

Abstract
The Escherichia coli RNA degradosome is a protein complex that plays a critical role in the turnover of numerous RNAs. The key component of the degradosome complex is the endoribonuclease RNase E, a multidomain protein composed of an N-terminal catalytic region and a C-terminal region that organizes the other protein components of the degradosome. Previously, the RNase E inhibitors RraA and RraB were identified genetically and shown to bind to the C-terminal region of RNase E, thus affecting both the protein composition of the degradosome and the endonucleolytic activity of RNase E. In the present work, we investigated the transcriptional regulation of rraB. rraB was shown to be transcribed constitutively from its own promoter, PrraB. Transposon mutagenesis and screening for increased β-galactosidase activity from a chromosomal PrraB-lacZ transcriptional fusion resulted in the isolation of a transposon insertion in glmS, encoding the essential enzyme glucosamine-6-phosphate synthase that catalyzes the first committed step of the uridine 5′-diphospho-N-acetyl-glucosamine (UDP-GlcNAc) pathway, which provides intermediates for peptidoglycan biogenesis. The glmS852::Tn5 allele resulted in an approximately 50% lower intracellular concentration of UDP-GlcNAc and conferred a fivefold increase in the level of rraB mRNA. This allele also mediated a twofold increase in β-galactosidase activity from a chromosomal fusion of the 5′ untranslated region of the rne gene to lacZ, suggesting that a reduction in cellular concentration of UDP-GlcNAc and the resulting increased expression of RraB might modulate the action of RNase E.
The endoribonuclease RNase E plays a central role in RNA metabolism, including the processing of rRNAs and tRNAs (30, 42, 43); the degradation of small regulatory RNAs; and, most importantly, the turnover of numerous cellular mRNAs in Escherichia coli (5, 13, 23, 33). Homologous RNase E has been identified in more than 50 bacteria, archaea, and plants (28). The 1,061-amino-acid E. coli RNase E protein can be divided into functional portions, from the N terminus to the C terminus, as follows. The N-terminal half (amino acid residues 1 to 529) contains the endonuclease active site (amino acid residues 1 to 395) and a zinc finger region (amino acid residues 400 to 415) (8, 9). The central region includes the membrane anchoring segment A (amino acid residues 565 to 582) and its flanking portions (25) as well as an arginine-rich RNA binding site (amino acid residues 604 to 688) (34). The C-terminal half (amino acid residues 734 to 1061) is an unstructured scaffold domain that contains binding sites for the other core degradosome components, namely, polynucleotide phosphorylase, the RhlB helicase, and the glycolytic enzyme enolase (11, 35, 45, 46, 53). Previous studies indicate that the assembled degradosome complex is necessary for normal mRNA degradation and the degradosome components functionally interact during decay of at least some RNAs in E. coli (4, 24).
The cellular level and activity of RNase E in E. coli are under complex regulation. First, the efficiency of RNase E cleavage depends on structural features of RNA substrates and the factors that affect the accessibility of putative cleavage sites. A 5′ monophosphate in substrate RNAs serves as an allosteric activator of the endonuclease activity (10, 21). Interactions of RNA substrates with Hfq and small RNAs exert an important role on the cleavage of certain mRNAs (1, 7, 39). Second, RNase E autoregulates its synthesis by modulating the decay of its own mRNA. Previous studies showed that the 5′ untranslated region (UTR) of rne mRNA is subject to RNase E degradation; i.e., a higher cellular level of RNase E results in faster degradation of its transcript, which in turn leads to a reduction in the RNase E level (21, 40). Third, the membrane localization of RNase E and its association with the bacterial cytoskeleton may affect its function through various mechanisms (25, 31, 51). Finally, our lab has shown that the activity of RNase E is affected globally by the endoribonuclease binding proteins RraA and RraB, which control the decay and abundance of large numbers of bacterial mRNAs in trans (16, 29, 54, 56).
RraB, previously annotated as YjgD in the NCBI database, is a 15.6-kDa protein that interacts with RNase E and inhibits RNase E endonucleolytic cleavages in E. coli. In contrast to RraA homologues, which exist in numerous bacterial genomes, RraB is found only in gammaproteobacteria, suggesting that the latter protein may have a more specialized role in modulating RNA degradation. Similar to RraA, RraB does not alter RNase E cleavage site specificity or interact detectably with the substrate RNAs. RraB also possesses the same affinity for RNase E as does RraA. However, RraB exhibits key differences in its mode of action and its effects on the transcript profile. RraB interacts with residues 694 to 727 at the C-terminal half of RNase E (16). Overexpression of RraB from a high-copy plasmid induces specific changes in degradosome composition that are distinct from those mediated by RraA overexpression. Importantly, the action of RraB results in a dramatic change in the global abundance of mRNAs that is different from that obtained through the action of RraA (16). The existence of two cellular proteins that exert differential effects on RNA decay via their interactions with RNase E and degradosome remodeling argues that modulation of RNA stability may be a mechanism for global control of transcript abundance in response to dynamic changes in the environment.
The E. coli rraB gene is 417 bp in length and is located at 96.4 min on the chromosome, adjacent to argI, which is transcribed from the opposite strand and codes for ornithine carbamoyltransferase I. Here, we show that rraB is transcribed from its own promoter (PrraB), which is divergent from the argI promoter and overlaps with the arginine repressor binding site (ARG box) (32) located in the argI-rraB intergenic region. However, we found no evidence that the promoter of rraB is regulated by the transcription factor ArgR or the availability of arginine. A screen for transposon insertions that enhance the β-galactosidase activity from a Φ(PrraB-lacZ) transcriptional fusion led to the isolation of a glmS852::Tn5 allele that upregulated rraB transcription by about fivefold. The glmS852::Tn5 allele resulted in a decrease in the cellular uridine 5′-diphospho-N-acetyl-glucosamine (UDP-GlcNAc) level to less than 50% of the level observed in the parental strain. Furthermore, β-galactosidase activity from an rne-lacZ fusion increases in the glmS852::Tn5 mutant. Since the rne-lacZ transcript is a substrate of RNase E, the increase in β-galactosidase activity indicates that the rne-lacZ fusion is stabilized as a result of a reduction in RNase E activity, presumably a consequence of enhanced inhibition by the increased expression of RraB.
MATERIALS AND METHODS
Strains and plasmids.
The strains, plasmids, and phage vectors used in this study are listed in Table 1. The plasmid pBAD30-glmS was constructed as follows. The glmS gene coding sequence was amplified by PCR from genomic DNA of strain MC4100 by the use of the primers glmS5 (5′-CGGCGGGGTACCAGGAGGTTACGATGTGTGGAATTGTTGGC-3′) and glmS3 (5′-CGGCCCAAGCTTTTACTCAACCGTAACCGATTTTGCCAGGTT-3′). The PCR product was digested with KpnI and HindIII and then ligated into the pBAD30 plasmid, which was similarly digested.
TABLE 1.
Strains, plasmids and phage vectors
| Strain, plasmid, or phage vector | Description | Reference or source |
|---|---|---|
| BW21116 | Δlac-169 creC510 hsdR514 uidA(ΔMluI)::pir+ | 19 |
| CAG51025 | MC1061 φλ[rpoH P3::lacZ] ΔrseB nadB3140::Tn10 ΔdegS arg::Tn5 | 19 |
| CJ1825 | MC1061 (λez1) | 16 |
| DHB4 | (ara-leu)7697 araD139 ΔlacX74 galE galK rpsL phoR (phoA)PvuII malF3 thi/F lac-pro lacIq | 14 |
| EC-O | thi-1 relA1 Δ(pro-lac)X113 supE44/F42-114(FTs) lac | 49 |
| JCB570 | MC1000 phoR zih12::Tn10 | 3 |
| JCB571 | MC1000 phoR zih12::Tn10 dsbA::kan | 6 |
| KS474 | F−lacX74 galE galK thi rpsL(strR) ΔphoA(PvuII) degP41(ΔPstl)::ΩkanR | 50 |
| LZ001 | P90C λ (−152 to −1 nt)rraB-lacZ | This study |
| LZ002 | P90C λ (−152 to −1 nt)rraB-lacZ glmS852::Tn5 | This study |
| LZ003 | CJ1825 glmS852::Tn5 | This study |
| MC4100 | F−araD139 Δ(argF-lac) U169 flbB(flhD)5301 deoC1 ptsF(fruA)25 relA1 rbsR22 rpsL150 thiA | 2 |
| Mjf256.10 | DHB4 gshA::Kmr ΔtrxB::CmrahpC(V164G) | 15 |
| MZB001 | EC-O λ p0-lacZ | This study |
| MZB002 | EC-O λ (−239 to −1 nt)rraB-lacZ | This study |
| MZB003 | EC-O λ (−152 to −1 nt)rraB-lacZ | This study |
| MZB004 | EC-O λ (−239 to −153 nt)rraB-lacZ | This study |
| P90C | [ara Δ(lac-pro) thi] | 38 |
| pBAD30 | pACYA184 ori; Ampr pBAD | 18 |
| pBAD30-glmS | pACYA184 ori; Ampr, glmS under pBAD promoter | This study |
| pSP417 | pBR322 ori; Ampr, multiple cloning sites (8 restriction sites) upstream of a promoterless lacZ | 44 |
| pMZB002 | pSP417, (−239 to −1 nt)rraB-lacZ | This study |
| pMZB003 | pSP417, (−152 to −1 nt)rraB-lacZ | This study |
| pMZB004 | pSP417, (−239 to −153 nt)rraB-lacZ | This study |
| λRS45 | bla′-lacZsc imm21 ind22 | 49 |
| λRS74 | placUV5-lacZ imm21 ind22 | 49 |
| λMZB1 | Same as λRS74, but containing the p0-lacZ fusion | This study |
| λMZB3 | Same as λRS45, but containing the (−152 to −1 nt)rraB-lacZ fusion | This study |
Growth conditions.
Unless otherwise stated, cells were grown in Luria-Bertani (LB) broth under aeration at 37°C, and growth was monitored by measuring the turbidity at 600 nm (OD600). Minimal media contained M9 salts (BD, Sparks, MD), 50 μg/ml thiamine, 30 μg/ml proline, and 0.2% glucose or 20 μg/ml glucosamine-6-phosphate (GlcN-6-P) or 0.2% glycerol. Media were supplemented with antibiotics as required (50 μg/ml ampicillin, 25 μg/ml kanamycin, or 10 μg/ml chloramphenicol).
RNA methods.
For reverse transcriptase PCR (RT-PCR) analysis, total RNA was isolated with the RNeasy kit (Qiagen, Valencia, CA) and treated with RNase-free DNase (Ambion, Austin, TX). Fifty nanograms of total RNA was subjected to RT-PCR analysis using the one-step RNA PCR kit (TaKaRa, New York, NY). Northern blot analyses were performed using total RNA isolated from E. coli JCB570 grown in LB medium under aeration at 37°C. Samples were collected in 1-hour intervals throughout the exponential and stationary phases. Five micrograms of total RNA per lane was loaded onto a denaturing gel containing formaldehyde and then transferred to a positively charged nylon membrane (Hybond N+, Amersham, United Kingdom). The AlkPhos Direct nucleic acid labeling and detection system (Amersham, United Kingdom) was used to synthesize the oligonucleotide probe specific to the rraB gene (5′-AACAACCTGATGACGCATGGCA-3′). Hybridization, washing of the membranes, and detection of signals were carried out according to the manufacturer's instructions.
Primer extension.
For primer extension, a 5′ 32P-labeled primer (5′-ACACCACTGACATTGCCTCCACCTTT-3′) was used in the RT reaction with 5 μg of total RNA (purified using RNeasy kit, as described above) and SuperScript III RNase H− reverse transcriptase (Invitrogen, Carlsbad, CA). The primer extension products were separated onto 6% polyacrylamide-7 M urea gels. The dideoxy-DNA sequence ladder from the same primer was prepared using the fmol DNA cycle sequencing system (Promega, Madison, WI).
Real-time RT-PCR.
Cells were grown in M9 minimal media with 0.2% glucose and collected at an absorbance at 600 nm of 0.2. Total RNA was isolated with the RNeasy kit (Qiagen, Valencia, CA) and treated with RNase-free DNase (Ambion, Austin, TX). Reverse transcription of 0.2 μg of total RNA was performed with random hexanucleotides and SuperScript III RNase H− reverse transcriptase (Invitrogen, Carlsbad, CA). The RT-PCR was performed using primers 5′-TCGTGATTTGCTGCGACATC-3′ and 5′-ACCTGGGCATCGATCAGATC-3′ for the rraB gene in the experiment. Primers 5′-TCGAACAGGTGGCGTTAAATG-3′ and 5′-GGAGCGCAAATGCAGACAT-3′ were used for the dnaX gene as a control. Real-time PCRs were performed using the ABI Prism 7900 sequence detection system (Applied Biosystems) under universal cycling conditions (2 min at 50°C, 10 min at 95°C, 40 cycles of 15 s at 95°C, and 1 min at 60°C). The reaction mixture (20 μl) contained 2× SYBR green PCR master mix (PE Applied Biosystems), 10 pmol of forward and reverse primers, and 5 μl of cDNA (0.1 to 10 ng). Sequence-specific standard curves were generated by using serial dilutions of cDNA, and the quantity of rraB cDNA was normalized relative to the quantity of dnaX cDNA in each sample. Cycle threshold (CT) values were determined by automated threshold analysis with ABI Prism version 2.3 software. Each experiment was performed at least three times with two independent RNA preparations.
lacZ fusions.
PCR amplification was used to generate DNA fragments between nucleotides (nt) −239 and −1 in rraB. These fragments were cloned upstream of the lacZ gene in the multicopy transcriptional fusion vector, pSP417. The lacZ fusions were transferred into λRS45, and the negative control in pSP417 was transferred into λRS74 via double recombination (49). Blue plaques containing the recombinant lambda phages were isolated and used to lysogenize strain EC-O (Δlac) to generate strains MZB001 to MZB004. All lysogens were tested for monolysogenization by PCR. A similar approach was employed for the transfer of the Φ(−152 to −1 nt)rraB-lacZ fusion into the strain P90C (Δlac) to generate LZ001.
To measure β-galactosidase activity, cultures were grown with aeration at 37°C in M9 medium overnight and subcultured into the fresh medium. To detect the effect of tryptophan or arginine limitation, cultures were grown in the appropriate media as described in the supplemental material. Aliquots were collected, centrifuged at 4°C, and resuspended in an appropriate volume of ice-cold Z buffer (60 mM Na2HPO4, 40 mM NaH2PO4, 10 mM KCl, 1 mM MgSO4; pH 7.0) (38) to give an OD600 value in the range from 0.6 to 0.9. β-Galactosidase activities were determined from at least three independent experiments (37).
Transposon mutagenesis.
Fifty nanograms of transposome (EZ-Tn5; Epicentre, Madison, WI) was electroporated into 50 μl (108 cells) competent cells of E. coli strain LZ001. Transformed cells were plated onto LB plates containing 25 μg/ml kanamycin and 25 μg/ml X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) and incubated at 37°C overnight. Out of about 32,000 colonies that were screened, six showed an intense blue color consistently. Of these, one strain gave markedly higher β-galactosidase activity in liquid cultures. Genomic DNA from this clone was extracted using a DNeasy kit (Qiagen, Valencia, CA), fragmented by EcoRI digestion, and self-ligated using T4 DNA ligase (Roche, Switzerland). The ligation product was transformed into the E. coli pir+ strain BW21116 (19), and kanamycin-resistant colonies were selected. Those colonies contained plasmids formed by circularized ligation products, made from partially digested genomic DNA containing the R6Kγori and kanamycin resistance cassette. Plasmid DNA was purified and sequenced using the primers R6KAN-2 and KAN-2 FP-1 provided by the EZ-Tn5 kit.
Determination of UDP-GlcNAc concentration.
The UDP-GlcNAc concentration in strains LZ001 and LZ002 was determined by high-pressure liquid chromatography (HPLC). Cultures were grown in M9 minimal media to an OD600 of 0.2 to 0.3, and the cell pellets were collected and deproteinized with 5% trichloroacetic acid. The supernatants were neutralized with 2.5 M potassium hydroxide-1.5 M K2HPO4 and stored at −20°C until further use. UDP-GlcNAc concentration was determined by HPLC, using a CarboPac PA1 column (250 mm × 4 mm; Dionex) and a guard column (4 mm × 50 mm) of the same material. Analytes were eluted using an ammonium acetate gradient (55) with detection at 262 nm. Under these conditions, UDP-GlcNAc elutes with a retention time of 15.3 min (41). The UDP-GlcNAc concentrations in cell extracts were calculated from the integrated peak areas by using appropriate standards, and the data were normalized based on total cell dry weight measured according to Namboori and Graham (41). Experiments were carried out in triplicates with independently prepared cell extracts.
RESULTS
Identification of the PrraB promoter.
Analysis of the DNA sequence upstream of the rraB gene using the GENETYX-MAC (11.2.5) software suggested the existence of two putative promoters that match the σ70 consensus −35 and −10 sequences. The proximal promoter has −35 and −10 sequences centered at 128 and 104 nucleotides, respectively, upstream of the rraB translation start site (Fig. 1A). The upstream region containing this putative promoter sequence is well conserved (≥80% sequence identity) among gammaproteobacteria, including Escherichia coli, Shigella flexneri, and Salmonella enterica (Fig. 1B).
FIG. 1.

Identification of PrraB. (A) Two pairs of putative promoters that match the σ70 consensus −35 and −10 sequences were identified using GENETYX-MAC 11.2.5. The arginine repressor binding sites (ARG boxes) are shown boxed, the argI promoter sequence is underlined, and the rraB coding sequence is highlighted. (B) Multiple sequence alignment of the argI-rraB intergenic sequence. B171, Escherichia coli B171; O157:H7-1, Escherichia coli O-157; K12, Escherichia coli K-12; CFT073, Escherichia coli CFT073. The transcription start codon, upstream putative −10 site, proximal −35, and −10 sites are shown in boxes. (C) Primer extension analysis. The asterisk and arrow show the transcription initiation site and the reverse transcription product, respectively.
Northern blot analysis with rraB-specific probes revealed that the rraB mRNA is transcribed throughout the exponential and stationary phase (see Fig. S1 in the supplemental material). A single band corresponding to a transcript of approximately 500 bases was detected, suggesting that transcription of rraB occurs from the putative proximal promoter and gives rise to a monocistronic mRNA. Primer extension of RNA isolated from E. coli JCB570 revealed a single rraB transcript that is initiated at a T residue 88 bp upstream from the ATG codon (Fig. 1C). This promoter was designated PrraB.
The promoter activity of PrraB was further analyzed using lacZ transcriptional fusions. The different regions upstream of rraB extending up to the putative distal promoter, as shown in Fig. 2A, were amplified by PCR and cloned upstream of the lacZ gene in the low-copy transcriptional fusion vector pSP417, resulting in plasmids pMZB002 [Φ(−239 to −1)rraB-lacZ], pMZB003 [Φ(−152 to −1)rraB-lacZ], and pMZB004 [Φ(−239 to −153)rraB-lacZ]. To rule out the possibility that differences in β-galactosidase activity expressed from the above-described transcriptional fusions might be partially due to the effects of plasmid copy number, the lacZ fusions were inserted into the chromosome (49). First, the lacZ fusions were transferred into either λRS74 or λRS45 via a double recombination event, and then E. coli EC-O was lysogenized with the recombinant lambda phages to generate strains carrying a single copy of the respective promoter-lacZ fusion. As shown in Fig. 2B, strain MZB003 [EC-O λ Φ(−152 to −1)rraB-lacZ], carrying the rraB upstream region that includes the proximal promoter fused to lacZ, showed levels of β-galactosidase activity similar to those observed in MZB002 [EC-O λ Φ(−239 to −1)rraB-lacZ], which contains a fusion to the rraB upstream region including both the proximal and the distal putative promoter sequences. In contrast, MZB004 [EC-O λ Φ(−239 to −153)rraB-lacZ], containing a fusion to the rraB upstream region that lacks the proximal promoter, gave background activity. These results further support the notion that rraB is transcribed from the proximal promoter, PrraB. In the studies described below, the transcriptional fusion λ Φ(−152 to −1)rraB-lacZ is now designated Φ(PrraB-lacZ).
FIG. 2.

Transcriptional LacZ fusions. (A) Schematic of the transcriptional rraB-lacZ fusions used in this study. (B and C) β-Galactosidase activities in MZB001, MZB002, MZB003, and MZB004 cells. Cells were grown in LB medium under aeration at 37°C and harvested in either log phase (OD600 = 0.5; black bars) or stationary phase (OD600 = 1.6; white bars). Samples were normalized by OD, and enzymatic activities were measured in Miller units. The data presented are the averages of the results for at least three independent determinations, and the error bars correspond to the standard deviation.
The expression of the rraA gene was previously shown to be induced in stationary phase (56). Northern blot analysis (see Fig. S1 in the supplemental material) and measurements of the β-galactosidase activity expressed from the Φ(PrraB-lacZ) fusion in exponential- and stationary-phase cells revealed that the transcription level of rraB is not affected by the growth phase (Fig. 2C). The E. coli rraB gene is adjacent to argI, which is transcribed from the opposite strand and encodes ornithine carbamoyltransferase I, a key enzyme in arginine biosynthesis. Earlier microarray analyses had indicated that the expression of rraB is correlated with that of argI and it is increased upon arginine deprivation (26, 47). Interestingly, the genomic organization of rraB and argI is conserved in other gammaproteobacteria, such as S. enterica and Yersinia pestis. In Vibrio cholerae, the rraB homologue VC0424 is located in a similar head-to-head orientation with VC0423 (see Fig. S2 in the supplemental material), which codes for arginine deiminase that is involved in arginine catabolism. Inspection of the nucleotide sequence in the argI-rraB intergenic region revealed the presence of a putative arginine repressor (ArgR) binding site (ARG box), which consists of two neighboring 18-bp palindromic sequences separated by 3 nucleotides (Fig. 1A). The influence of arginine availability on rraB transcription was examined as follows. First, excess arginine (20 mM) was added to cultures growing in minimal medium. Second, cells grown in LB medium were transferred into medium without arginine for 2 hours to achieve transient arginine starvation. Neither excess nor depletion of arginine exerted any significant effect on the β-galactosidase activity of MZB003 cells carrying the Φ(PrraB-lacZ) fusion (see Fig. S3 in the supplemental material).
Khodursky et al. reported that the expression of the rraB gene is repressed in response to tryptophan starvation (26). To evaluate the effect of tryptophan on the transcriptional activity from PrraB, we determined the β-galactosidase activity in MZB003 grown either in minimal media with excess tryptophan or under conditions of tryptophan starvation (see Fig. S4 in the supplemental material). None of these conditions affected the level of β-galactosidase activity expressed from the Φ(PrraB-lacZ) fusion, indicating that PrraB promoter does not respond to tryptophan availability.
Transposon mutagenesis.
Transposon mutagenesis and screening for colonies displaying a higher level of β-galactosidase activity from the Φ(PrraB-lacZ) fusion were employed to isolate chromosomal lesions that might result in increased transcription from the PrraB. First, the Φ(PrraB-lacZ) fusion was transferred into the Δlac strain P90C, resulting in strain LZ001, which gave pale-blue colonies on LB agar plates containing 25 μg/ml X-Gal. Following transposon mutagenesis using the EZ-Tn5 transposome, six dark-blue colonies were isolated from the approximately 32,000 colonies tested. One of these colonies, designated LZ002, reproducibly produced the dark-blue phenotype on X-Gal plates and also resulted in higher β-galactosidase activity in lysates from cells grown in liquid cultures. Genomic DNA from LZ002 was purified, digested with EcoRI, self-ligated, and transformed into the E. coli pir+ strain BW21116 (19). The colonies formed on kanamycin plates carry plasmids made of circular genomic DNA fragments containing the Tn5 insertion. Plasmid DNA was isolated and the site of the transposon insertion was determined by DNA sequencing using transposon-specific primers. This analysis revealed that the transposon insertion occurred at 3910475 bp of the E. coli chromosome, which corresponds to nucleotide 852 in glmS (Fig. 3A). glmS encodes the enzyme l-glutamine d-fructose-6-phosphate amidotransferase (EC 2.6.1.16), also known as GlcN-6-P synthase. GlcN-6-P synthase is a critical metabolic control point in the biosynthesis of amino sugar-containing macromolecules. It catalyzes the first committed step in the pathway leading to the formation of UDP-GlcNAc, a universal GlcNAc donor for the biosynthesis of cell walls, extracellular matrix, glycolipids, and the protein posttranslational modifications (36, 41).
FIG. 3.

A transposon insertion in glmS upregulates rraB. (A) Location of the transposon insertion in the glmS gene. glmS is cotranscribed with upstream glmU from the promoter glmUp1. (B) Growth curve of strains LZ001 (solid line) and LZ002 (glmS852::Tn5; broken line) cultured in M9 media with 0.2% glucose. (C) β-Galactosidase activities in strains LZ001 (white bars) and LZ002 (black bars) in the presence or absence of 20 μg/ml GlcN-6-P were measured in cells grown in M9 media and harvested at an OD600 of 0.2 to 0.3. (D) Complementation of the glmS852::Tn5 mutation with plasmid pBAD-glmS. E. coli strains LZ001 (white bars) and LZ002 (black bars) transformed with plasmid pBAD30 or pBAD30-glmS as indicated were grown overnight in M9 media with 0.2% glucose and subcultured into fresh M9 media with 0.2% glycerol. At an OD600 of 0.4 to 0.5, 0.2% arabinose was added. Cells were harvested 2 h after induction, and the β-galactosidase activity was determined.
The glmS852::Tn5 strain LZ002 grew slower than the parental wild-type strain in both rich (data not shown) and minimal media (Fig. 3B). LZ002 displayed fourfold-higher β-galactosidase activity than did its isogenic control, LZ001, in exponential phase (Fig. 3C). Consistent with this result, the steady-state level of rraB mRNA as determined by real-time RT-PCR was fivefold greater in glmS852::Tn5 cells than in the parental strain, LZ001 (Table 2). To rule out the possibility that the increase in transcription from the PrraB promoter in cells containing the glmS852::Tn5 allele was related to their lower growth rate, we also compared the steady-state rraB transcript level in strain Mjf256.10 (15), which grows at a comparable rate to that of LZ002, as a result of mutations that affect the cytoplasmic reduction pathways and are unrelated to UDP-GlcNAc metabolism. In contrast to the fivefold increase observed in the rraB mRNA levels of LZ002 relative to its parental strain, the rraB transcription in strain Mjf256.10 was slightly lower (67%) than that in its parental strain, DHB4, as determined by real-time RT-PCR.
TABLE 2.
Real-time PCR quantification of rraB mRNA in LZ001 and LZ002 strains
| Strain | Total RNA (ng) |
rraB mRNA normalized relative to dnaX mRNA | rraB mRNA relative to wild type | |
|---|---|---|---|---|
| rraB | dnaX | |||
| LZ001 | 13.47 ± 0.55 | 58.77 ± 9.77 | 0.23 ± 0.03 | 1 |
| LZ002 | 101.23 ± 0.29 | 84.54 ± 1.89 | 1.20 ± 0.14 | 5.23 |
To test whether specific complementation of glmS can reverse the effect of glmS852::Tn5 mutation, glmS was cloned into a low-copy plasmid, pBAD30, and expressed from an arabinose-inducible promoter. When grown in M9 minimal medium supplemented with 0.2% glucose but without arabinose, LZ002 cells transformed with pBAD30-glmS exhibited β-galactosidase activity similar to that of LZ002 cells without plasmid (data not shown). However, when the strain LZ002 containing pBAD30-glmS was grown in the presence of 0.2% arabinose, the β-galactosidase activity was reduced to the level observed in the parental strain, LZ001. Under these conditions, cells containing pBAD-glmS accumulate GlmS at a level comparable to that obtained from the chromosomal copy of glmS in the parental strain, LZ001, as monitored by Western blotting using anti-GlmS antibodies (data not shown). As a control, transferring the empty pBAD30 plasmid did not reverse the effect of glmS852::Tn5 mutation under both growth conditions (Fig. 3D). Moreover, consistent with the physiological role of GlcN-6-P synthase, addition of its enzymatic reaction end product (GlcN-6-P) to LZ002 cells resulted in a normal growth rate (see Fig. S5 in the supplemental material) and basal β-galactosidase activity (Fig. 3C). Finally, the glmS852::Tn5 allele did not affect the LacZ activity from a chromosomal Φ(PrraA-lacZ) fusion (56), indicating that the impairment of glmS specifically enhances the transcription of rraB but not that of the other RNase E inhibitor, rraA (data not shown).
Because of the importance of glmS in cell wall synthesis and its related pathways, we speculated that rraB is involved in cell envelope stress response. We tested the effect of mutations in cellular envelope stress response, such as dsbA, degP, and rseB/degS (6, 50, 17), on PrraB transcription by introducing these mutations to strain LZ001 by the use of P1 transduction. None of them shows a significant effect on the PrraB activity based on β-galactosidase activity (see Fig. S6 in the supplemental material).
Effect of glmS852::Tn5 mutation on rne-lacZ activity.
RNase E autoregulates its expression in cis by cleaving its own mRNA near the 5′ UTR. Consequently, the production of RNase E is inversely affected by changes in the catalytic activity of the enzyme. Strain CJ1825 (MC1060 λez1) contains a chromosomal fusion of the truncated rne gene (the 5′ UTR of rne gene, and the N-terminal 181 codons) linked to the lacZ reporter. Due to the self-cleavage of the rne-lacZ transcript, a higher β-galactosidase activity in strain CJ1825 indicates a decrease in RNase E activity and vice versa (20). We previously showed that overexpression of RraB conferred increased β-galactosidase activity from rne-lacZ of this strain (16). We examined whether the glmS852::Tn5 allele and the resulting upregulation of rraB also inhibit RNase E and, in turn, give rise to higher β-galactosidase activity. The glmS852::Tn5 mutation was introduced into strain CJ1825 via P1 transduction to give rise to E. coli LZ003. The glmS852::Tn5 allele in E. coli LZ003 also caused a growth defect in both rich and minimal media (data not shown). β-Galactosidase activity was increased by more than twofold in LZ003 relative to that in the wild type (Fig. 4). Notably, the observed increase in β-galactosidase activity from the rne-lacZ fusion in strain LZ003 is comparable to that obtained through overexpression of rraB transcribed from a trc promoter in plasmid pTrc99a (16).
FIG. 4.
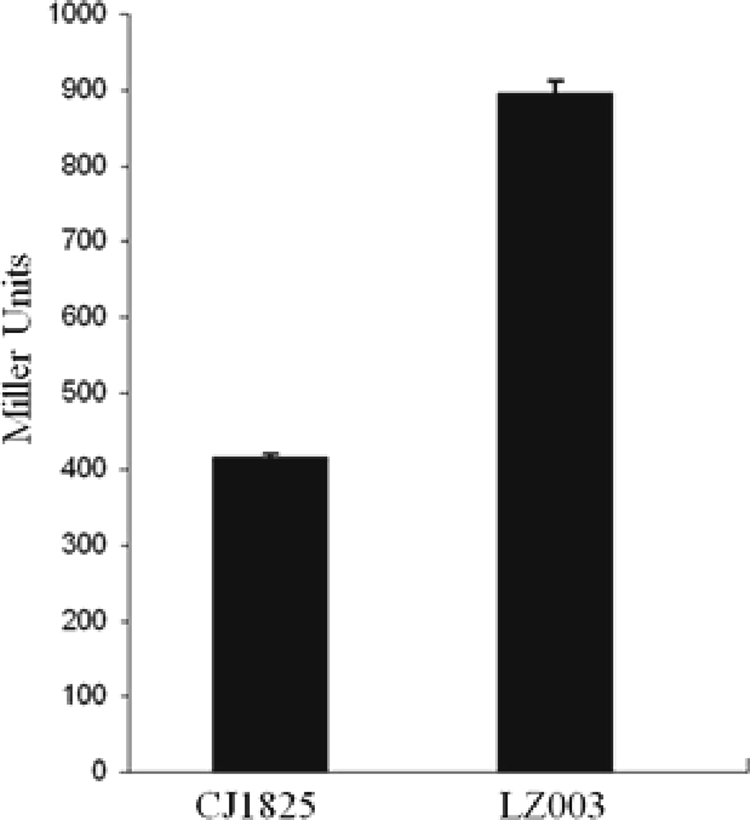
The glmS852::Tn5 allele reduces β-galactosidase activity from the rne-lacZ fusion. E. coli strains CJ1825 and LZ003 were cultured in LB media. At log phase, cells were harvested, and the β-galactosidase activities were determined.
DISCUSSION
In earlier studies, we showed that RraB is a protein inhibitor that interacts with RNase E, modulates the composition of the degradosome, and affects the ability of the RNA-degrading machinery to recognize and cleave numerous RNAs. Studying the regulation of rraB gene expression is important for understanding its physiological significance. Primer extension and lacZ transcriptional fusion experiments demonstrated that rraB is transcribed from its own promoter (PrraB) located 88 bp upstream from the translation start codon. Northern blot analysis and lacZ transcriptional fusion experiments demonstrated that, unlike for rraA, the transcription of rraB is not growth phase dependent. The E. coli rraB gene and its homologues in other bacteria, such as Salmonella enterica serovar Typhimurium, Y. pestis, and V. cholerae, are arranged in a head-to-head alignment with the adjacent gene, argI. Simultaneous regulation of two divergent promoters by ArgR has been described for a number of adjacent gene pairs, such as argE and argC or argG and metY (27). While the existence of a putative ArgR binding site suggests that transcription from PrraB might be regulated by the availability of l-arginine, we observed that the transcriptional activity, as determined from the level of β-galactosidase, was unaffected by the presence or absence of l-arginine. Similarly, we found that transcription of PrraB is not affected by tryptophan starvation, inconsistent with the results of earlier microarray analysis (26, 47). Because in microarray studies the steady-state mRNA level is the outcome of both transcriptional regulation and posttranscriptional effects, the observed correlation between the mRNA levels of tryptophan/arginine metabolic genes and rraB in those studies could be related to rraB mRNA itself or other posttranscriptional regulations, rather than its promoter.
Transposon mutagenesis was employed to identify transcriptional regulators of the rraB promoter by screening for enhanced β-galactosidase activity from the Φ(PrraB-lacZ) transcriptional fusion. Screening of over 30,000 colonies led to the isolation of one clone (LZ002), which displayed fourfold-higher β-galactosidase activity, and moreover, a fivefold-greater steady-state accumulation of the rraB transcript. Surprisingly, the transposon insertion was located in the glmS gene encoding the essential enzyme GlcN-6-P synthase, a ubiquitous protein, present in all known organisms. Both prokaryotic and eukaryotic enzymes contain an N-terminal glutamine binding domain and a C-terminal d-fructose-6-phosphate binding domain, with the eukaryotic enzymes having an additional 40 to 90 amino acid residues linking the two. Except for the eukaryotic linker region, the N-terminal and C-terminal domains are highly conserved, including all the residues involved in substrate binding and catalysis (36). We found that in strain LZ002, the Tn5 transposon was inserted in the C terminus after the conserved N-terminal domain. Upon further inspection, we noticed the existence of a putative start codon at the 3′ end of Tn5, which is preceded by an AU-rich sequence 15 nt upstream that might serve as a ribosomal binding site for the translation of a chimera comprising the C-terminal part of GlmS. According to this hypothesis, the transposon insertion might result in the synthesis of two GlmS polypeptides, one comprising the N-terminal 284 amino acids and the second comprising amino acids 285 to 620. Early studies of GlmS showed that chymotrypsin cleaves this protein at Tyr240 within a flexible loop to produce an N-terminal glutamine hydrolase domain and a C-terminal fructose-6-phosphate isomerase domain; however, the reconstituted protein fragments do not catalyze the full GlcN-6-P synthase reaction (14). The additional amino acids 241 to 284 in the present construct form α-helices 7 and 8, which could interact with helices of the isomerase fragment to form a stable SIS domain (52). Interaction between the two subunits would form an ammonia channel, conferring weak GlcN-6-P synthase activity, in turn enabling the synthesis of essential UDP-GlcNAc. Because the cellular level of GlcN-6-P is beyond the detection limit, to evaluate this hypothesis, we determined the cellular concentration of UDP-GlcNAc in the glmS852::Tn5 strain LZ002 and its isogenic control, LZ001, using HPLC (41). As expected, LZ002 was found to synthesize UDP-GlcNAc, but at a reduced level. The concentration of UDP-GlcNAc in the mutant strain was 53% lower than that in the parent strain (LZ002 versus LZ001, respectively) with a statistically significant difference of P < 0.01 (two-tailed t test).
These results indicate that these cells have a functional GlcN-6-P synthase, possibly resulting from the translation of the C-terminal fragment, which in combination with the N-terminal polypeptide can reconstitute the active enzyme. Regardless of the mechanism that mediates the synthesis of GlcN-6-P, our results indicate that the transcription of rraB is modulated either by GlcN-6-P or by other metabolites derived from GlcN-6-P. This effect is not the result of the reduced growth rate conferred by the glms852::Tn5 allele; it does not affect the other RNase E inhibitor rraA, indicating that it is specific to rraB.
As the enzyme catalyzing the first committed step of the UDP-GlcNAc pathway that provides the building blocks for the formation of cell wall macromolecules, GlcN-6-P synthase is an obvious point of metabolic control. According to Collins et al. (12), in Bacillus subtilis, the 5′ UTR of the glmS mRNA contains a metabolite-sensing ribozyme. Binding of GlcN-6-P to the ribozyme stimulates autocatalytic site-specific cleavage near the 5′ end of the transcript, resulting in the degradation of the transcript by RNase J1. Interestingly, RNase J1 is the endoribonuclease in B. subtilis with functional homology to RNase E in E. coli, except that RNase J1 specifically cleaves transcripts with 5′ hydroxyl groups (12). Kalamorz et al. (23) reported that the regulation of glmS by GlcN-6-P in E. coli differs substantially from that in B. subtilis; in E. coli, when the intracellular GlcN-6-P concentration decreases, the short form of small RNA GlmY (GlmY*) accumulates and acts to stabilize a second small RNA, GlmZ, which is a homologue of GlmY. GlmZ is derived from the processing of the glmUS primary transcripts by RNase E and can stabilize glmS trancripts in concert with the protein YhbJ (23, 48). Moreover, the glmS mRNA generated by RNase E from glmUS transcript is highly susceptible to poly(A)-dependent degradation (22). We found that the glmS852::Tn5 allele increased rraB transcription, thereby lowering the RNase E activity. These findings raise the possibility that upregulation of rraB in response to changes in metabolites of the GlcN-6-P pathway results in a reduction in RNase E activity, which in turn affects the processing of the glmUS primary transcripts. Thus, as with other posttranscriptional mechanisms of regulation, RraB may facilitate rapid alterations in RNA decay and/or processing in response to specific environmental stimuli, such as a reduced cellular level of GlcN-6-P. The mechanism by which mutations that reduce GlcN-6-P synthase activity affect RraB expression and its detailed effect on the processing of the glmUS transcript are subjects of ongoing studies.
Supplementary Material
Acknowledgments
We are grateful to Yasuaki Kawarasaki for help with Northern blotting and Teresa N. Giles for reading the manuscript.
This work was supported in part by grants from the Welch Foundation and NIH grants GM-55090 (to G.G.) and AI-064444 (to D.E.G.).
Footnotes
Published ahead of print on 28 August 2009.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Aiba, H. 2007. Mechanism of RNA silencing by Hfq-binding small RNAs. Curr. Opin. Microbiol. 10:134-139. [DOI] [PubMed] [Google Scholar]
- 2.Baker, K., N. Mackman, M. Jackson, and I. B. Holland. 1987. Role of SecA and SecY in protein export as revealed by studies of TonA assembly into the outer membrane of Escherichia coli. J. Mol. Biol. 198:693-703. [DOI] [PubMed] [Google Scholar]
- 3.Bardwell, J. C., K. McGovern, and J. Beckwith. 1991. Identification of a protein required for disulfide bond formation in vivo. Cell 67:581-589. [DOI] [PubMed] [Google Scholar]
- 4.Bernstein, J. A., P. H. Lin, S. N. Cohen, and S. Lin-Chao. 2004. Global analysis of Escherichia coli RNA degradosome function using DNA microarrays. Proc. Natl. Acad. Sci. USA 101:2758-2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bernstein, J. A., A. B. Khodursky, P.-H. Lin, S. Lin-Chao, and S. N. Cohen. 2002. Global analysis of mRNA decay and abundance in Escherichia coli at single-gene resolution using two-color fluorescent DNA microarrays, Proc. Natl. Acad. Sci. USA 99:9697-97026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bessette, P. H., J. Qiu, J. A. Bardwell, J. R. Swartz, and G. Georgiou. 2001. Effect of sequences of the active-site dipeptides of DsbA and DsbC on in vivo folding of multidisulfide proteins in Escherichia coli. J. Bacteriol. 183:980-988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brennan, R. G., and T. M. Link. 2007. Hfq structure, function and ligand binding. Curr. Opin. Microbiol. 10:125-133. [DOI] [PubMed] [Google Scholar]
- 8.Callaghan, A. J., M. J. Marcaida, J. A. Stead, K. J. McDowall, W. G. Scott, and B. F. Luisi. 2005. Structure of Escherichia coli RNase E catalytic domain and implications for RNA turnover. Nature 437:1187-1191. [DOI] [PubMed] [Google Scholar]
- 9.Caruthers, J. M., Y. Feng, D. B. McKay, and S. N. Cohen. 2006. Retention of core catalytic functions by a conserved minimal ribonuclease E peptide that lacks the domain required for tetramer formation. J. Biol. Chem. 281:27046-27051. [DOI] [PubMed] [Google Scholar]
- 10.Celesnik, H., A. Deana, and J. G. Belasco. 2007. Initiation of RNA decay in Escherichia coli by 5′ pyrophosphate removal. Mol. Cell 27:79-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coburn, G. A., and G. A. Mackie. 1999. Degradation of mRNA in Escherichia coli: an old problem with some new twists. Prog. Nucleic Acid Res. Mol. Biol. 62:55-108. [DOI] [PubMed] [Google Scholar]
- 12.Collins, J. A., I. Irnov, S. Baker, and W. C. Winkler. 2007. Mechanism of mRNA destabilization by the glmS ribozyme. Genes Dev. 21:3356-3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coros, C. J., C. L. Piazza, V. R. Chalamcharla, and M. Belfort. 2008. A mutant screen reveals RNase E as a silencer of group II intron retromobility in Escherichia coli. RNA 14:2634-2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Denisot, M. A., F. Le Goffic, and B. Badet. 1991. Glucosamine-6-phosphate synthase from Escherichia coli yields two proteins upon limited proteolysis: identification of the glutamine amidohydrolase and 2R ketose/aldose isomerase-bearing domains based on their biochemical properties. Arch. Biochem. Biophys. 288:225-230. [DOI] [PubMed] [Google Scholar]
- 15.Faulkner, M. J., K. Veeravalli, S. Gon, G. Georgiou, and J. Beckwith. 2008. Functional plasticity of a peroxidase allows evolution of diverse disulfide-reducing pathways. Proc. Natl. Acad. Sci. USA 105:6735-6740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao, J., K. Lee, M. Zhao, J. Qiu, X. Zhan, A. Saxena, C. J. Moore, S. N. Cohen, and G. Georgiou. 2006. Differential modulation of E. coli mRNA abundance by inhibitory proteins that alter the composition of the degradosome. Mol. Microbiol. 61:394-406. [DOI] [PubMed] [Google Scholar]
- 17.Grigorova, I. L., R. Chaba, H. J. Zhong, B. M. Alba, V. Rhodius, C. Herman, and C. A. Gross. 2004. Fine-tuning of the Escherichia coli sigmaE envelope stress response relies on multiple mechanisms to inhibit signal-independent proteolysis of the transmembrane anti-sigma factor, RseA. Genes Dev. 18:2686-2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guzman, L. M., D. Belin, M. J. Carson, and J. Beckwith. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177:4121-4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haldimann, A., L. L. Daniels, and B. L. Wanner. 1998. Use of new methods for construction of tightly regulated arabinose and rhamnose promoter fusions in studies of the Escherichia coli phosphate regulon. J. Bacteriol. 180:1277-1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jain, C., and J. G. Belasco. 1995. RNase E autoregulates its synthesis by controlling the degradation rate of its own mRNA in Escherichia coli: unusual sensitivity of the rne transcript to RNase E activity. Genes Dev. 9:84-96. [DOI] [PubMed] [Google Scholar]
- 21.Jiang, X., and J. G. Belasco. 2004. Catalytic activation of multimeric RNase E and RNase G by 5′-monophosphorylated RNA. Proc. Natl. Acad. Sci. USA 101:9211-9216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Joanny, G., J. Le Derout, D. Bréchemier-Baey, V. Labas, J. Vinh, P. Régnier, and E. Hajnsdorf. 2007. Polyadenylation of a functional mRNA controls gene expression in Escherichia coli. Nucleic Acids Res. 35:2494-2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kalamorz, F., B. Recichenbach, W. Marz, B. Rak, and B. Gorke. 2007. Feedback control of glucosamine-6-phosphate synthase GlmS expression depends on the small RNA GlmZ and involves the novel protein YhbJ in Escherichia coli. Mol. Microbiol. 65:1518-1533. [DOI] [PubMed] [Google Scholar]
- 24.Khemici, V., and A. J. Carpousis. 2004. The RNA degradosome and poly(A) polymerase of Escherichia coli are required in vivo for the degradation of small mRNA decay intermediates containing REP-stabilizers. Mol. Microbiol. 51:777-790. [DOI] [PubMed] [Google Scholar]
- 25.Khemici, V., L. Poljak, B. F. Luisi, and A. J. Carpousis. 2008. The RNase E of Escherichia coli is a membrane-binding protein. Mol. Microbiol. 70:799-813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khodursky, A. B., B. J. Peter, N. R. Cozzarelli, D. Botstein, P. O. Brown, and C. Yanofsky. 2000. DNA microarray analysis of gene expression in response to physiological and genetic changes that affect tryptophan metabolism in Escherichia coli. Proc. Natl. Acad. Sci. USA 97:12170-12175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krin, E., C. L. Winter, P. N. Bertin, A. Danchin, and A. Kolb. 2003. Transcription regulation coupling of the divergent argG and metY promoters in Escherichia coli K-12. J. Bacteriol. 185:3139-3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee, K., and S. N. Cohen. 2003. A Streptomyces coelicolor functional orthologue of Escherichia coli RNase E shows shuffling of catalytic and PNPase-binding domains. Mol. Microbiol. 48:349-360. [DOI] [PubMed] [Google Scholar]
- 29.Lee, K., X. Zhan, J. Gao, J. Qiu, Y. Feng, R. Meganathan, S. N. Cohen, and G. Georgiou. 2003. RraA: a protein inhibitor of RNase E activity that globally modulates RNA abundance in E. coli. Cell 114:623-634. [PubMed] [Google Scholar]
- 30.Li, Z., and M. P. Deutscher. 2002. RNase E plays an essential role in the maturation of Escherichia coli tRNA precursors. RNA 8:97-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liou, G.-G., W.-N. Jane, S. N. Cohen, N.-S. Lin, and S. Lin-Chao. 2001. RNA degradosomes exist in vivo in Escherichia coli as multicomponent complexes associated with the cytoplasmic membrane via the N-terminal region of ribonuclease E. Proc. Natl. Acad. Sci. USA 98:63-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Makarova, K. S., A. A. Mironov, and M. S. Gelfand. 2001. Conservation of the binding site for the arginine repressor in all bacterial lineages. Genome Biol. 2:research0013. [DOI] [PMC free article] [PubMed]
- 33.Maki, K., K. Uno, T. Morita, and H. Aiba. 2008. RNA, but not protein partners, is directly responsible for translational silencing by a bacterial Hfq-binding small RNA. Proc. Natl. Acad. Sci. USA 105:10332-10337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McDowall, K. J., and S. N. Cohen. 1996. The N-terminal domain of the rne gene product has RNase E activity and is non-overlapping with the arginine-rich RNA-binding site. J. Mol. Biol. 255:349-355. [DOI] [PubMed] [Google Scholar]
- 35.Miczak, A., V. R. Kaberdin, C.-L. Wei, and S. Lin-Chao. 1996. Proteins associated with RNase E in a multicomponent ribonucleolytic complex. Proc. Natl. Acad. Sci. USA 93:3865-3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Milewski, S. 2002. Glucosamine-6-phosphate synthase—the multi-facets enzyme. Biochim. Biophys. Acta 1597:173-192. [DOI] [PubMed] [Google Scholar]
- 37.Miller, J. H. 1992. A short course in bacterial genetics: a laboratory manual and handbook for Escherichia coli and related bacteria. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 38.Miller, J. H. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 39.Moll, I., T. Afonyushkin, O. Vytvytska, V. R. Kaberdin, and U. D. O. Blasi. 2003. Coincident Hfq binding and RNase E cleavage sites on mRNA and small regulatory RNAs. RNA 9:1308-1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mudd, E. A., H. M. Krisch, and C. F. Higgins. 1990. RNase E, an endoribonuclease, has a general role in the chemical decay of Escherichia coli mRNA: evidence that rne and ams are the same genetic locus. Mol. Microbiol. 4:2127-2135. [DOI] [PubMed] [Google Scholar]
- 41.Namboori, S. C., and D. E. Graham. 2008. Enzymatic analysis of uridine diphosphate N-acetyl-d-glucosamine. Anal. Biochem. 381:94-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ow, M. C., and S. R. Kushner. 2002. Initiation of tRNA maturation by RNase E is essential for cell viability in E. coli. Genes Dev. 16:1102-1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Perwez, T., D. Hami, V. F. Maples, M. Zhao, B. C. Wang, and S. R. Kushner. 2008. Intragenic suppressors of temperature-sensitive rne mutations lead to the dissociation of RNase E activity on mRNA and tRNA substrates in Escherichia coli. Nucleic Acids Res. 36:5306-5318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Podkovyrov, S. M., and T. J. Larson. 1995. A new vector-host system for construction of lacZ transcriptional fusions where only low-level gene expression is desirable. Gene 156:151-152. [DOI] [PubMed] [Google Scholar]
- 45.Py, B., H. Causton, E. A. Mudd, and C. F. Higgins. 1994. A protein complex mediating mRNA degradation in Escherichia coli. Mol. Microbiol. 14:717-729. [DOI] [PubMed] [Google Scholar]
- 46.Py, B., C. F. Higgins, H. M. Krisch, and A. J. Carpousis. 1996. A DEAD-box RNA helicase in the Escherichia coli RNA degradosome. Nature 381:169-172. [DOI] [PubMed] [Google Scholar]
- 47.Ramelot, T. A., S. Ni, S. Goldsmith-Fischman, J. R. Cort, B. Honig, and M. A. Kennedy. 2003. Solution structure of Vibrio cholerae protein VC0424: a variation of the ferredoxin-like fold. Protein Sci. 12:1556-1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reichenbach, B., A. Maes, F. Kalamorz, E. Hajnsdorf, and B. Gorke. 2008. The small RNA GlmY acts upstream of the sRNA GlmZ in the activation of glmS expression and is subject to regulation by polyadenylation in Escherichia coli. Nucleic Acids Res. 36:2570-2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Simons, R. W., F. Houman, and N. Kleckner. 1987. Improved single and multicopy lac-based cloning vectors for protein and operon fusions. Gene 53:85-96. [DOI] [PubMed] [Google Scholar]
- 50.Strauch, K. L., and J. Beckwith. 1988. An Escherichia coli mutation preventing degradation of abnormal periplasmic proteins. Proc. Natl. Acad. Sci. USA 85:1576-1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Taghbalout, A., and L. Rothfield. 2008. RNaseE and RNA helicase B play central roles in the cytoskeletal organization of the RNA degradosome. J. Biol. Chem. 283:13850-13855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Teplyakov, A., G. Obmolova, B. Badet, and M. A. Badet-Denisot. 2001. Channeling of ammonia in glucosamine-6-phosphate synthase. J. Mol. Biol. 313:1093-1102. [DOI] [PubMed] [Google Scholar]
- 53.Vanzo, N. F., Y. S. Li, B. Py, E. Blum, C. F. Higgins, L. C. Raynal, H. M. Krisch, and A. J. Carpousis. 1998. Ribonuclease E organizes the protein interactions in the Escherichia coli RNA degradosome. Genes Dev. 12:2770-2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yeom, J. H., and K. Lee. 2006. RraA rescues Escherichia coli cells over-producing RNase E from growth arrest by modulating the ribonucleolytic activity. Biochem. Biophys. Res. Commun. 45:1372-1376. [DOI] [PubMed] [Google Scholar]
- 55.Zhang, H., Y. Zhou, H. Bao, and H. Liu. 2006. Vi antigen biosynthesis in Salmonella typhi: characterization of UDP-N-acetylglucosamine C-6 dehydrogenase (TviB) and UDP-N-acetylglucosaminuronic acid C-4 epimerase (TviC). Biochemistry 45:8163-8173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhao, M., L. Zhou, Y. Kawarasaki, and G. Georgiou. 2006. Regulation of RraA, a protein inhibitor of RNase E-mediated RNA decay. J. Bacteriol. 188:3257-3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.