

Abstract
We report about an infant affected by dilated cardiomyopathy (CMP) in whom metabolic investigations evidenced medium-chain-acyl-CoA dehydrogenase deficiency (MCADD), that is one of three types of inherited disorders of mitochondrial fatty-acid β-oxidation. Long-chain and very long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficits are recognized as responsible of hypertrophic or, less frequently, dilated cardiomyopathy (CMP) in childhood. Otherwise, to our knowledge, no case of MCADD associated to dilated CMP has been reported in literature.
1. Case Report
A 2-month-old female infant with severe dilated CMP, put on medication with digitalis and diuretics, was referred to our Institution to undergo heart transplant. Her twin sister died suddenly in the first few days of life, and a clear cause of death was not demonstrated. Initial physical examination revealed growth retardation, due to failure to thrive, hypotonia, S3 gallop, tachypnea, and liver enlargement. The echocardiogram showed severe left ventricular dilatation (end diastolic diameter = 45 mm) with global hypokinesis (Figure 1); furthermore an ejection fraction of 20% was noted (Figure 2).
Figure 1.
Figure 2.
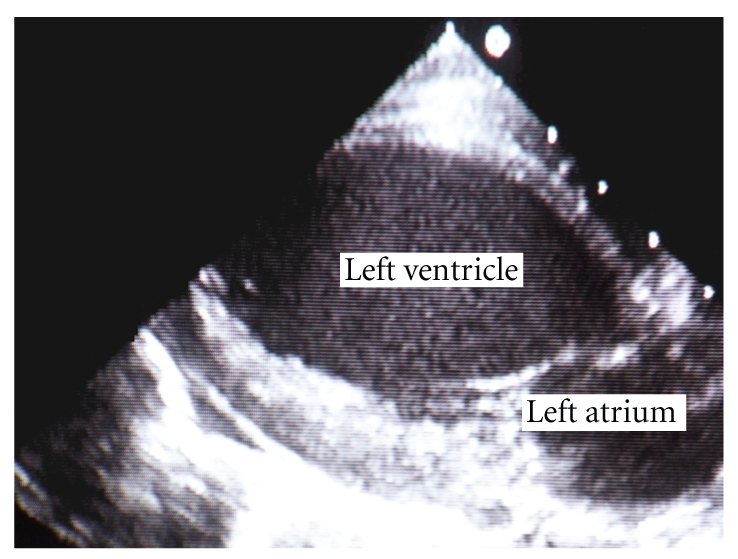
In addition thoracic radiograph evidenced severe cardiomegaly. No significant ventricular arrhythmias were detected. Heart failure required medical treatment with diuretics and digitalis.
Detection of elevated urinary dicarboxylic acids prompted biochemical studies of fatty acid metabolism, which disclosed a normal carnitine level (24 umol/L). Moreover the serum acylcarnitine profile showed elevated levels of C6, C8, C10, and C10.1, that were consistent with diagnosis of MCADD.
After initial treatment with intravenous glucose, the patient was fed with a low-fat diet and carnitine supplementation. Over the next six weeks this treatment led to a dramatic clinical improvement, and she became more active and gained 400 g. Repeated echocardiogram indicated progressive enhancement of ejection fraction to 35%; in addition left ventricular end diastolic diameter reduced to 40 mm. The patient was discharged home on 90th day.
2. Discussion
Despite advances in drug therapy dilated CMP remains one of the leading cause of death and heart transplant in childhood.
Its incidence in infancy is estimated in about 4.4 cases per 100 000 per year, with a high mortality: 50%–60% at two years. Moreover about one third of patients dies or undergoes heart transplant in the first year [1, 2]. Although the most of cases (66%) are defined “idiopathic”, some CMP may have an identifiable cause [1–4]. Identification of underlying cause of CMP in children may improve outcome after specific treatment [1, 3, 4]. Positive family history of heart failure, sudden death, or genetic syndrome is usually predictive of a probable cause for CMP [4].
With the exception of medium chain CoA acyl dehydrogenase deficiency, inborn disorders of fatty-acid β-oxygenation, have been identified as possible causes of CMP in infancy [2]. Alteration of mitochondrial beta-oxidation could be responsible of about 15% of CMP [5]. Congestive heart failure is determined by reduced energetic production due to defect of fatty-acids metabolism that reactivates the less effective foetal glycolytic pathway [6, 7]. Furthermore accumulation of nonesterified fatty acids has toxic effects on ionic channel of cardiac myocytes [5] that may trigger ventricular arrhythmias even fatal [5, 8, 9].
Fatty-acid β-oxygenation disorders are determined by an autosomal recessively inherited deficiency of a single enzyme in the mitochondrial metabolism [4, 6]. The β-oxidation shortens progressively very-long-chain acyl-CoA through a four-step enzymatic process: carnitine cycle, the beta-oxidation cycle, the electron transfer pathway, and the synthesis of ketone bodies. Firstly the carnitine cycle allows the long chain fatty acids to get into the mitochondria; otherwise short- and medium-chain fatty acids may enter directly. Successively the acyl-CoA, produced by the beta-oxidation cycle, can be utilized in the Kreb's cycle as well as in the production of ketone bodies.
The metabolic disorder can involve any of the four different acyl-CoA dehydrogenases, which are of primary importance for beta-oxidation cycle. Clinical manifestations of fatty-acid β-oxygenation deficiencies are usually precipitated by fasting, such as infectious disease, and vary widely according to the specific enzimatic defect [2, 10–12]. MCADD is the most common error in mitochondrial beta-oxidation [13], with a prevalence of 1 case per 15 000 in USA [8] and about 4.5 cases/100 000 live births in England [14]. Its clinical consequences are variable; patients usually manifest before two years of age with life-threatening hypoketotic hypoglycaemia that can progress to coma and death [2, 8, 10].
Clinical presentation of MCADD may be dramatic, even in neonatal age [8, 14], with lethargy, hypoglycaemia, acidosis, and hepatomegaly. Although ventricular tachyarrhythmias and cardiac arrest may occur, no abnormalities of myocardial function have been revealed in these patients. Mortality rate of MCADD ranges from 20% to 25% in undiagnosed patients [13]. Because prompt diagnosis and causal therapy may considerably improve outcome, diagnosis should be considered in neonates and infants with “idiopathic” CMP or life-threatening ventricular arrhythmias [5, 8], especially if family history is suggestive of unexplained sudden infant death. Treatment of fatty-acid oxidation defects includes avoiding fasting and replacement of dietary long chain by low dose of medium or short-chain fatty-acids, providing most of the calories intake as carbohydrates to reduce lipolysis, whereas L-carnitine supplementation is controversial [11].
The case described by us is unusual because a comprehensive review of literature indicates that MCADD has never been reported in association with dilated or hypertrophic CMP.
References
- 1.Towbin JA, Lowe AM, Colan SD, et al. Incidence, causes, and outcomes of dilated cardiomyopathy in children. Journal of the American Medical Association. 2006;296(15):1867–1876. doi: 10.1001/jama.296.15.1867. [DOI] [PubMed] [Google Scholar]
- 2.Kelly DP, Strauss AW. Inherited cardiomyopathies. The New England Journal of Medicine. 1994;330(13):913–919. doi: 10.1056/NEJM199403313301308. [DOI] [PubMed] [Google Scholar]
- 3.Felker GM, Thompson RE, Hare JM, et al. Underlying causes and long-term survival in patients with initially unexplained cardiomyopathy. The New England Journal of Medicine. 2000;342(15):1077–1084. doi: 10.1056/NEJM200004133421502. [DOI] [PubMed] [Google Scholar]
- 4.Cox GF, Sleeper LA, Lowe AM, et al. Factors associated with establishing a causal diagnosis for children with cardiomyopathy. Pediatrics. 2006;118(4):1519–1531. doi: 10.1542/peds.2006-0163. [DOI] [PubMed] [Google Scholar]
- 5.Bonnet D, Martin D, de Lonlay P, et al. Arrhythmias and conduction defects as presenting symptoms of fatty acid oxidation disorders in children. Circulation. 1999;100(22):2248–2253. doi: 10.1161/01.cir.100.22.2248. [DOI] [PubMed] [Google Scholar]
- 6.Mathur A, Sims HF, Gopalakrishnan D, et al. Molecular heterogeneity in very-long-chain acyl-CoA dehydrogenase deficiency causing pediatric cardiomyopathy and sudden death. Circulation. 1999;99(10):1337–1343. doi: 10.1161/01.cir.99.10.1337. [DOI] [PubMed] [Google Scholar]
- 7.Derks TGJ, Reijngoud D-J, Waterham HR, et al. The natural history of medium-chain acyl CoA dehydrogenase deficiency in the Netherlands: clinical presentation and outcome. The Journal of Pediatrics. 2006;148(5):665–670. doi: 10.1016/j.jpeds.2005.12.028. [DOI] [PubMed] [Google Scholar]
- 8.Rice G, Brazelton T, III, Maginot K, Srinivasan S, Hollman G, Wolff JA. Medium chain acyl-coenzyme A dehydrogenase deficiency in a neonate. The New England Journal of Medicine. 2007;357(17):p. 1781. doi: 10.1056/NEJMc071277. [DOI] [PubMed] [Google Scholar]
- 9.Yang Z, Lantz PE, Ibdah JA. Post-mortem analysis for two prevalent β-oxidation mutations in sudden infant death. Pediatrics International. 2007;49(6):883–887. doi: 10.1111/j.1442-200X.2007.02478.x. [DOI] [PubMed] [Google Scholar]
- 10.Vianey-Saban C, Divry P, Brivet M, et al. Mitochondrial very-long-chain acyl-coenzyme A dehydrogenase deficiency: clinical characteristics and diagnostic considerations in 30 patients. Clinica Chimica Acta. 1998;269(1):43–62. doi: 10.1016/s0009-8981(97)00185-x. [DOI] [PubMed] [Google Scholar]
- 11.Dyke PC, II, Konczal L, Bartholomew D, McBride KL, Hoffman TM. Acute dilated cardiomyopathy in a patient with deficiency of long-chain 3-hydroxyacyl-CoA dehydrogenase. Pediatric Cardiology. 2009;30(4):523–526. doi: 10.1007/s00246-008-9351-8. [DOI] [PubMed] [Google Scholar]
- 12.Roe CR, Sweetman L, Roe DS, David F, Brunengraber H. Treatment of cardiomyopathy and rhabdomyolysis in long-chain fat oxidation disorders using an anaplerotic odd-chain triglyceride. The Journal of Clinical Investigation. 2002;110(2):259–269. doi: 10.1172/JCI15311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilson CJ, Champion MP, Collins JE, Clayton PT, Leonard JV. Outcome of medium chain acyl-CoA dehydrogenase deficiency after diagnosis. Archives of Disease in Childhood. 1999;80(5):459–462. doi: 10.1136/adc.80.5.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pollitt RJ, Leonard JV. Prospective surveillance study of medium chain acyl-CoA dehydrogenase deficiency in the UK. Archives of Disease in Childhood. 1998;79(2):116–119. doi: 10.1136/adc.79.2.116. [DOI] [PMC free article] [PubMed] [Google Scholar]