. Nonalcoholic fatty liver disease in patients with type 2 diabetes. Clin Gastroenterol Hepatol. 2004;2:262–265. doi: 10.1016/s1542-3565(04)00014-x. [DOI] [PubMed] [Google Scholar]
- 129.El-Serag HR, Tran T, Everhart JE. Diabetes increases the risk of chronic liver disease and hepatocellular carcinoma. Gastroenterology. 2004;126:460–468. doi: 10.1053/j.gastro.2003.10.065. [DOI] [PubMed] [Google Scholar]
- 130.Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, et al. Endoplasmic reticulum stress links obesity, insulin action and type 2 diabetes. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 131.Muoio DM. Insulin resistance takes a trip through the ER. Science. 2004;306:4285–4426. doi: 10.1126/science.1104680. [DOI] [PubMed] [Google Scholar]
- 132.George DK, Goldwurm S, MacDonald GA, Cowley LL, Walker NI, Ward PJ, et al. Increased hepatic iron concentration in nonalcoholic steatohepatitis is associated with increased fibrosis. Gastroenterology. 1998;114:311–318. doi: 10.1016/s0016-5085(98)70482-2. [DOI] [PubMed] [Google Scholar]
- 133.Bonkovsky HL, Jawaid Q, Tortorelli K, LeClair P, Cobb J, Lambrecht RW, et al. Nonalcoholic steatohepatitis and iron increased prevalence of mutations of HFE gene in nonalcoholic steatohepatitis. J Hepatol. 1999;31:421–429. doi: 10.1016/s0168-8278(99)80032-4. [DOI] [PubMed] [Google Scholar]
- 134.Bacon BR, Farahvash MJ, Janney CG, Neuschwander-Tetri BA. Nonalcoholic steatohepatitis: an expanded clinical entity. Gastroenterology. 1994;107:1103–1109. doi: 10.1016/0016-5085(94)90235-6. [DOI] [PubMed] [Google Scholar]
- 135.Angulo P, Keach JC, Batts KP, Lindor KD. Independent predictors of liver fibrosis in patients with nonalcoholic steatohepatitis. Hepatology. 1999;30:1356–1362. doi: 10.1002/hep.510300604. [DOI] [PubMed] [Google Scholar]
- 136.Younossi ZM, Gramlich T, Bacon BR, Matteoni CA, Boparai N, O’Neill R, et al. Hepatic iron and nonalcoholic fatty liver disease. Hepatology. 1999;30:847–850. doi: 10.1002/hep.510300407. [DOI] [PubMed] [Google Scholar]
- 137.Chitturi S, Weltman M, Farrell GC, McDonald D, Kench J, Liddle C, et al. HFE mutations, hepatic iron, and fibrosis: ethnic-specific association of NASH with C282Y but not with fibrotic severity. Hepatology. 2002;36:142–149. doi: 10.1053/jhep.2002.33892. [DOI] [PubMed] [Google Scholar]
- 138.Bugianesi E, Manzini P, D’Antico S, Vanni E, Longo F, Leone N, et al. Relative contribution of iron burden, HFE mutations, and insulin resistance to fibrosis in nonalcoholic fatty liver. Hepatology. 2004;39:179–187. doi: 10.1002/hep.20023. [DOI] [PubMed] [Google Scholar]
- 139.Fargion S, Mattioli M, Fracanzani AL, Sampietro M, Tavazzi D, Fociani P, et al. Hyperferritinemia, iron overload, and multiple metabolic alterations identify patients at risk for nonalcoholic steatohepatitis. Am J Gastroenterol. 2001;96:2448–2455. doi: 10.1111/j.1572-0241.2001.04052.x. [DOI] [PubMed] [Google Scholar]
- 140.Fernández-Real JM, Ricart-Engel W, Arroyo E, Balançá R, Casamitjana-Abella R, Cabrero D, et al. Serum ferritin as a component of the insulin resistance syndrome. Diabetes Care. 1998;21:62–68. doi: 10.2337/diacare.21.1.62. [DOI] [PubMed] [Google Scholar]
- 141.Mendler MH, Turlin B, Moirand R, Jouanolle AM, Sapey T, Guyader D, et al. Insulin resistance-associated hepatic iron overload. Gastroenterology. 1999;117:1115–1163. doi: 10.1016/s0016-5085(99)70401-4. [DOI] [PubMed] [Google Scholar]
- 142.Macdonald GA, Powell LW. More clues to the relationship between hepatic iron and steatosis: an association with insulin resistance? Gastroenterology. 1999;117:1241–1244. doi: 10.1016/s0016-5085(99)70412-9. [DOI] [PubMed] [Google Scholar]
- 143.Dinneen SF, Silverberg JD, Batts KP, O’Brien PC, Ballard DJ, Rizza RA. Liver iron stores in patients with non-insulin-dependent diabetes mellitus. Mayo Clin Proc. 1994;69:13–15. doi: 10.1016/s0025-6196(12)61605-x. [DOI] [PubMed] [Google Scholar]
- 144.Rauen U, Petrat F, Sustmann R, Groot H. Iron-induced mitochondrial permeability transition in cultured hepatocytes. J Hepatol. 2004;40:607–615. doi: 10.1016/j.jhep.2003.12.021. [DOI] [PubMed] [Google Scholar]
- 145.Bulteau AL, O’Neill HA, Kennedy MC, Ikeda-Saito M, Isaya G, Szweda LI. Frataxin acts as an iron chaperone protein to modulate mitochondrial aconitase activity. Science. 2004;305:242–245. doi: 10.1126/science.1098991. [DOI] [PubMed] [Google Scholar]