

Abstract
We are exploring cell-based vaccines as a treatment for the 50% of patients with large primary uveal melanomas who develop lethal metastatic disease. MHC II uveal melanoma vaccines are MHC class I+ uveal melanoma cells transduced with CD80 genes and MHC II genes syngeneic to the recipient. Previous studies demonstrated that the vaccines activate tumor-specific CD4+ T cells from patients with metastatic uveal melanoma. We have hypothesized that vaccine potency is due to the absence of the MHC II-associated invariant chain (Ii). In the absence of Ii, newly synthesized MHC II molecules traffic intracellularly via a non-traditional pathway where they encounter and bind novel tumor peptides. Using confocal microscopy, we now confirm this hypothesis and demonstrate that MHC II molecules are present in both the endosomal and secretory pathways in vaccine cells. We also demonstrate that uveal melanoma MHC II vaccines activate uveal melanoma-specific, cytolytic CD8+ T cells that do not lyse normal fibroblasts or other tumor cells. Surprisingly, the CD8+ T cells are cytolytic for HLA-A syngeneic and MHC I-mismatched uveal melanomas. Collectively, these studies demonstrate that MHC II uveal melanoma vaccines are potent activators of tumor-specific CD4+ and CD8+ T cells and suggest that the non-conventional intracellular trafficking pattern of MHC II may contribute to their enhanced immunogenicity. Since MHC I compatibility is unnecessary for the activation of cytolytic CD8+ T cells, the vaccines could be used in uveal melanoma patients without regard to MHC I genotype.
Keywords: Tumor immunology, Antigen presentation, MHC II intracellular trafficking
Introduction
Uveal melanoma is the most common intra-ocular malignancy. Although there are effective treatments for primary tumors in the eye, 50% of patients who develop large primary tumors develop metastatic disease and die within 6–15 months of diagnosis [20, 24]. Because there are no effective therapies for metastatic disease, we are exploring cell-based vaccines as a therapeutic option. An optimal vaccine will activate tumor-specific immunity that eliminates or controls existing metastatic disease and provides long-term protection against the outgrowth of latent metastatic cells. Because of the critical role of CD4+ T cells in facilitating the development of cytotoxic CD8+ T cells and immune memory, we have designed vaccines that specifically target the activation of tumor-reactive CD4+ T lymphocytes [13].
Our MHC II vaccines are tumor cells that constitutively express MHC class I (MHC I), but not MHC class II (MHC II) molecules, and are transduced with constructs encoding the CD80 molecule and an MHC II allele matched to the patient’s HLA haplotype. Expression of these transgenes enables vaccine cells to present MHC II-restricted peptides and the requisite co-stimulatory signal to activate T cells and function as antigen presenting cells (APC) [2, 3]. Professional APC, such as dendritic cells (DC), contain the accessory molecule invariant chain (Ii), which binds to the peptide binding cleft of newly synthesized MHC II molecules. Newly synthesized MHC II molecules with their associated Ii have two alternative intracellular trafficking patterns. Their predominant pathway is to traffic directly to endosomal compartments. However, they may also first transit to the plasma membrane where they are then endocytosed into endosomal vesicles. In either case, Ii occupies the MHC II binding groove during trafficking so that peptides are unable to bind until the MHC II molecules reach endosomal compartments where the acidic environment degrades Ii and the binding cleft becomes accessible to peptides [5, 10, 14, 22, 32, 39].
In contrast, MHC II vaccines do not contain Ii and, therefore, peptides can bind to MHC II molecules in locations other than endocytic compartments. As a result, MHC II vaccines have the potential to present novel tumor-encoded MHC II-restricted peptides that are not presented by professional APC. These novel peptides may have heightened antigenicity because they have not previously been seen by the host’s immune system. In vitro studies with peripheral blood mononuclear cells (PBMC) from healthy donors and cancer patients demonstrated that MHC II vaccines activate a diverse repertoire of IFNγ-secreting, tumor-specific CD4+ T cells, and that the repertoire of CD4+ T cells activated by MHC II vaccines is distinct from the repertoire activated by Ii+ APC [31, 34].
We now report that, in addition to being potent activators of CD4+ T cells, MHC II uveal melanoma vaccines also activate tumor-specific CD8+ T cells that are cytotoxic for wild-type uveal melanoma tumor cells. Confocal microscopy studies indicate that MHC II molecules in the Ii− vaccine cells preferentially traffic through Rab3b+ secretory vesicles, while MHC II molecules of Ii+ cells are mostly absent from these vesicles. As a result, in Ii− vaccine cells, MHC II molecules have an altered intracellular trafficking pattern relative to MHC II molecules in Ii+ cells, consistent with our hypothesis that the absence of Ii facilitates T cell activation by re-directing MHC II molecules to different intracellular compartments where they bind atypical peptides.
Materials and methods
Cell lines, PBMC
Primary (MEL202, MEL270, MEL205) and metastatic (OMM2.3) uveal melanoma cell lines were established from uveal melanoma patients and cultured as described [37]. OMM2.3 and MEL270 were derived from the same patient. SUM159PT mammary carcinoma cells were cultured as described [13]. HS68 normal human foreskin fibroblasts were obtained from the American Type Tissue Culture Collection (ATCC) and cultured as recommended by ATCC. Blood samples were obtained from healthy donors and uveal melanoma patients by venipuncture as described [9, 33]. All cell lines and procedures with human materials were approved by the Institutional Review Boards of the participating institutions.
HLA typing
HLA typing was performed as described [9]. HLA genotypes are referred to by their short-hand form (i.e. HLA-DRB1*0101 is DR1).
Retroviral constructs, transductions, and drug selection
To generate the MHC II-eGFP (enhanced green fluorescence protein) construct, the β chain of the pLNCX2/DR1 plasmid [13] was replaced with the DR1β-eGFP gene from DR1β-eGFP pcDNA3 [41]. DR1β-eGFP pcDNA3 and pLNCX2/DR1 were digested separately with NotI (Fermentas Inc., Glen Burnie, MD) and BspEI (New England Biolabs, Ipswich, MA), which created identical and annealing 5′ ends. The cleaved pLNCX2–DR1 vector was dephosphorylated for 1 h at 37°C using 1.5 units of shrimp alkaline phosphatase (USB, Cleveland, OH). The reaction was stopped by heating to 65°C for 15 min and the digested plasmids were run on a 0.7% agarose gel. The appropriate bands were excised, purified using a QIAEX II Gel Extraction Kit (Qiagen, Valencia, CA) and ligated with T4 DNA ligase (Invitrogen, Carlsbad, CA) at a ratio of 2:1 of vector to insert. Ligated constructs were transformed into DH5α cells by electroporation and transformants were selected on LB-agar plates containing 100 μg/ml ampicillin. Positive clones were identified by digestion with BspEI and NotI.
The pLHCX/CD80 and pLPCX/Ii retroviral constructs, retrovirus production, transductions, and drug selections for DR1 and DR1-eGFP (600 μg/ml G418, Sigma, St. Louis, MO), CD80 (75 μg/ml hygromycin, Calbiochem, San Diego, CA) and Ii (0.2 μg/ml puromycin, Clontech) were performed as described [13, 34]. Transduced cells were grown in the same medium as their parental cells [9] and have been maintained in culture for >1 year.
Fluorescence microscopy, flow cytometry
For fluorescence microscopy, tumor cells were cultured to ~75% confluency in six-well plates containing no. 1 coverslips at 2.5 × 106 cells/5 ml/well. Coverslip-adherent cells were labeled for cell surface molecules with primary and secondary antibodies for 30 min to 1 h with gentle agitation on a shaker platform and washed with excess PBS–10% FCS before, between, and after mAb incubations. All procedures subsequent to cell culture were carried out on ice with ice-cold reagents. For internal staining, the cells were fixed with 2% formaldehyde, blocked with PBS containing 10% FCS, and then stained. All reagents for internal staining contained 0.2% saponin. After staining, some samples were incubated with 4′,6-diamidino-2-phenylindole (DAPI) for 10 min at 4°C with gentle agitation to visualize the nuclei. Coverslips were mounted on a 1 mm glass microscope slide using Fluoromount G (Southern Biotech, Birmingham, AL), sealed with nail polish, and observed using an Olympus IX81 (Olympus, Center Valley, PA) fluorescent or Leica SP5 confocal microscope. Background fluorescence was established using unstained and isotype control stained slides. Three to five fields per slide containing ~30–50 cells per field were observed and images were analyzed using Volocity or Leica image analysis software or NIH-ImageJ freeware. Fluorescence staining for flow cytometry was performed as described [9].
Antibodies, fluorescent probes, and western blots
Mouse anti-human CD63 (IgG1; late endosome MIIC marker) and mouse anti-human Rab5 (IgG2a; early endosome marker) mAbs were used at 1 μg/ml and 0.5 μg/ml, respectively, and were purchased from BD Biosciences (San Jose, CA). Mouse anti-human Rab3b (IgG2a; secretory vesicle marker) mAb was used at 3 μg/ml and was from Abcam (Cambridge, MA). Isotype control (anti-mouse IgG1 and IgG2a) mAbs were used at the same dilution as their corresponding specific mAb. Second step antibodies for microscopy (goat-anti-mouse IgG-ALEXA 488 and goat-anti-mouse IgG-ALEXA 568) and Lysotracker Red were from Molecular Probes (Carlsbad, CA). Culture supernatants of mAbs to HLA-DR (mAb L243), HLA-ABC (mAb W6/32), and Ii (mAb PIN1.1) were prepared as described [13]. Western blots were done as described [27, 33] using culture supernatant from hybridoma PIN1.1 at a 1:100 dilution followed by sheep-anti-mouse-horseradish peroxidase (Amersham, Piscataway, NJ) at a 1:10,000 dilution.
PBMC priming with MHC II tumor cell vaccines
Peripheral blood mononuclear cells were primed and expanded in vitro with vaccine cells as previously described [9] with the following modifications: cells were expanded with IL-15 (20 ng/ml; PeproTech, Rocky Hill, NJ) for 7 days instead of with IL-2 for 5 days. The resulting primed PBMC were immediately used or frozen and stored in liquid nitrogen.
Lactate dehydrogenase (LDH) release assay
Titered numbers of primed PBMC (2 × 104 to 4 × 103/200 μl total volume) and transductants or parental cells (2 × 103/200 μl total volume T cell medium) [9] were co-cultured in U bottom 96-well plates at 37°C and 5% CO2. After 16 h of culture, the 96-well plates were centrifuged, and the supernatants were collected and assayed for LDH using a Promega Cytotox 96 Kit (Promega, Madison, WI) according to the manufacturer’s instructions. Maximum LDH release from detergent lysed target cells and spontaneous release from effector and target cells served as controls. Percent cytotoxicity = [experimental − (effector spontaneous + target spontaneous)/(target maximum − target spontaneous)] × 100%.
51Cr release assay
Chromium release assays were performed as described [26] with the following modifications: target cells were incubated at up to 107 cells in 100 μl FCS with 150 μC 51Cr (Perkin-Elmer, Shelton, CN) at 37°C 1 h, washed with excess serum-free medium, resuspended in complete medium with 10% FCS, incubated an additional 30 min at 37°C, washed with excess medium and resuspended in T cell medium. Labeled target cells (1 × 104 per well) were co-cultured with varying numbers of effector cells in a total volume of 200 μl T cell medium/well in U bottom 96-well plates and cultured for 16 h after which the plates were centrifuged (1,200 rpm for 3 min), and 50 μl of supernatant from each well were mixed with 250 μl of Optiphase aqueous scintillation fluid (Perkin-Elmer) and counted in a Wallac Trilux Microbeta scintillation counter. Data points are the average ± SD of triplicate wells. Spontaneous release was measured in the absence of effectors; total release was measured as cpm released in the presence of 100 μl of 10% NP-40 per well. Spontaneous release ranged from 10 to 25%. For some experiments, PBMC were depleted for CD8+ and/or CD56+ cells as described [9, 13] prior to the 51Cr release assay. Depleted populations contained less than 2 and 1% of CD8+ T cells and NK cells, respectively.
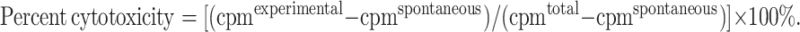 |
CD107a mobilization assay
Effector (2 × 105 per well) and target cells (2 × 104 per well) were co-cultured at a ratio of 1:10 in the presence of mAb CD107a-PE (eBioscience) in 200 μl T cell medium/well in U bottom 96-well plates at 37°C and 5% CO2. After 1 h, monensin (Golgistop, BD Biosciences) was added to each well and the cells cultured for an additional 5 h. Six hours later, cells were harvested, washed and stained for cell surface markers CD3-biotin, CD4-FITC, CD8-FITC or CD56-FITC and SA-PerCP, or isotype controls, and analyzed by flow cytometry (Epics XL flow cytometer and Expo 32 software, Beckman/Coulter, Fullerton, CA). For some experiments, cells were fixed in 4% formaldehyde, stored at 4°C, and analyzed the next day.
Statistical analysis
Standard deviations were calculated using Excel v2002.
Results
HLA-DR is stably expressed at the cell surface of uveal melanoma cells in the absence of Ii
We hypothesized that in the absence of Ii the intracellular trafficking pattern of newly synthesized MHC II molecules is changed as compared to the classical trafficking pathway in Ii+ APC. To test this hypothesis, we have generated uveal melanoma transductants containing HLA-DR1 fused to green fluorescence protein (HLA-DR1-eGFP) and used fluorescence microscopy to localize the recombinant proteins. A MHC II-eGFP retroviral plasmid was constructed by replacing the HLA-DRβ1 gene of the pLNCX2/DR1 plasmid [13] with the HLA-DRβ1 gene fused at its 3′ end to the eGFP gene [41] (Fig. 1a). Retroviruses were then prepared containing the MHC II-eGFP construct and MHC II-Ii− human primary uveal melanoma cells (MEL202) were transduced with the retroviruses to generate MEL202/DR1-eGFP cells. To study the effects of Ii on intracellular trafficking of HLA-DR molecules, MEL202/DR1-eGFP cells were further transduced with retroviruses containing the pLPCX/Ii construct [34] to generate Ii+ MEL202/DR1-eGFP/Ii cells. MEL202/DR1 cells [9] were similarly transduced to generate MEL202/DR1/Ii cells. The resulting cells were selected on the appropriate selection drug and have been maintained in culture for more than 1 year.
Fig. 1.

MHC II-eGFP- and HLA-DR1-transduced uveal melanoma cells express conformationally correct HLA-DR in the absence of invariant chain. a The MHC II-eGFP construct was generated by replacing the wild-type DRB1 gene in the pLNCX2/DR1 construct with the wild-type DRB1 gene fused at its 3′ end to the gene encoding eGFP. b Primary uveal melanoma MEL202 cells transduced with MHC II-eGFP, HLA-DR1, and/or Ii retroviruses were stained with mAb L243-PE which detects conformationally correct HLA-DR molecules, mAb W6/32-FITC which detects HLA-A, B, and C molecules, isotype control mAbs. eGFP cells were unstained when analyzed for eGFP. All samples were analyzed by flow cytometry. c Detergent lysates of MEL202 parental cells and transductants and control Sweig and Jurkat cells were electrophoresed on 10% SDS-PAGE gels under non-reducing conditions and transferred to nitrocellulose. Blots were stained for Ii (mAb PIN1.1) or β-tubulin (mAb anti-β-tubulin clone 2.1). Sweig and Jurkat cells are Ii+ and Ii− control cells, respectively. Data for b and c are from one of three and two independent experiments, respectively
To ascertain that the retrovirally transduced uveal melanoma cells contain properly conformed HLA-DR1 molecules, the transductants were stained with the HLA-DR-specific, conformation-dependent L243 mAb [35] and analyzed by flow cytometry (Fig. 1b). This mAb only recognizes mature MHC II-peptide complexes and does not recognize MHC II-Ii+ complexes [4, 39]. The HLA-DR1 and HLA-DR1-eGFP transductants express cell surface HLA-DR and co-expression of Ii does not significantly alter HLA-DR levels. As expected, transduction with HLA-DR1 or Ii does not affect expression of MHC class I molecules as detected by the pan HLA-A, B, C mAb W6/32 (Fig. 1b). These findings are in agreement with previous studies showing that MHC II-eGFP fusion molecules behave in a similar fashion to wild-type MHC II molecules [8, 41].
Ii expression in the transductants was confirmed by western blotting (Fig. 1c). Therefore, HLA-DR1 wild-type and HLA-DR1-eGFP fusion proteins are stably expressed in the uveal melanoma transductants, and the absence of Ii does not significantly impact MHC II cell surface expression.
MHC II molecules traffic intracellularly via the endocytic and secretory pathways in the absence of Ii
We have used intracellular staining to localize HLA-DR molecules in the uveal melanoma transductants to test the hypothesis that the absence of Ii alters the intracellular trafficking pattern of HLA-DR. As expected, HLA-DR associates with Ii in MEL202/DR1-eGFP cells as shown by co-localization of eGFP (HLA-DR1) and Ii (PIN1.1 mAb) (Fig. 2a). Ii and to a lesser extent the carboxyl end of the HLA-DRβ chain contain trafficking signals that ultimately direct stable MHC II complexes to endocytic compartments. To determine if HLA-DR1 molecules are present in endosomal and/or secretory pathways, MHC II/DR1-eGFP and MHC II/DR1-eGFP/Ii cells were intracellularly stained with mAbs specific for organelles of these pathways. In both Ii+ and Ii− cells, HLA-DR1 (eGFP) co-localizes with CD63 (Fig. 2b), a marker for late endosomal MHC II compartments (MIIC) [42] where exogenously derived peptides are loaded onto MHC II molecules [10]. MHC II molecules of Ii+ and Ii− cells are also present in early endosomes as seen by co-localization of eGFP with Rab5, a marker of early endosomes [40] (Fig. 2b). Therefore, MHC II molecules traffic via the endocytic pathway in both Ii+ and Ii− cells.
Fig. 2.

MHC II molecules traffic via the endocytic and secretory pathways in the absence of invariant chain. Cells in all panels were fixed and permeabilized before antibody staining and observed by confocal microscopy. a MHC II and Ii co-localize in MEL202/DR1-eGFP/Ii+ cells. MEL202/DR1-eGFP/Ii+ cells stained for Ii (mAb PIN1.1). b MHC II molecules localize to endocytic compartments in Ii+ and Ii− MEL202 transductants. MEL202/DR1-eGFP and MEL202/DR1-eGFP/Ii cells stained for endocytic markers (mAbs Rab5 and CD63). c MHC II molecules frequently localize to Rab3b+ secretory vesicles in Ii−, but not in Ii+, MEL202/DR1-eGFP cells. MEL202/DR1-eGFP and MEL202/DR1-eGFP/Ii cells stained for a secretory vesicle marker (mAb Rab3b). Arrows indicate areas of co-localized endosomal staining. a–c Each image is representative of 3–5 fields per slide in two independent experiments in which a total of ~30–50 cells per field were observed
To assess if MHC II molecules also transit via the secretory pathway, Ii+ and Ii− transductants were intracellularly stained with Rab3b, a marker for secretory vesicles [16] (Fig. 2c). eGFP (MHC II) is frequently present in Rab3b+ secretory vesicles of MEL202/DR1-eGFP cells, but is rarely present in Rab3b+ secretory vesicles in MEL202/DR1-eGFP/Ii cells. Therefore, in the absence of Ii, MHC II molecules traffic via the endocytic and secretory routes, while in the presence of Ii, MHC II molecules are predominant in the endocytic pathway.
MHC II uveal melanoma vaccines activate uveal melanoma-specific cytolytic effectors from healthy donors and uveal melanoma patients
Previous studies established that the MHC II uveal melanoma vaccines expressing high levels of HLA-DR and CD80 [9] are strong activators of tumor-specific CD4+ T cells provided the vaccines and PBMC share MHC II alleles [9]. Since MHC II uveal melanoma vaccines also express MHC I alleles, we hypothesized that the vaccines may also activate tumor-specific cytotoxic CD8+ T cells.
To test this hypothesis, A3,A24+ DR1+ PBMC from healthy donors were primed with vaccine cells that were MHC I and II matched to the PBMC (MEL202/DR1/CD80 vaccines; A1,3+ DR1+), and the primed PBMC tested for cytotoxic activity against MHC I-matched (MEL202: A1,3+) and -mismatched (OMM2.3, MEL270: both A11,29+) targets (Fig. 3a). MEL202 and MEL270 (primary) and OMM2.3 (metastatic) cells were used as targets to determine if the vaccines were equally effective against primary and metastatic uveal melanoma cells. Maximum cytotoxicity was obtained against unmodified, MHC I-matched MEL202 cells; however, significant cytotoxicity was also seen against MHC I-mismatched MEL270 and OMM2.3 targets. These results suggest that at least some of the cytotoxic PBMC effectors activated by the uveal melanoma vaccines are not MHC I restricted.
Fig. 3.

MHC II uveal melanoma vaccines activate tumor-specific, MHC I-unrestricted, cytolytic effector cells from healthy donors and uveal melanoma patients. HLA-A3,24+ DR1+ (a), HLA-A74+ DR1+ (b), HLA-A2,74+ DR1+ (c) PBMC from healthy donors, or HLA-A33+ DR1+ PBMC from metastatic patient M-185 (d) were primed with MEL202/DR1/CD80 vaccine cells (HLA-A1,A3+ DR1+) and tested by LDH release (c) or 51Cr release (a, b, d) for cytotoxicity against primary (MEL202: HLA-A1,A3+; MEL270: HLA-A11,29+) or metastatic (OMM2.3: HLA-A11,29+) uveal melanoma, or mammary carcinoma (SUM159PT: HLA-A2,24+) target cells. Data for a, b, c, and d are from one of four, three, three, and two independent experiments, respectively
To clarify if the vaccine cells and PBMC must share MHC I alleles, A74+ DR1+ PBMC from healthy donors were primed with fully MHC I-mismatched A1,3+ DR1+ vaccine cells (MEL202/DR1/CD80), and the primed PBMC were then tested for cytolytic activity against fully mismatched uveal melanomas (Fig. 3b). As measured by 51Cr release, primed HLA-A74+ DR1+ PBMC lysed fully mismatched wild-type HLA-A1,3+ MEL202 as well as OMM2.3 and MEL270 (both HLA-A11,29+) targets, confirming that the vaccines activate MHC I-non-restricted effectors. To determine if vaccine-activated effectors were uveal melanoma-specific, HLA-A2,74+ DR1+ PBMC were primed with MEL202/DR1/CD80 vaccines (HLA-A1,3+) and tested for cytotoxic activity against uveal melanoma and MHC I-matched mammary carcinoma cells (SUM159PT: HLA-A2,24+) (Fig. 3c). As seen previously, MHC I compatibility was not needed for cytotoxicity against parental primary and metastatic uveal melanoma (MEL202 and OMM2.3, respectively). Interestingly, mammary carcinoma cells were not significantly lysed even though they shared one MHC I allele (A2) with the primed PBMC. These data demonstrate that MHC II uveal melanoma vaccines activate effector cells that are cytotoxic for wild-type primary and metastatic uveal melanoma targets regardless of their MHC I genotype, and do not react with non-uveal melanoma targets.
To determine if the vaccines also activate effector cells from uveal melanoma patients, HLA-A33+ DR1+ PBMC from metastatic patient M-185 were primed with MHC class I-mismatched MEL202/DR1/CD80 vaccine cells and tested for cytotoxicity against primary and metastatic uveal melanoma cells (Fig. 3d). Vaccine-activated patient’s PBMC showed similar cytolytic activity for MHC I-mismatched uveal melanoma targets from either primary or metastatic tumors. Therefore, MHC II uveal melanoma vaccines activate cytolytic effector cells from healthy donors and uveal melanoma patients.
MHC II uveal melanoma vaccines activate tumor-specific CD8+ T cells
We next identified the effector cells activated by the MHC II uveal melanoma vaccines. CD107a is an epitope of the Lamp-1 protein contained within cytolytic vesicles. It is used as a marker of cytotoxic CD8+ effector T cells [29]. HLA-A2,74+ DR1+ PBMC from a healthy donor were primed with MEL202/DR1/CD80 vaccine cells and subsequently co-cultured with primary (MEL202, MEL270) or metastatic (OMM2.3) uveal melanoma cells in the presence of fluorescently labeled CD107a antibodies. Resulting cells were stained for CD3, CD8, CD4, and NK (CD56) markers and analyzed by flow cytometry. CD107a was expressed by a subset of CD3+CD8+ T cells (Fig. 4a) and NK cells (data not shown), but not by CD3+CD4+ T cells (data not shown), suggesting that both CD8+ T cells and NK cells were activated by the vaccines. To determine which effector population(s) was cytolytic for uveal melanoma targets, aliquots of the vaccine-primed PBMC were depleted for CD56+ or CD8+ plus CD56+ cells after priming and subsequently tested for cytolytic activity against primary (MEL202, MEL270, MEL205) and metastatic (OMM2.3) uveal melanoma and non-malignant human foreskin fibroblasts (HS68) (Fig. 4b). Combined depletion of CD8+ and CD56+ cells eliminated target cell lysis, while depletion of NK (CD56+) cells only slightly reduced lysis. Normal fibroblasts were not killed despite their expression of MHC I alleles shared with the vaccine cells (HLA-A1) or with the uveal melanoma targets (HLA-A29). Therefore, MHC II uveal melanoma vaccines activate CD8+ T cells that are cytolytic for unmodified, MHC I-matched and -mismatched uveal melanoma cells, and do not kill non-malignant cells.
Fig. 4.

MHC II uveal melanoma vaccines activate tumor-specific, CD8+ T cells that are cytolytic for MHC I-matched and -mismatched wild-type uveal melanoma targets. a HLA-A2,74+ DR1+ PBMC from a healthy donor were primed with MEL202/DR1/CD80 (HLA-A1,3+) vaccine cells, co-cultured with mAbs to CD107a and primary (MEL202, MEL270) or metastatic (OMM2.3) uveal melanoma targets, and subsequently labeled with mAbs to CD3 or CD8. CD3+CD8+ T cells were gated and analyzed for CD107a expression relative to primed PBMC cultured in the absence of target cells. b HLA-A2,A74+ DR1+ PBMC from a healthy donor were primed with MEL202/DR1/CD80 vaccine cells and tested for cytotoxic activity against 51Cr-labeled primary (MEL202, MEL270, MEL205: HLA-A2, 24+) or metastatic (OMM2.3) uveal melanoma or non-malignant human foreskin fibroblasts (HS68: HLA-A1,29+). PBMC were undepleted or depleted for CD8+ and CD56+ or CD56+ cells after priming and before the cytotoxic assay. CD56 and CD8 plus CD56-depleted PBMC contained <2 and 1% of CD8+ T cells and NK cells. Data for a are from one of three independent experiments. Data for CD56 and CD56 plus CD8 depletions of b are one of four and two independent experiments, respectively
Discussion
MHC II uveal melanoma vaccines were designed to activate the host’s immune system to a large repertoire of tumor peptides. They were based on the concept that by presenting novel MHC II-restricted peptides they would activate CD4+ T cells that would not be activated by professional APC. The activated CD4+ T cells would then facilitate the activation of tumor-specific CD8+ T cells and promote long-term immunity against metastatic disease [13]. We hypothesized that the ability of the vaccines to present novel MHC II-restricted peptides would result from an altered intracellular trafficking pathway of MHC II molecules due to the absence of Ii chain [9, 34]. The confocal experiments presented here demonstrate that the absence of Ii allows MHC II molecules to preferentially enter Rab3b+ secretory vesicles in addition to trafficking through the endocytic pathway. Since these secretory vesicles are distinct from the endocytic pathway and contain peptides derived from different sources, our results support the concept that vaccine MHC II molecules are likely to bind peptides that are not traditionally presented by MHC II molecules of professional APC. The vaccines also activate CD8+ cytolytic T cells that are specific for uveal melanoma targets and are not cytolytic for non-uveal melanoma tumor targets or for non-malignant fibroblasts.
Early studies in Ii chain knock-out mice indicated that MHC II molecules are unstable or dysfunctional in the absence of Ii [7, 15, 38], while other reports demonstrated that Ii was not necessary for correct MHC II expression [19, 23, 30]. Instability in the absence of Ii appears relevant only to some MHC II alleles [6], and may be limited to mouse MHC II molecules, since many human MHC II alleles have been successfully expressed in the absence of Ii (J. Thompson, M. Srivastava, and S. Ostrand-Rosenberg, unpublished). Ii is not essential for expression of functional MHC II molecules, but constitutive expression of CLIP, the portion of Ii that binds to the MHC II-peptide binding site, has been associated with poor prognosis of patients with myeloid leukemia [11, 12], and heightened Ii or CLIP expression is associated with the inability to make a Th1 response [28, 36] that favors anti-tumor immunity. In fact, down-regulation of Ii by RNAi in myeloid leukemic blasts facilitates activation of type 1 CD4+ T cells (van Luijn et al., Invariant chain and CLIP down-modulation enhances the immunogenicity of myeloid leukemic blasts resulting in increased CD4+ T cell responses; submitted for publication). In addition, similar results have been obtained using MHC II vaccines prepared from lung and breast cancer cells [31, 34]. Therefore, Ii expression is not essential for MHC II expression and function, and its absence may facilitate anti-tumor immunity through multiple mechanisms.
Although MHC II molecules of professional Ii+ APC typically present peptides derived from endocytosed, exogenously synthesized molecules, a subset of their peptides are derived from endogenous sources [25]. Acquisition of these peptides has been ascribed to three mechanisms. (a) Binding of ER-resident peptides to MHC II molecules prior to binding of Ii; (b) binding of unfolded ER-resident peptides to MHC II molecules prior to binding of Ii and subsequent processing of the peptides in endosomal compartments; and (c) transport of cytosolic peptides to endosomal/lysosomal compartments by autophagy and their subsequent binding to MHC II molecules [1, 21].
Our results suggest that the absence of Ii facilitates the presentation of endogenous peptides by two mechanisms. In the absence of Ii, the MHC II-peptide binding groove is not occupied, thus newly synthesized MHC II molecules can readily bind endogenous peptides in the ER or other pre-endocytic compartments. Secondly, since newly synthesized MHC II molecules traffic through Rab3b+ secretory compartments, they encounter a repertoire of endogenous peptides that are not encountered by MHC II molecules that do not enter these secretory vesicles. Therefore, the absence of the Ii chain not only facilitates the presentation of peptides derived from endogenous sources, but also re-directs MHC II molecules to a variant pathway where they potentially encounter and bind a different repertoire of peptides. The variant trafficking pattern combined with the advantages of vaccine cells made from tumors from an immune privileged site may explain the immunogenicity of the MHC II uveal melanoma vaccines.
Previous experiments established that MHC II uveal melanoma vaccines must be HLA-DR matched to patients to induce tumor-specific CD4+ T cells [9]. Our finding that CD8+ T cells activated by the vaccines are cytolytic for MHC I mismatched, as well as syngeneic, uveal melanoma targets indicates that MHC I matching is not necessary. Kan-Mitchell [17, 18] also found that CD8+ T cells from patients with uveal melanoma recognized targets independent of MHC class I genotype. Whether uveal melanomas are unusual in inducing this type of allogeneic cytotoxicity and how the activated CD8+ T cells recognize MHC I-mismatched targets are unclear. However, allogeneic reactivity simplifies translation of the vaccines to the clinic. Since matching for MHC I alleles is not necessary, and only one of the patient’s MHC II alleles must be matched to the vaccine [9], it will not be necessary to generate a specific vaccine for each patient. Rather, a frozen bank of established vaccines expressing the most common HLA-DR alleles could be generated, and a patient’s HLA-DR genotype would determine which vaccine would be appropriate. This “semi-customized” approach would limit the diversity of vaccines needed, and would allow for treatment of most uveal melanoma patients.
Acknowledgments
We thank Dr. J. Neefjes for his kind gift of the DR1β-eGFP pcDNA3 plasmid, Dr. J. Leach for use of her fluorescent microscope, Ms. C. Petty for assistance with the confocal microscopy, Ms. V. Clements for her excellent technical assistance, and Dr. P. Chen for his helpful discussions. These studies were supported by NIH R01CA52527, R01CA84232 (SOR), and NIH R01EY016486 (BRK). JJB was partially supported by post-doctoral fellowships from Fight for Sight, Inc., Rotterdamse Vereniging Blindenbelangen, Stichting Blindenhulp, Stichting Blinden-Penning, Stichting Dondersfonds, Stichting Nelly Reef Fund, Gratama Stichting, Stichting Admiraal van Kinsbergen Fonds, and Foundation ‘De Drie Lichten’. UKI was supported by a MARC-U-STAR training grant (NIH/NIGMS GM08663).
References
- 1.Aichinger G, Karlsson L, Jackson MR, Vestberg M, Vaughan JH, Teyton L, Lechler RI, Peterson PA. Major histocompatibility complex class II-dependent unfolding, transport, and degradation of endogenous proteins. J Biol Chem. 1997;272:29127–29136. doi: 10.1074/jbc.272.46.29127. [DOI] [PubMed] [Google Scholar]
- 2.Armstrong TD, Clements VK, Martin BK, Ting JP, Ostrand-Rosenberg S. Major histocompatibility complex class II-transfected tumor cells present endogenous antigen and are potent inducers of tumor-specific immunity. Proc Natl Acad Sci USA. 1997;94:6886–6891. doi: 10.1073/pnas.94.13.6886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Armstrong TD, Clements VK, Ostrand-Rosenberg S. MHC class II-transfected tumor cells directly present antigen to tumor-specific CD4+ T lymphocytes. J Immunol. 1998;160:661–666. [PubMed] [Google Scholar]
- 4.Benaroch P, Yilla M, Raposo G, Ito K, Miwa K, Geuze HJ, Ploegh HL. How MHC class II molecules reach the endocytic pathway. EMBO J. 1995;14:37–49. doi: 10.1002/j.1460-2075.1995.tb06973.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berger AC, Roche PA. MHC class II transport at a glance. J Cell Sci. 2009;122:1–4. doi: 10.1242/jcs.035089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bikoff EK, Germain RN, Robertson EJ. Allelic differences affecting invariant chain dependency of MHC class II subunit assembly. Immunity. 1995;2:301–310. doi: 10.1016/1074-7613(95)90054-3. [DOI] [PubMed] [Google Scholar]
- 7.Bikoff EK, Huang LY, Episkopou V, van Meerwijk J, Germain RN, Robertson EJ. Defective major histocompatibility complex class II assembly, transport, peptide acquisition, and CD4+ T cell selection in mice lacking invariant chain expression. J Exp Med. 1993;177:1699–1712. doi: 10.1084/jem.177.6.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boes M, Cerny J, Massol R, Op den Brouw M, Kirchhausen T, Chen J, Ploegh HL. T-cell engagement of dendritic cells rapidly rearranges MHC class II transport. Nature. 2002;418:983–988. doi: 10.1038/nature01004. [DOI] [PubMed] [Google Scholar]
- 9.Bosch JJ, Thompson JA, Srivastava MK, Iheagwara UK, Murray TG, Lotem M, Ksander BR, Ostrand-Rosenberg S. MHC class II-transduced tumor cells originating in the immune-privileged eye prime and boost CD4(+) T lymphocytes that cross-react with primary and metastatic uveal melanoma cells. Cancer Res. 2007;67:4499–4506. doi: 10.1158/0008-5472.CAN-06-3770. [DOI] [PubMed] [Google Scholar]
- 10.Busch R, Rinderknecht CH, Roh S, Lee AW, Harding JJ, Burster T, Hornell TM, Mellins ED. Achieving stability through editing and chaperoning: regulation of MHC class II peptide binding and expression. Immunol Rev. 2005;207:242–260. doi: 10.1111/j.0105-2896.2005.00306.x. [DOI] [PubMed] [Google Scholar]
- 11.Chamuleau ME, Ossenkoppele GJ, van de Loosdrecht AA. MHC class II molecules in tumour immunology: prognostic marker and target for immune modulation. Immunobiology. 2006;211:619–625. doi: 10.1016/j.imbio.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 12.Chamuleau ME, Souwer Y, Van Ham SM, Zevenbergen A, Westers TM, Berkhof J, Meijer CJ, van de Loosdrecht AA, Ossenkoppele GJ. Class II-associated invariant chain peptide expression on myeloid leukemic blasts predicts poor clinical outcome. Cancer Res. 2004;64:5546–5550. doi: 10.1158/0008-5472.CAN-04-1350. [DOI] [PubMed] [Google Scholar]
- 13.Dissanayake SK, Thompson JA, Bosch JJ, Clements VK, Chen PW, Ksander BR, Ostrand-Rosenberg S. Activation of tumor-specific CD4(+) T lymphocytes by major histocompatibility complex class II tumor cell vaccines: a novel cell-based immunotherapy. Cancer Res. 2004;64:1867–1874. doi: 10.1158/0008-5472.CAN-03-2634. [DOI] [PubMed] [Google Scholar]
- 14.Dodi AI, Brett S, Nordeng T, Sidhu S, Batchelor RJ, Lombardi G, Bakke O, Lechler RI. The invariant chain inhibits presentation of endogenous antigens by a human fibroblast cell line. Eur J Immunol. 1994;24:1632–1639. doi: 10.1002/eji.1830240727. [DOI] [PubMed] [Google Scholar]
- 15.Elliott EA, Drake JR, Amigorena S, Elsemore J, Webster P, Mellman I, Flavell RA. The invariant chain is required for intracellular transport and function of major histocompatibility complex class II molecules. J Exp Med. 1994;179:681–694. doi: 10.1084/jem.179.2.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fukuda M. Regulation of secretory vesicle traffic by Rab small GTPases. Cell Mol Life Sci. 2008;65:2801–2813. doi: 10.1007/s00018-008-8351-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang XQ, Mitchell MS, Liggett PE, Murphree AL, Kan-Mitchell J. Non-fastidious, melanoma-specific CD8+ cytotoxic T lymphocytes from choroidal melanoma patients. Cancer Immunol Immunother. 1994;38:399–405. doi: 10.1007/BF01517210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kan-Mitchell J, Liggett PE, Harel W, Steinman L, Nitta T, Oksenberg JR, Posner MR, Mitchell MS. Lymphocytes cytotoxic to uveal and skin melanoma cells from peripheral blood of ocular melanoma patients. Cancer Immunol Immunother. 1991;33:333–340. doi: 10.1007/BF01756599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kenty G, Bikoff EK. BALB/c invariant chain mutant mice display relatively efficient maturation of CD4+ T cells in the periphery and secondary proliferative responses elicited upon peptide challenge. J Immunol. 1999;163:232–241. [PubMed] [Google Scholar]
- 20.Kujala E, Makitie T, Kivela T. Very long-term prognosis of patients with malignant uveal melanoma. Invest Ophthalmol Vis Sci. 2003;44:4651–4659. doi: 10.1167/iovs.03-0538. [DOI] [PubMed] [Google Scholar]
- 21.Lechler R, Aichinger G, Lightstone L. The endogenous pathway of MHC class II antigen presentation. Immunol Rev. 1996;151:51–79. doi: 10.1111/j.1600-065X.1996.tb00703.x. [DOI] [PubMed] [Google Scholar]
- 22.Long EO, LaVaute T, Pinet V, Jaraquemada D. Invariant chain prevents the HLA-DR-restricted presentation of a cytosolic peptide. J Immunol. 1994;153:1487–1494. [PubMed] [Google Scholar]
- 23.Miller J, Germain RN. Efficient cell surface expression of class II MHC molecules in the absence of associated invariant chain. J Exp Med. 1986;164:1478–1489. doi: 10.1084/jem.164.5.1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mooy CM, De Jong PT. Prognostic parameters in uveal melanoma: a review. Surv Ophthalmol. 1996;41:215–228. doi: 10.1016/S0039-6257(96)80024-5. [DOI] [PubMed] [Google Scholar]
- 25.Muntasell A, Carrascal M, Alvarez I, Serradell L, van Veelen P, Verreck FA, Koning F, Abian J, Jaraquemada D. Dissection of the HLA-DR4 peptide repertoire in endocrine epithelial cells: strong influence of invariant chain and HLA-DM expression on the nature of ligands. J Immunol. 2004;173:1085–1093. doi: 10.4049/jimmunol.173.2.1085. [DOI] [PubMed] [Google Scholar]
- 26.Ostrand-Rosenberg S, Grusby MJ, Clements VK. Cutting edge: STAT6-deficient mice have enhanced tumor immunity to primary and metastatic mammary carcinoma. J Immunol. 2000;165:6015–6019. doi: 10.4049/jimmunol.165.11.6015. [DOI] [PubMed] [Google Scholar]
- 27.Qi L, Rojas JM, Ostrand-Rosenberg S. Tumor cells present MHC class II-restricted nuclear and mitochondrial antigens and are the predominant antigen presenting cells in vivo. J Immunol. 2000;165:5451–5461. doi: 10.4049/jimmunol.165.10.5451. [DOI] [PubMed] [Google Scholar]
- 28.Rohn TA, Boes M, Wolters D, Spindeldreher S, Muller B, Langen H, Ploegh H, Vogt AB, Kropshofer H. Upregulation of the CLIP self peptide on mature dendritic cells antagonizes T helper type 1 polarization. Nat Immunol. 2004;5:909–918. doi: 10.1038/ni1108. [DOI] [PubMed] [Google Scholar]
- 29.Rubio V, Stuge TB, Singh N, Betts MR, Weber JS, Roederer M, Lee PP. Ex vivo identification, isolation and analysis of tumor-cytolytic T cells. Nat Med. 2003;9:1377–1382. doi: 10.1038/nm942. [DOI] [PubMed] [Google Scholar]
- 30.Sekaly RP, Tonnelle C, Strubin M, Mach B, Long EO. Cell surface expression of class II histocompatibility antigens occurs in the absence of the invariant chain. J Exp Med. 1986;164:1490–1504. doi: 10.1084/jem.164.5.1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Srivastava MK, Bosch JJ, Thompson JA, Ksander BR, Edelman MJ, Ostrand-Rosenberg S. Lung cancer patients’ CD4(+) T cells are activated in vitro by MHC II cell-based vaccines despite the presence of myeloid-derived suppressor cells. Cancer Immunol Immunother. 2008;57:1493–1504. doi: 10.1007/s00262-008-0490-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stumptner-Cuvelette P, Benaroch P. Multiple roles of the invariant chain in MHC class II function. Biochim Biophys Acta. 2002;1542:1–13. doi: 10.1016/S0167-4889(01)00166-5. [DOI] [PubMed] [Google Scholar]
- 33.Thompson JA, Dissanayake SK, Ksander BR, Knutson KL, Disis ML, Ostrand-Rosenberg S. Tumor cells transduced with the MHC class II transactivator and CD80 activate tumor-specific CD4+ T cells whether or not they are silenced for invariant chain. Cancer Res. 2006;66:1147–1154. doi: 10.1158/0008-5472.CAN-05-2289. [DOI] [PubMed] [Google Scholar]
- 34.Thompson JA, Srivastava MK, Bosch JJ, Clements VK, Ksander BR, Ostrand-Rosenberg S. The absence of invariant chain in MHC II cancer vaccines enhances the activation of tumor-reactive type 1 CD4+ T lymphocytes. Cancer Immunol Immunother. 2008;57:389–398. doi: 10.1007/s00262-007-0381-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tomazin R, Hill AB, Jugovic P, York I, van Endert P, Ploegh HL, Andrews DW, Johnson DC. Stable binding of the herpes simplex virus ICP47 protein to the peptide binding site of TAP. EMBO J. 1996;15:3256–3266. [PMC free article] [PubMed] [Google Scholar]
- 36.Topilski I, Harmelin A, Flavell RA, Levo Y, Shachar I. Preferential Th1 immune response in invariant chain-deficient mice. J Immunol. 2002;168:1610–1617. doi: 10.4049/jimmunol.168.4.1610. [DOI] [PubMed] [Google Scholar]
- 37.Verbik DJ, Murray TG, Tran JM, Ksander BR. Melanomas that develop within the eye inhibit lymphocyte proliferation. Int J Cancer. 1997;73:470–478. doi: 10.1002/(SICI)1097-0215(19971114)73:4<470::AID-IJC3>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 38.Viville S, Neefjes J, Lotteau V, Dierich A, Lemeur M, Ploegh H, Benoist C, Mathis D. Mice lacking the MHC class II-associated invariant chain. Cell. 1993;72:635–648. doi: 10.1016/0092-8674(93)90081-Z. [DOI] [PubMed] [Google Scholar]
- 39.Walseng E, Bakke O, Roche PA. Major histocompatibility complex class II-peptide complexes internalize using a clathrin- and dynamin-independent endocytosis pathway. J Biol Chem. 2008;283:14717–14727. doi: 10.1074/jbc.M801070200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Woodman PG. Biogenesis of the sorting endosome: the role of Rab5. Traffic. 2000;1:695–701. doi: 10.1034/j.1600-0854.2000.010902.x. [DOI] [PubMed] [Google Scholar]
- 41.Wubbolts R, Fernandez-Borja M, Oomen L, Verwoerd D, Janssen H, Calafat J, Tulp A, Dusseljee S, Neefjes J. Direct vesicular transport of MHC class II molecules from lysosomal structures to the cell surface. J Cell Biol. 1996;135:611–622. doi: 10.1083/jcb.135.3.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zwart W, Griekspoor A, Kuijl C, Marsman M, van Rheenen J, Janssen H, Calafat J, van Ham M, Janssen L, van Lith M, Jalink K, Neefjes J. Spatial separation of HLA-DM/HLA-DR interactions within MIIC and phagosome-induced immune escape. Immunity. 2005;22:221–233. doi: 10.1016/j.immuni.2005.01.006. [DOI] [PubMed] [Google Scholar]