

Abstract
Average human life expectancy has progressively increased over many decades largely due to improvements in nutrition, vaccination, antimicrobial agents, and effective treatment/prevention of cardiovascular disease, cancer, etc. Maximal life span, in contrast, has changed very little. Caloric restriction (CR) increases maximal life span in many species, in concert with improvements in mitochondrial function. These effects have yet to be demonstrated in humans, and the duration and level of CR required to extend life span in animals is not realistic in humans. Physical activity (voluntary exercise) continues to hold much promise for increasing healthy life expectancy in humans, but remains to show any impact to increase maximal life span. However, longevity in Caenorhabditis elegans is related to activity levels, possibly through maintenance of mitochondrial function throughout the life span. In humans, we reported a progressive decline in muscle mitochondrial DNA abundance and protein synthesis with age. Other investigators also noted age-related declines in muscle mitochondrial function, which are related to peak oxygen uptake. Long-term aerobic exercise largely prevented age-related declines in mitochondrial DNA abundance and function in humans and may increase spontaneous activity levels in mice. Notwithstanding, the impact of aerobic exercise and activity levels on maximal life span is uncertain. It is proposed that age-related declines in mitochondrial content and function not only affect physical function, but also play a major role in regulation of life span. Regular aerobic exercise and prevention of adiposity by healthy diet may increase healthy life expectancy and prolong life span through beneficial effects at the level of the mitochondrion.
Keywords: Mitochondria, Obesity, Aging, Cellular response, Cell death
Introduction
The question of how and why we age continues to puzzle biologists in spite of important advances in our understanding of underlying molecular and cellular mechanisms. Postreproductive life makes little sense if one accepts the view that the purpose of life is to spawn and carry forward a lineage to promote the long-term survival of the species. According to the above view, aging is an essential process to cull organisms that are not capable of reproduction and would sap resources that could otherwise be available to reproducing progeny. In contradiction to the above view, women have longer life span than men in spite of a definitive cessation of reproductive capacity by approximately 50 years of age. In contrast, men live shorter lives although their reproductive age outlasts that of women. One also could pose the following question: Why have billions of years of evolution not selected for longer reproductive phases and therefore longer life spans? Again from an evolutionary perspective, it seems advantageous for a species to have relatively short periods for reproduction, followed by death, to allow for more rapid selection of beneficial traits compared to a long-lived organism, which would continue to pass along its genetic code and slow the process of selection. Most humans, even after fulfilling our obligation and capacity for reproduction, have want to continue living. The average life expectancy of humans has increased remarkably over the past 100 years, largely as a result of advances in modern medicine for treatment of diseases such as diabetes, cancer, and cardiovascular disease. Thus, although human interventions to improve health resulted in avoidance of premature deaths of humans and extended the life expectancy, the maximum life span of humans has changed very little. It seems that we are, at this point, helpless to many deleterious cellular changes that ultimately lead to a senescent phenotype and eventually death. The whimsical notion of cheating the aging process has spawned an enormous amount of research aimed at understanding the mechanisms of cellular aging. It is doubtful that any single process could entirely account for the emergence of the senescent phenotype, but the purpose of this review is to highlight substantial evidence to implicate the mitochondrion as a major factor in this process.
A leading hypothesis of aging is based on free radical theory of aging by Harman [1]. Harman argued that oxygen-free radicals (reactive oxygen species) produced during normal cellular respiration would cause cumulative damage to molecules which would eventually lead to organismal loss of functionality and ultimately, death. Since free radicals or reactive oxygen species are produced in mitochondria during electron transport, substantial attention has been focused on mitochondria and aging.
Overview of mitochondrial physiology
The modern day mitochondrion is believed to have evolved over a billion or more years, originating as an invading Eubacterium in early eukaryotic cells. Of the 1,000 or so mitochondrial proteins, only 13 are encoded by the mitochondrial genome while the remainder are transcribed and translated from the nuclear genome and transported into inner mitochondrial membrane [2]. Over time, the cell has come to rely on mitochondria to maintain energetic homeostasis. Indeed, these organelles are a major source of chemical energy in the form of adenosine triphosphate (ATP), which is required to fuel many thermodynamically unfavorable processes within cells (e.g., ion transport against electrochemical gradients, protein synthesis, and contractility). The process of mitochondrial oxidative phosphorylation is responsible for conversion of macronutrient energy to ATP through a set of exquisitely coupled and coordinated reactions where macronutrients are oxidized (e.g., glucose, fatty acids, and amino acids), oxygen is reduced to water, and adenosine diphosphate (ADP) is phosphorylated to ATP (Fig. 1). The process begins when carbon substrates enter the tricarboxylic acid cycle either through acetyl CoA or anaplerotic reactions. Oxidation of these substrates generates reducing equivalents in the form of NADH and FADH2, which provide electron flow though respiratory chain complexes I (NADH dehydrogenase) and II (succinate dehydrogenase), respectively. Electron flow through complexes I and II converges on complex III (ubiquinone–cytochrome c reductase), along with electrons shuttled in from electron transferring flavoproteins (beta oxidation), though the mobile electron carrier coenzyme Q. A second mobile electron carrier transfers electrons on to complex IV (cytochrome c oxidase) where they are finally transferred to oxygen, yielding water. A proton gradient across the inner mitochondrial membrane is generated by the action of electron transport through complexes I, III, and IV. The potential energy of this gradient is harnessed by complex V (ATP synthase) to phosphorylate ADP to ATP. Thus, the maintenance of the mitochondrial membrane potential by electron transport is critical to proper function of the organelle, and therefore, the cell.
Fig. 1.
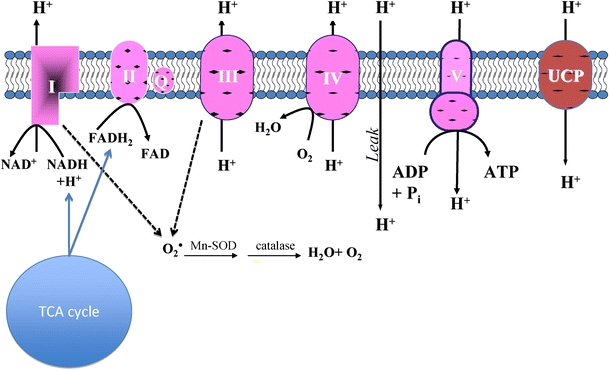
ADP is phosphorylated to ATP at complex V (ATP synthase) at the expense of the proton gradient maintained as electrons are passed from reducing equivalents (NADH, FADH2) to cytochromes along the inner mitochondrial membrane. These reducing equivalents are generated as carbon substrates are oxidized in the tricarboxylic acid cycle (TCA). Single electrons interact with molecular oxygen at complexes I and III to generate the superoxide radical (O•2). The sequential actions of superoxide dismutase (SOD) and catalase convert superoxide into oxygen and water
Evidence of altered mitochondrial function with aging
The role of mitochondria in the aging process has been a topic of intense interest for many years. In humans, studies have focused largely on skeletal muscle because it is a postmitotic tissue, tissue samples are relatively easy to acquire, and it is a determinant of physical function which is known to decline dramatically with aging [3, 4]. Skeletal muscle is also a highly metabolically active tissue, accounting for roughly 65% of glucose disposal following a meal and vital for peripheral glucose disposal. Age-related changes in mitochondrial content and function are well documented. Electron microscopy has been used to demonstrate that mitochondrial volume density decreases with aging in skeletal muscle [5]. Less abundant mitochondria would logically lead to decreased capacity for oxidative phosphorylation. Indeed, we find that the maximal rate of mitochondrial ATP synthesis declined over the life span, measured by recombinant firefly luciferase in the presence of ADP and substrate combinations specific to distinct respiratory chain enzymes [6]. Mitochondrial oxidative capacity decreased by about 8% per decade using substrates providing electron flow into complex I, complex II, and electron-transferring flavoprotein [6]. Since these rates were expressed relative to tissue mass, the age-related decline in mitochondrial capacity may reflect reduced content of the organelle in skeletal muscle. However, when ATP production rates were expressed per unit of mitochondrial protein, which accounts for differences in mitochondrial content, there were persisting age effects (5% per decade). Thus, the effects of age on mitochondrial function are compounded by reduced mitochondrial content as well as impaired intrinsic activity of the mitochondrial machinery [6]. Polarographic-based measurements of mitochondrial function are generally in agreement with the concept that mitochondrial function declines with aging [7, 8], although some reports show that the effects of old age are more modest [9]. These types of in vitro measurements in isolated mitochondria permit functional assessment of distinct levels of the respiratory chain and tricarboxylic acid cycle, however, in vivo assessment of mitochondrial function by magnetic resonance spectroscopy is advantageous from a standpoint of physiological relevance (intact circulatory and regulatory systems). Numerous in vivo studies also find that oxidative capacity is reduced in older adults [5, 10, 11], although several others find that oxidative capacity is similar in young and older adults with similar physical activity levels [12–15]. The importance of physical (in)activity as a determinant of the aging phenotype will be discussed later.
In an effort to understand the mechanisms responsible for this age-related decline in mitochondrial function, numerous investigators have examined various molecular and cellular disturbances with aging. Age-related changes at multiple levels between the expression of genes and the assembly of the functional organelle appear to be responsible for the overall decline in mitochondrial function. Aging affects the expression of genes encoding mitochondrial proteins, evidenced by decreased messenger RNA (mRNA) transcript levels [6, 16–18], possibly due to reduced gene transcription or mRNA instability with aging. Mitochondrial DNA copy number decreases with age [6, 16, 19, 20], which could account for the reduction of mitochondrial gene transcripts and therefore, the proteins encoded by these genes. We recently used mass spectrometry to identify and quantify the expression of numerous skeletal muscle proteins involved in fuel metabolism [6, 19]. We found aging significantly decreased the expression of numerous nuclear and mitochondrial-encoded proteins involved in mitochondrial function [6, 19]. Protein expression is not only affected by mRNA template availability, but also by the rate of protein synthesis, which we find to decline with aging [21]. Whether aging affects the synthesis rates of mitochondrial proteins encoded by both genomes in the same way has yet to be determined. This question is now addressable, thanks to recent advances in measuring the synthesis rates of individual mitochondrial proteins [22]. Specific rates of mitochondrial protein breakdown have yet to be measured in vivo, but observations that Lon protease expression, a key enzyme for mitochondrial proteolysis, is reduced in older mice suggests that mitochondrial protein turnover is likely to be reduced with aging [23]. Indeed, we recently reported that whole body proteolysis decreases with aging [24]. An overall decrease in protein expression may result from the mismatch between rates of protein synthesis and breakdown. Importantly, decreased protein turnover may lead to accumulation of oxidatively damaged dysfunctional proteins, such as protein carbonylation [25] and nitrotyrosine-modified proteins [26]. Nucleic acids also demonstrate increased levels of oxidative damage with aging [6, 27], particularly mitochondrial DNA (mtDNA), whose susceptibility is increased by its proximity to the source of damaging reactive oxygen species and the lack of protection by histones. Mitochondrial fusion and fission appear to be critical for the maintenance of mtDNA [28], but a causal link to aging and mtDNA damage remains in its nascent stages [29]. There is some data to indicate that the effects of aging on mtDNA abundance, gene expression, and the aforementioned downstream effects may be traced back to alterations and mutations to portions of DNA that encode mitochondrial proteins. Although mtDNA deletions and point mutations increase with age based on cross-sectional studies [30–33], the prevalence of these alterations are generally less than 1%; a number that is far less the 50–80% that is purported to be required to induce any physiological effect [34]. It is important, however, to consider that the DNA mutations that occur with aging are widespread and inconsistent such that the frequency of mutations may be underestimated if only a few marker genes are assessed. Furthermore, even relatively modest degrees of DNA mutations may be selectively amplified [35, 36]. Using long-chain polymerase chain reaction (PCR), several studies have shown that full-length mtDNA is reduced with old age [37, 38]. In sum, there are multiple factors at the levels of gene expression, protein synthesis, protein quality, and mitochondrial dynamics that can account for reductions in mitochondrial number and function with aging.
It is clear that mitochondrial function is altered with old age and may underlie age-related changes in physical function, protein synthesis, and muscle mass. A remaining question is whether mitochondria regulate the senescent phenotype (i.e., is mitochondrial dysfunction cause or consequence of aging?). Although this question remains largely unanswered, there is some evidence to directly implicate mitochondria as a cause of cellular aging. For example; an accelerated aging phenotype is evident in mice with increased mtDNA mutations and genetic mitochondrial diseases [39–42]. The remainder of this review will appraise evidence to suggest that mitochondria play a major role in regulating cellular senescence.
Mitochondria and oxidants
The beneficial role of mitochondria in supplying high-energy phosphates generally overshadows its less favorable role in production of reactive oxygen species (ROS). Mitochondria are responsible for the majority of cellular ROS, although nonmitochondrial sources such as cycloogygenases, NADPH oxidase, and peroxisomes also contribute a modest amount. The interaction of single electrons with molecular oxygen results in the formation of superoxide anions at respiratory chain complexes I and III [42, 43]. The rate of superoxide production is generally regarded as being equivalent to about 3% of total oxygen reduced by cytochrome c oxidase, however, there is some evidence that this number may be much lower [44] and that ROS production rate is largely dependent on the mitochondrial membrane potential (ΔΨ). A higher membrane potential such as during state 2 or state 4 respiration would increase the redox potential and perhaps promote backflow of electrons through complexes I and III, whereas state 3 respiration minimizes this effect by increasing the forward flux of electrons along the cytochromes, providing less opportunity for generation of superoxide. Reactive oxygen species are generally regarded as exerting deleterious effects on vital components of cells though oxidation of proteins, lipids, and nucleic acids. Protein modifications may change the structure and catalytic activity of key enzymes, lipid oxidation may alter the fluidity of the membranes, and DNA oxidation may interfere with the proper transcription of genes. Thus, it seems that mitochondria may pose a serious liability to the well-being of the cell. Notwithstanding, the cell seems adept at withstanding this potential threat through the action of various antioxidant scavenging systems. Superoxide is converted to hydrogen peroxide (H2O2) by the action of superoxide dismutase, which is found in two forms: manganese superoxide dismutase (SOD1) in the mitochondrial matrix and copper–zinc superoxide dismutase (SOD2) in the cytosol. Hydrogen peroxide is also considered a reactive oxygen species and is further deactivated by one of three enzymes (catalase, glutathione peroxidase, and peroxiredoxins) [43]. These antioxidant enzyme systems allow the cell to reap the energenic benefits of mitochondria while minimizing the potentially damaging effects of ROS, provided that the buffering capacity is greater than or equal to the rate of ROS production. However, some degree of oxidative damage is likely even with sufficient antioxidant systems, particularly in mitochondrial proteins, lipids, and DNA that are proximal to the source of superoxide. Furthermore, mitochondrial DNA, unlike nuclear DNA, is not afforded the protection of histones making it more susceptible to the damaging effects of ROS. Indeed, the mutation rate of mtDNA is nearly 20-fold higher than nuclear DNA [45].
With amazing prescience and little supporting evidence, Harman put forth the “free radical theory of aging” that implicated free radicals in the aging process and associated deleterious effects [46]. Harman drew inspiration from a notion that life span is an inverse function of metabolic rate, which is proportional to oxygen consumption, and that hyperbaric oxygen toxicity and radiation toxicity could be explained by the same underlying phenomenon: oxygen free radicals. There is some uncertainty concerning the relevance of reactive oxygen species to the aging process. Levels of damaged DNA, evident from increased levels of 8-oxo-2′-deoxyguanosine, increase with aging in skeletal muscle [6] and brain tissue [47], both of which are mostly postmitotic. Skeletal muscle from older humans also exhibits higher levels of oxidation to proteins (carbonylation) and lipids (peroxidation) [48]. A link between cellular ROS and life span is suggested from studies where oxidant scavenging systems are either enhanced or decreased. Life span of cultured fibroblasts increases in the presence of elevated superoxide dismutase [49]. Similarly, overexpression of superoxide dismutase and catalase significantly prolongs the life span of drosophila [50, 51]. It is, however, important to point out that this effect could not be reproduced in longer-lived strains of the fly [52]. Further, supporting evidence comes from long-lived AGE-1 mutants of the nematode worm Caenorhabditis elegans. Life span extension in these worms is accompanied by higher than normal expression of SOD and catalase [53, 54]. Although it seems that enhancing the antioxidant buffering capacity extends life span in several species, the reverse is not always true (i.e., decreasing ROS scavenging systems does not shorten life span). Although RNAi knockdown of SOD accelerates aging in human fibroblasts [55], life span is not significantly altered in mice with knockouts of glutathione peroxidase, SOD1, or SOD2 [56–58]. The link between aging and cellular ROS primarily stems from their potential damaging effects on proteins, nucleic acids, and lipids, but there is intriguing evidence that ROS may also act on a number of signaling pathways involved in development (GTPase Ras), aging, and cell stress (JNK, p38, p53; see [43] for review).
Regardless of the effects of artificially altering ROS scavenging systems, it is clear that the senescent phenotype is characterized by evidence of ROS-induced damage to vital cellular components. The balance between the production and scavenging of ROS ultimately determines the overall level of oxidative stress to the organism. The current opinion seems to favor the notion that aging is accompanied by an increase in ROS production rather than decreased antioxidant enzyme systems. Although SOD2 activity decreases with age in humans [59], both SOD1 and catalase activities have been shown to increase with age in skeletal muscle [37, 59, 60]. These studies cast doubt on the notion that age-related oxidative stress could result from blunted oxidant scavenging capacity and suggest that upregulation of multiple oxidant buffering systems with age may represent an adaptive response to elevated ROS production. Given that the overwhelming majority of cellular ROS are of mitochondrial origin, it is reasonable to posit that some age-related derangement to mitochondrial physiology may be to blame for elevated ROS production.
Can a link be established between elevated ROS production and mitochondrial dysfunction with aging? The commensurate increase in cellular markers of oxidative damage and decreased respiratory chain function with age seem at odds with the “rate of living hypothesis,” which posits that longevity and metabolic rate are inversely related. Based on that theory and the notion of tight stoichiometry between oxygen consumption and superoxide production, one could predict that age-related declines in mitochondrial function may actually reduce the production superoxide anions. However, the rate of living hypothesis has fallen by the wayside in favor of a more contemporary view of ROS production being controlled by several factors including (1) the redox potential for donating a single electron to oxygen, (2) the redox potential of electron donors at various points along the cytochrome chain, and (3) cellular oxygen tension [43]. In short, altering the redox potential to a more reduced state without a corresponding increase in phosphorylation (i.e., state 4 respiration) would increase the mitochondrial membrane potential, promote backflow of electrons, and increase the production of ROS. Based on this logic, dissipation of the ΔΨ by allowing proton leakage in the absence of ADP phosphorylation (i.e., upcoupling) should decrease ROS production [61]. This concept was first illustrated in isolated mitochondria that were treated with chemical uncouplers [62]. These mitochondria exhibited decreased ROS production when ΔΨ was decreased by uncoupling. In nature, a class of proteins known as uncoupling proteins (UCPs) appears to play an important role in the process of uncoupling, with homologs UCP1 expressed in brown adipose tissue mitochondria and UCP2 and UCP3 in skeletal muscle [63]. Similar to the effects of uncoupling in isolated mitochondria, UCP3 knockout mice exhibit increased markers of oxidative stress [64, 65], consistent with the notion that mitochondrial coupling is a major determinant of ROS production.
The importance of mitochondrial membrane potential and modulators such as UCPs has prompted some investigators to pursue the idea that mitochondrial coupling, rather than capacity, may be a key determinant of cellular aging through an influence on ROS production. A recent study in humans used a combination of in vivo methods to assess mitochondrial coupling in skeletal muscles of young and older humans [10]. The first dorsal interosseus muscle was highly coupled but exhibited an age-related decline in mitochondrial function and myocellular ATP concentrations. In contrast, the tibalis anterior muscle exhibited mild uncoupling and exhibited no evidence of age-related mitochondrial dysfunction. The authors proposed that mild mitochondrial uncoupling partially dissipates the mitochondrial membrane potential, reduces ROS production, and helps preserve the functionality of skeletal muscle mitochondria [10]. Although the mechanism behind this effect remains speculative, there is some evidence that UCP3 expression decreases with aging [66] and could be muscle specific. Transgenic mice overexpressing UCP3 in skeletal muscle are characterized by increased state 4 respiration, which prevents the increase in ROS production evident with aging in wild-type animals [67]. These and numerous other studies have fueled the emergence of the “uncoupling to survive” hypothesis, which suggests that dissipating the mitochondrial membrane potential will protect cells from the ravages of reactive oxygen species [68]. Indeed, survival appears to be increased in organisms that are inherently uncoupled. Once again, the long-lived AGE-1 mutants of C. elegans provide some valuable insight as these worms show lower than normal mitochondrial membrane potential [69] and among numerous other reports [70, 71], provide a tantalizing link between mitochondria, ROS, and life span.
As discussed in the preceding paragraphs, much attention has been focused on oxidative stress as a determinant of life span. Studies of naked mole rats add an interesting twist to the free radical theory of aging. These animals are the longest living rodents with life spans of 30 years or longer. In spite of their longevity, naked mole rats exhibit higher levels of lipid peroxidation, protein carbonylation, and DNA oxidative damage than mice at comparable points in their life span, even as young animals [72]. These data are in sharp contrast with the theory that oxidative stress causes senescence. Clues into the mechanisms underlying their paradoxical longevity come from observations that these animals have higher proteasome activity and less protein ubiquitination, which remained remarkably constant over two decades of life. Thus, although naked mole rats exhibit higher than normal reversible cysteine oxidation at a young age, these rodents appear to achieve their exceptional longevity through increased proteolytic activity, which maintains a functional proteome by minimizing accrual of irreversible oxidative damage [72]. These data point to protein homeostasis (i.e., degradation of damaged proteins and replacement with new proteins) as a factor that can enhance life span, even under conditions where oxidative stress may be elevated.
Mitochondria regulate programmed cell death
Programmed cell death (i.e., apoptosis) is the end-stage of the cell cycle when cellular structures are degraded by proteases such as caspases and nucleases. In addition to receptor-mediated apoptosis, this process seems to be triggered by factors such as steroid hormones and DNA damage. Mitochondria also play a role in regulating these processes [73]. Cytochrome c is a mobile electron carrier that functions to shuttle electrons from complexes I and II to complex III and thus, plays a vital role in mitochondrial ATP synthesis. Ironically, a protein that is so vital to life is also a trigger for apoptosis. Under normal conditions, cytochrome c resides within the inner mitochondrial membrane, associated with cardiolipin, a membrane phospholipid [74]. However, certain stimuli, such as DNA damage, protein damage, or perturbation of metabolic homeostasis, initiate the release of cytochrome c into the cytosol which activates caspases [75]. Several factors, such as increased cytosolic calcium and ROS, have been proposed to weaken the interaction between cytochrome c and its cardiolipin anchor and initiate the detachment and mobilization of cytochrome c [76]. The outer mitochondrial membrane is ordinarily impermeable to proteins. In fact, addition of exogenous cytochrome c to mitochondrial preparations is a common test for outer membrane integrity [77]. Cytochrome c is believed to exit into the cytosol through pores that form in a process known as mitochondrial outer membrane permeabilization. The exact mechanisms of outer membrane permeabilization are still being investigated, although several likely candidates have emerged such as B-cell lymphoma protein-2, voltage-dependent anion channels, ceramide channels, and formation of permeability transition pores (for review see [76]). Once in the cytosol, cytochrome c initiates the formation of apoptosomes and activation of a series of caspases that begin the process of demolishing the cell.
The link between mitochondria and apoptosis goes beyond caspase-dependent pathways of cellular destruction. Apoptosis-inducing factor (AIF) is another intermembrane flavoprotein that induces apoptosis when it is released into the cytosol [78]. Unlike cytochrome c release, AIF induces apoptosis through caspase-independent mechanisms. AIF translocates to the nucleus and induces DNA fragmentation and chromatin condensation [78]. Furthermore, a recent study in yeast demonstrates that mitochondrial DNA regulates nuclear genome stability [79]. Depletion of mitochondrial DNA by aging, ethidum bromide, or genetic mutation causes cell-cycle arrest and loss of heterozygosity of marker genes on chromosomes IV and XII. The authors constructed an intriguing model whereby loss of mtDNA reduces the mitochondrial membrane potential and reduced the biosynthesis of iron–sulfur clusters which are directly linked with stability of the nuclear genome [79].
Programmed cell death, when properly regulated, is a vital mechanism to regulate development, cell numbers, and prevent the accumulation perilous tumor cells. However, when improperly activated, apoptosis may contribute to the demise of the organism, such as in neurons in Alzheimer's disease [80], cardiac myocytes in myocardial infarction [81], and skeletal muscle of HIV-infected patients [82] and people with muscular dystrophy [83]. Given that muscle wasting is a hallmark of senescence, it is possible that apoptotic processes are upregulated with aging. Support for this possibility comes from reports that aged rats exhibited increased caspase-3 levels and elevated apoptosis compared to young rats [84]. Although these and other findings [85, 86] implicate elevated apoptosis in the senescent phenotype, there is contrasting evidence to suggest otherwise. The family of bcl2 proteins act to inhibit caspase activation during apoptosis, but mice that do not express these proteins appear to age normally [87], suggesting that apoptosis may not be an important determinant of the aging phenotype. Furthermore, one could question whether decreasing apoptosis would confer any beneficial effects on life span. Apoptosis is a uniquely adapted process to degrade distressed cells. In light of the cellular stresses that induce apoptosis, inhibition of this process may be detrimental to the organism unless healthy, viable cells are being destroyed as a consequence of dysregulation of apoptotic signaling with aging. At this point, the dysregulation of apoptosis does not seem to be a candidate mechanism underlying the finite life span of all organisms.
Countermeasures for aging
Evidence supporting a role for mitochondria, not only as custodians of energetic homeostasis, but also as mediators of life span and senescence, has generated an abundance of interest in mitochondrial-targeted therapeutics to prevent, reverse, or delay age-related detriments and perhaps extend life span. Exercise is perhaps one of the most straightforward and effective ways to increase the content and overall function of mitochondria. The mid-1960s marked a major breakthrough where treadmill running in rats was shown to increase the content and activities of key mitochondrial proteins, which, for the first time, linked specific mitochondrial adaptations to increased capacity to perform aerobic work as a result of exercise training [88]. Since the publication of Holloszy's pioneering work, numerous studies have touted the beneficial effects of exercise. Endurance exercise stimulates mitochondrial biogenesis, increases the expression and activities of mitochondrial enzymes, and increases the overall tissue capacity for oxidative metabolism [88–100]. Microarray analyses in mice revealed that these exercise-induced adaptations occurred in parallel with increased mRNA transcripts corresponding to mitochondrial proteins encoded by the nuclear and mitochondrial genomes [91]. Exercise training also significantly increased mtDNA copy number in mice [91] and humans [19]. Activation of peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) is believed to be a major driving force in the stimulation of mitochondrial biogenesis with exercise. Briefly, exercise increases the activity (acute) and expression (chronic) of PGC-1α, which stimulates transcription of nuclear-encoded mitochondrial genes through nuclear respiratory factors (NRF-1, NRF-2) [101] and stimulated the transcription of mitochondrial-encoded genes through mitochondrial transcription factor A (TFAM) [102]. During exercise, cellular signals such as cytosolic calcium and AMP activate several protein kinases that phosphorylate and activate PGC-1α [103]. For a comprehensive review of the topic of cellular signaling for mitochondrial biogenesis, the interested reader is referred to several excellent review articles [104–106]. In addition to expanding mitochondrial volume and increasing the capacity for oxidative ATP synthesis, endurance exercise has also been shown to reduce mitochondrial ROS production and protect against mitochondrial-mediated apoptosis [107]. Others find that although exercise actually increases the formation of ROS, the net cellular ROS load is reduced by upregulated oxidant scavenging systems [108].
Given that many documented adaptations to endurance exercise are the same factors that are impaired with aging, there has been much interest in the utility of exercise to attenuate the deterioration of mitochondrial function with aging. Beneficial adaptations to endurance training seem to be maintained across the life span, as evidenced by robust increases in VO2 peak [98, 109], mitochondrial enzyme activities [20, 98, 109], mitochondrial content [20, 110], protein synthesis rates [111], mtDNA copy number [20], and gene transcripts for mitochondrial proteins [98]. Studies in older rodents provides evidence that exercise training is able to decrease ROS production [112], attenuate DNA oxidative damage, increase the activity of DNA repair processes [113], and increase proteasome activity to aid in the removal of oxidatively damaged proteins [114]. In some cases, these training adaptations in older adults seem to be blunted in comparison to young [115, 116], but several cross-sectional studies of masters level athletes show that markers of muscle mitochondrial function are unchanged with age [19, 117, 118]. Although exercise delays the onset of many mitochondrial changes associated with aging, there are several factors that cannot be attenuated even by vigorous endurance exercise programs. We recently found that chronic (more than 4 years) vigorous endurance training (more than 5 h per week) increased mitochondrial ATP production capacity, increased mitochondrial enzyme activities, increased abundance of mitochondrial proteins, and increased mtDNA abundance [19]. In spite of this high level of physical activity, there remained substantial age-related declines in mtDNA copy number and expression of several mitochondrial respiratory chain proteins. Thus, it seems that exercise can help delay the onset of many age-related detriments, but there is a component of mitochondrial aging that is an inevitable function of chronological age. The aforementioned studies of exercise adaptations with age and other studies involving careful control of health and physical activity patterns [9, 13–15, 117, 119] indicate that environmental and lifestyle factors can account for much of the aging phenotype. We proposed that voluntary physical activities increase muscle mitochondrial capacity which in turn enhances spontaneous activity [120]. In mice, an aerobic exercise program increased muscle mitochondrial DNA abundance and ATP production rate [91] which was associated with increased activity. It appears that maintaining activity by mutation of AGE1 gene increases life span of C. elegans [121]. A similar phenomenon may occur in humans (Fig. 2), but remains to be proven by experimental data. However, although exercise seems to increase average life expectancy by decreasing the incidence of age-related comorbidities, at this point, there is no evidence that exercise increases maximal life span [122–124].
Fig. 2.
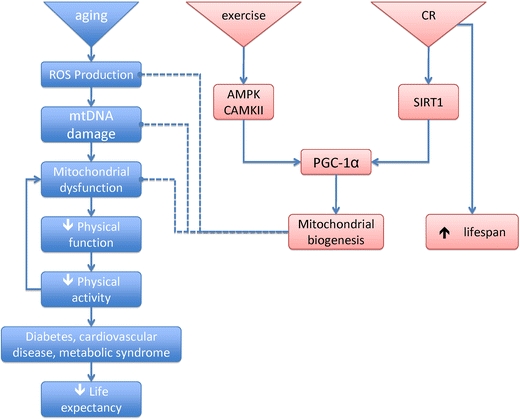
The free radical theory of aging posits that a senescent phenotype is induced by accumulation of oxidative damage resulting from reactive oxygen species. Exercise and caloric restriction are two interventions that induce mitochondrial biogenesis through PGC-1α. Although exercise and CR increase average life expectancy by protecting against age-related comorbidities, only CR has been shown to increase maximal life span; an effect that seems to require the activation of sirtuins
At this point, the only known intervention that increases maximal life span is restriction of caloric intake by 30–40% of ad libitum intake [125–127]. The proposed mechanisms underlying this extension of life span by CR involve a shift from a state of growth and proliferation to maintenance and repair [125–127]. Initially, CR was believed to extend life span by decreasing metabolic rate, decreasing mitochondrial oxygen consumption, and therefore, attenuating oxidative stress. However, the exact opposite appears to be true, evidenced by increases in mitochondrial content and oxygen consumption in response to CR [128]. The fact that CR does not increase life span when the gene encoding cytochrome c is deleted [129] or in the presence of electron transport inhibitors [130, 131] suggests that mitochondria are critical factors in the phenomenon of life span extension by CR. There are several mechanisms by which mitochondria may be responsible for the life span-enhancing effects of CR [1]. In contrast to what would be predicted by the rate of living hypothesis, increased mitochondrial respiration with CR is accompanied by decreased ROS production [126]. Expansion of the mitochondrial reticulum with CR could minimize ROS production by allowing electron flow from reducing equivalents (NADH, FADH2) to be distributed over more cytochromes, thus providing less opportunity for reverse electron flow [1]. Another possible mechanism for blunted ROS production involves a shift in substrate oxidation [126]. In an attempt to spare carbohydrate, CR may promote lipid oxidation, which shuttles electrons into the cytochrome chain through electron transferring flavoprotein, which bypasses complex I (one of the two sites for mitochondrial superoxide generation) and reduces the likelihood of ROS formation [1]. Mitochondrial coupling is a component of mitochondrial physiology that may be altered by CR. Mitochondrial coupling, through its influence on the mitochondrial membrane potential, could affect the production of ROS. The impact of CR on mitochondrial coupling is unclear. One report shows that CR decreases proton leak and ROS production [132], whereas others suggest that decreased ROS production occurs as a result of greater proton leak [133]. Another possible mechanism involves the possibility of increased mitochondrial autophagy with CR. Lysosomal degradation of damaged/dysfunctional organelles and replacement with newly formed organelles would be beneficial from a standpoint of cellular energetics and production of ROS. Studies in C. elegans indicate that genes regulating mitochondrial autophagy (bec-1 and Ce-atg7) are required for the longevity phenotype in dietary restriction mutant worms [134]. Others find that autophagic vacuoles were increased in the hearts of CR rats [135]. The link between mitochondrial autophagy and CR-induced life span extension is in its nascent stages.
Calorie restriction appears to exert its effects on mitochondria in mammals through a regulated process involving a class of proteins known as sirtuins, specifically SIRT1. SIRT1 deacetylates histones and proteins and is activated under certain physiological conditions where the NAD/NADH ratio is elevated (e.g., caloric restriction) [136]. Activation of SIRT1 may extend life span by removing acetyl groups from histones, thereby affecting global gene expression and perhaps silencing genes that otherwise induce a senescent phenotype when expressed [137]. Proteins are also targets for the deacetylating action of SIRT1, one of which is PGC-1α [138, 139]. The deacetylation of PGC-1α increases its activity and initiates downstream signaling involving transcription factor targets (NRF-1, TFAM), which, in turn, stimulate mitochondrial biogenesis. At the same time, SIRT1 also deacetylates proteins involved in mitochondrial autophagy [140], suggesting that SIRT1 helps maintain highly functional mitochondria by regulating the degradation of old, potentially dysfunctional mitochondria and synthesis of new mitochondria. Exogenous SIRT1 activators such as resveratrol and other small molecule activators have been shown to mimic the beneficial effects of CR and extend life span when given exogenously to several organisms [141–143]. However, a recent report shows that resveratrol decreases signs of aging but does not increase life span in mice [144]. Recent studies have demonstrated that resveratrol-treated aging mice on a high-calorie diet had increased insulin sensitivity and a trend toward increased survival [145]. These compounds have also been shown to reduce diet-induced obesity and insulin resistance and to increase mitochondria activity in a rodent model [141, 143, 146]. Data in humans is fairly limited. One study found that CR for 6 months increased SIRT1 expression, TFAM expression, and mitochondrial number [147]. These changes did not translate to increased mitochondrial respiration and actually decreased whole-body energy expenditure. The role of CR and CR mimetics continues to be an important area of research that may someday result in effective interventions to reduce age-related comorbidities and perhaps even extend life span in humans.
Concluding remarks
Advances in medicine and public heath have dramatically increased average life expectancy over the past 200 years. An enormous effort has recently been expended to understand how the aging process is regulated at the molecular and cellular levels with hopes to find a way to extend maximal life span; something that has remained fairly constant in humans. There are numerous determinants of life span, but one common thread that has emerged in a variety of species from yeast to rodents is regulation of life span by mitochondria. It is fascinating that an organelle believed to have once been an invading Eubacterium has acquired so much control over the fate of the cell. It is clear that mitochondria play a role in the cell that goes well beyond their dogmatic function as the “powerhouse of the cell.” A potential pathway for the role of mitochondria in extending life span is outlined (Fig. 2). It is likely that mitochondrial decay that occurs with age cannot be counteracted in humans who are not on caloric restriction unless physical activity is voluntarily enhanced. As we advance the frontiers of our understanding of how senescence and life span are regulated, it may become possible to not only decrease the incidence of age-related comorbidities, but also extend maximal life span. Addressing the latter without regard to the former would create an undesirable situation of a rapidly expanding population of individuals who would stress an already overburdened health-care industry with little contribution to society.
Acknowledgments
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Guarente L. Mitochondria—a nexus for aging, calorie restriction, and sirtuins? Cell. 2008;132:171–176. doi: 10.1016/j.cell.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJ, Staden R, Young IG. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- 3.Grimby G. Muscle performance and structure in the elderly as studied cross-sectionally and longitudinally. J Gerontol A Biol Sci Med Sci. 1995;50(Spec No:17-22):17–22. doi: 10.1093/gerona/50a.special_issue.17. [DOI] [PubMed] [Google Scholar]
- 4.Wolfson L, Judge J, Whipple R, King M. Strength is a major factor in balance, gait, and the occurrence of falls. J Gerontol A Biol Sci Med Sci. 1995;50(Spec No:64-7):64–67. doi: 10.1093/gerona/50a.special_issue.64. [DOI] [PubMed] [Google Scholar]
- 5.Conley KE, Jubrias SA, Esselman PC. Oxidative capacity and ageing in human muscle. J Physiol. 2000;526(Pt 1):203–210. doi: 10.1111/j.1469-7793.2000.t01-1-00203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Short KR, Bigelow ML, Kahl J, Singh R, Coenen-Schimke J, Raghavakaimal S, Nair KS. Decline in skeletal muscle mitochondrial function with aging in humans. Proc Natl Acad Sci U S A. 2005;102:5618–5623. doi: 10.1073/pnas.0501559102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boffoli D, Scacco SC, Vergari R, Solarino G, Santacroce G, Papa S. Decline with age of the respiratory chain activity in human skeletal muscle. Biochim Biophys Acta. 1994;1226:73–82. doi: 10.1016/0925-4439(94)90061-2. [DOI] [PubMed] [Google Scholar]
- 8.Trounce I, Byrne E, Marzuki S. Decline in skeletal muscle mitochondrial respiratory chain function: possible factor in ageing. Lancet. 1989;1:637–639. doi: 10.1016/s0140-6736(89)92143-0. [DOI] [PubMed] [Google Scholar]
- 9.Rasmussen UF, Krustrup P, Kjaer M, Rasmussen HN. Experimental evidence against the mitochondrial theory of aging. A study of isolated human skeletal muscle mitochondria. Exp Gerontol. 2003;38:877–886. doi: 10.1016/s0531-5565(03)00092-5. [DOI] [PubMed] [Google Scholar]
- 10.Amara CE, Shankland EG, Jubrias SA, Marcinek DJ, Kushmerick MJ, Conley KE. Mild mitochondrial uncoupling impacts cellular aging in human muscles in vivo. Proc Natl Acad Sci U S A. 2007;104:1057–1062. doi: 10.1073/pnas.0610131104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McCully KK, Fielding RA, Evans WJ, Leigh JS, Jr, Posner JD. Relationships between in vivo and in vitro measurements of metabolism in young and old human calf muscles. J Appl Physiol. 1993;75:813–819. doi: 10.1152/jappl.1993.75.2.813. [DOI] [PubMed] [Google Scholar]
- 12.Chilibeck PD, McCreary CR, Marsh GD, Paterson DH, Noble EG, Taylor AW, Thompson RT. Evaluation of muscle oxidative potential by 31P-MRS during incremental exercise in old and young humans. Eur J Appl Physiol Occup Physiol. 1998;78:460–465. doi: 10.1007/s004210050446. [DOI] [PubMed] [Google Scholar]
- 13.Kent-Braun JA, Ng AV. Skeletal muscle oxidative capacity in young and older women and men. J Appl Physiol. 2000;89:1072–1078. doi: 10.1152/jappl.2000.89.3.1072. [DOI] [PubMed] [Google Scholar]
- 14.Lanza IR, Befroy DE, Kent-Braun JA. Age-related changes in ATP-producing pathways in human skeletal muscle in vivo. J Appl Physiol. 2005;99:1736–1744. doi: 10.1152/japplphysiol.00566.2005. [DOI] [PubMed] [Google Scholar]
- 15.Lanza IR, Larsen RG, Kent-Braun JA. Effects of old age on human skeletal muscle energetics during fatiguing contractions with and without blood flow. J Physiol. 2007;583:1093–1105. doi: 10.1113/jphysiol.2007.138362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barazzoni R, Short KR, Nair KS. Effects of aging on mitochondrial DNA copy number and cytochrome c oxidase gene expression in rat skeletal muscle, liver, and heart. J Biol Chem. 2000;275:3343–3347. doi: 10.1074/jbc.275.5.3343. [DOI] [PubMed] [Google Scholar]
- 17.Calleja M, Pena P, Ugalde C, Ferreiro C, Marco R, Garesse R. Mitochondrial DNA remains intact during Drosophila aging, but the levels of mitochondrial transcripts are significantly reduced. J Biol Chem. 1993;268:18891–18897. [PubMed] [Google Scholar]
- 18.Welle S, Bhatt K, Thornton CA. High-abundance mRNAs in human muscle: comparison between young and old. J Appl Physiol. 2000;89:297–304. doi: 10.1152/jappl.2000.89.1.297. [DOI] [PubMed] [Google Scholar]
- 19.Lanza IR, Short DK, Short KR, Raghavakaimal S, Basu R, Joyner MJ, McConnell JP, Nair KS. Endurance exercise as a countermeasure for aging. Diabetes. 2008;57:2933–2942. doi: 10.2337/db08-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Menshikova EV, Ritov VB, Fairfull L, Ferrell RE, Kelley DE, Goodpaster BH. Effects of exercise on mitochondrial content and function in aging human skeletal muscle. J Gerontol A Biol Sci Med Sci. 2006;61:534–540. doi: 10.1093/gerona/61.6.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rooyackers OE, Adey DB, Ades PA, Nair KS. Effect of age on in vivo rates of mitochondrial protein synthesis in human skeletal muscle. Proc Natl Acad Sci U S A. 1996;93:15364–15369. doi: 10.1073/pnas.93.26.15364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jaleel A, Short KR, Asmann YW, Klaus KA, Morse DM, Ford GC, Nair KS. In vivo measurement of synthesis rate of individual skeletal muscle mitochondrial proteins. Am J Physiol Endocrinol Metab. 2008;295:E1255–E1268. doi: 10.1152/ajpendo.90586.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bota DA, Van Remmen H, Davies KJ. Modulation of Lon protease activity and aconitase turnover during aging and oxidative stress. FEBS Lett. 2002;532:103–106. doi: 10.1016/s0014-5793(02)03638-4. [DOI] [PubMed] [Google Scholar]
- 24.Henderson GC, Dhatariya K, Ford GC, Klaus KA, Basu R, Rizza RA, Jensen MD, Khosla S, O'Brien P, Nair KS. Higher muscle protein synthesis in women than men across the lifespan, and failure of androgen administration to amend age-related decrements. FASEB J. 2009;23:631–641. doi: 10.1096/fj.08-117200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hepple RT, Qin M, Nakamoto H, Goto S. Caloric restriction optimizes the proteasome pathway with aging in rat plantaris muscle: implications for sarcopenia. Am J Physiol Regul Integr Comp Physiol. 2008;295:R1231–R1237. doi: 10.1152/ajpregu.90478.2008. [DOI] [PubMed] [Google Scholar]
- 26.Fugere NA, Ferrington DA, Thompson LV. Protein nitration with aging in the rat semimembranosus and soleus muscles. J Gerontol A Biol Sci Med Sci. 2006;61:806–812. doi: 10.1093/gerona/61.8.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Michikawa Y, Mazzucchelli F, Bresolin N, Scarlato G, Attardi G. Aging-dependent large accumulation of point mutations in the human mtDNA control region for replication. Science. 1999;286:774–779. doi: 10.1126/science.286.5440.774. [DOI] [PubMed] [Google Scholar]
- 28.Herlan M, Vogel F, Bornhovd C, Neupert W, Reichert AS. Processing of Mgm1 by the rhomboid-type protease Pcp1 is required for maintenance of mitochondrial morphology and of mitochondrial DNA. J Biol Chem. 2003;278:27781–27788. doi: 10.1074/jbc.M211311200. [DOI] [PubMed] [Google Scholar]
- 29.Lee S, Jeong SY, Lim WC, Kim S, Park YY, Sun X, Youle RJ, Cho H. Mitochondrial fission and fusion mediators, hFis1 and OPA1, modulate cellular senescence. J Biol Chem. 2007;282:22977–22983. doi: 10.1074/jbc.M700679200. [DOI] [PubMed] [Google Scholar]
- 30.Linnane AW, Baumer A, Maxwell RJ, Preston H, Zhang CF, Marzuki S. Mitochondrial gene mutation: the ageing process and degenerative diseases. Biochem Int. 1990;22:1067–1076. [PubMed] [Google Scholar]
- 31.Melov S, Shoffner JM, Kaufman A, Wallace DC. Marked increase in the number and variety of mitochondrial DNA rearrangements in aging human skeletal muscle. Nucleic Acids Res. 1995;23:4122–4126. doi: 10.1093/nar/23.20.4122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Munscher C, Muller-Hocker J, Kadenbach B. Human aging is associated with various point mutations in tRNA genes of mitochondrial DNA. Biol Chem Hoppe Seyler. 1993;374:1099–1104. doi: 10.1515/bchm3.1993.374.7-12.1099. [DOI] [PubMed] [Google Scholar]
- 33.Zhang C, Linnane AW, Nagley P. Occurrence of a particular base substitution (3243 A to G) in mitochondrial DNA of tissues of ageing humans. Biochem Biophys Res Commun. 1993;195:1104–1110. doi: 10.1006/bbrc.1993.2158. [DOI] [PubMed] [Google Scholar]
- 34.Sciacco M, Bonilla E, Schon EA, DiMauro S, Moraes CT. Distribution of wild-type and common deletion forms of mtDNA in normal and respiration-deficient muscle fibers from patients with mitochondrial myopathy. Hum Mol Genet. 1994;3:13–19. doi: 10.1093/hmg/3.1.13. [DOI] [PubMed] [Google Scholar]
- 35.Brierley EJ, Johnson MA, Lightowlers RN, James OF, Turnbull DM. Role of mitochondrial DNA mutations in human aging: implications for the central nervous system and muscle. Ann Neurol. 1998;43:217–223. doi: 10.1002/ana.410430212. [DOI] [PubMed] [Google Scholar]
- 36.Schwarze SR, Lee CM, Chung SS, Roecker EB, Weindruch R, Aiken JM. High levels of mitochondrial DNA deletions in skeletal muscle of old rhesus monkeys. Mech Ageing Dev. 1995;83:91–101. doi: 10.1016/0047-6374(95)01611-3. [DOI] [PubMed] [Google Scholar]
- 37.Gianni P, Jan KJ, Douglas MJ, Stuart PM, Tarnopolsky MA. Oxidative stress and the mitochondrial theory of aging in human skeletal muscle. Exp Gerontol. 2004;39:1391–1400. doi: 10.1016/j.exger.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 38.Kovalenko SA, Kopsidas G, Kelso JM, Linnane AW. Deltoid human muscle mtDNA is extensively rearranged in old age subjects. Biochem Biophys Res Commun. 1997;232:147–152. doi: 10.1006/bbrc.1997.6251. [DOI] [PubMed] [Google Scholar]
- 39.Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, Morrow JD, Van Remmen H, Sedivy JM, Yamasoba T, Tanokura M, Weindruch R, Leeuwenburgh C, Prolla TA. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- 40.Trifunovic A, Hansson A, Wredenberg A, Rovio AT, Dufour E, Khvorostov I, Spelbrink JN, Wibom R, Jacobs HT, Larsson NG. Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc Natl Acad Sci U S A. 2005;102:17993–17998. doi: 10.1073/pnas.0508886102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly Y, Gidlof S, Oldfors A, Wibom R, Tornell J, Jacobs HT, Larsson NG. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 42.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 44.Staniek K, Nohl H. Are mitochondria a permanent source of reactive oxygen species? Biochim Biophys Acta. 2000;1460:268–275. doi: 10.1016/s0005-2728(00)00152-3. [DOI] [PubMed] [Google Scholar]
- 45.Johnson FB, Sinclair DA, Guarente L. Molecular biology of aging. Cell. 1999;96:291–302. doi: 10.1016/s0092-8674(00)80567-x. [DOI] [PubMed] [Google Scholar]
- 46.Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 47.Richter C, Park JW, Ames BN. Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc Natl Acad Sci U S A. 1988;85:6465–6467. doi: 10.1073/pnas.85.17.6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mecocci P, Fano G, Fulle S, MacGarvey U, Shinobu L, Polidori MC, Cherubini A, Vecchiet J, Senin U, Beal MF. Age-dependent increases in oxidative damage to DNA, lipids, and proteins in human skeletal muscle. Free Radic Biol Med. 1999;26:303–308. doi: 10.1016/s0891-5849(98)00208-1. [DOI] [PubMed] [Google Scholar]
- 49.Serra V, von Zglinicki T, Lorenz M, Saretzki G. Extracellular superoxide dismutase is a major antioxidant in human fibroblasts and slows telomere shortening. J Biol Chem. 2003;278:6824–6830. doi: 10.1074/jbc.M207939200. [DOI] [PubMed] [Google Scholar]
- 50.Orr WC, Sohal RS. Extension of life-span by overexpression of superoxide dismutase and catalase in Drosophila melanogaster. Science. 1994;263:1128–1130. doi: 10.1126/science.8108730. [DOI] [PubMed] [Google Scholar]
- 51.Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van Remmen H, Wallace DC, Rabinovitch PS. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308:1909–1911. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- 52.Orr WC, Mockett RJ, Benes JJ, Sohal RS. Effects of overexpression of copper-zinc and manganese superoxide dismutases, catalase, and thioredoxin reductase genes on longevity in Drosophila melanogaster. J Biol Chem. 2003;278:26418–26422. doi: 10.1074/jbc.M303095200. [DOI] [PubMed] [Google Scholar]
- 53.Larsen PL. Aging and resistance to oxidative damage in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 1993;90:8905–8909. doi: 10.1073/pnas.90.19.8905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vanfleteren JR, De Vreese A. Rate of aerobic metabolism and superoxide production rate potential in the nematode Caenorhabditis elegans. J Exp Zool. 1996;274:93–100. doi: 10.1002/(SICI)1097-010X(19960201)274:2<93::AID-JEZ2>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 55.Blander G, de Oliveira RM, Conboy CM, Haigis M, Guarente L. Superoxide dismutase 1 knock-down induces senescence in human fibroblasts. J Biol Chem. 2003;278:38966–38969. doi: 10.1074/jbc.M307146200. [DOI] [PubMed] [Google Scholar]
- 56.Ho YS, Magnenat JL, Bronson RT, Cao J, Gargano M, Sugawara M, Funk CD. Mice deficient in cellular glutathione peroxidase develop normally and show no increased sensitivity to hyperoxia. J Biol Chem. 1997;272:16644–16651. doi: 10.1074/jbc.272.26.16644. [DOI] [PubMed] [Google Scholar]
- 57.Melov S, Schneider JA, Day BJ, Hinerfeld D, Coskun P, Mirra SS, Crapo JD, Wallace DC. A novel neurological phenotype in mice lacking mitochondrial manganese superoxide dismutase. Nat Genet. 1998;18:159–163. doi: 10.1038/ng0298-159. [DOI] [PubMed] [Google Scholar]
- 58.Reaume AG, Elliott JL, Hoffman EK, Kowall NW, Ferrante RJ, Siwek DF, Wilcox HM, Flood DG, Beal MF, Brown RH, Jr, Scott RW, Snider WD. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Genet. 1996;13:43–47. doi: 10.1038/ng0596-43. [DOI] [PubMed] [Google Scholar]
- 59.Pansarasa O, Bertorelli L, Vecchiet J, Felzani G, Marzatico F. Age-dependent changes of antioxidant activities and markers of free radical damage in human skeletal muscle. Free Radic Biol Med. 1999;27:617–622. doi: 10.1016/s0891-5849(99)00108-2. [DOI] [PubMed] [Google Scholar]
- 60.Pansarasa O, Castagna L, Colombi B, Vecchiet J, Felzani G, Marzatico F. Age and sex differences in human skeletal muscle: role of reactive oxygen species. Free Radic Res. 2000;33:287–293. doi: 10.1080/10715760000301451. [DOI] [PubMed] [Google Scholar]
- 61.Skulachev VP. Why are mitochondria involved in apoptosis? Permeability transition pores and apoptosis as selective mechanisms to eliminate superoxide-producing mitochondria and cell. FEBS Lett. 1996;397:7–10. doi: 10.1016/0014-5793(96)00989-1. [DOI] [PubMed] [Google Scholar]
- 62.Boveris A, Chance B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem J. 1973;134:707–716. doi: 10.1042/bj1340707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bouillaud F, Couplan E, Pecqueur C, Ricquier D. Homologues of the uncoupling protein from brown adipose tissue (UCP1): UCP2, UCP3, BMCP1 and UCP4. Biochim Biophys Acta. 2001;1504:107–119. doi: 10.1016/s0005-2728(00)00241-3. [DOI] [PubMed] [Google Scholar]
- 64.Brand MD, Pamplona R, Portero-Otin M, Requena JR, Roebuck SJ, Buckingham JA, Clapham JC, Cadenas S. Oxidative damage and phospholipid fatty acyl composition in skeletal muscle mitochondria from mice underexpressing or overexpressing uncoupling protein 3. Biochem J. 2002;368:597–603. doi: 10.1042/BJ20021077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vidal-Puig AJ, Grujic D, Zhang CY, Hagen T, Boss O, Ido Y, Szczepanik A, Wade J, Mootha V, Cortright R, Muoio DM, Lowell BB. Energy metabolism in uncoupling protein 3 gene knockout mice. J Biol Chem. 2000;275:16258–16266. doi: 10.1074/jbc.M910179199. [DOI] [PubMed] [Google Scholar]
- 66.Kerner J, Turkaly PJ, Minkler PE, Hoppel CL. Aging skeletal muscle mitochondria in the rat: decreased uncoupling protein-3 content. Am J Physiol Endocrinol Metab. 2001;281:E1054–E1062. doi: 10.1152/ajpendo.2001.281.5.E1054. [DOI] [PubMed] [Google Scholar]
- 67.Nabben M, Hoeks J, Briede JJ, Glatz JF, Moonen-Kornips E, Hesselink MK, Schrauwen P. The effect of UCP3 overexpression on mitochondrial ROS production in skeletal muscle of young versus aged mice. FEBS Lett. 2008;582:4147–4152. doi: 10.1016/j.febslet.2008.11.016. [DOI] [PubMed] [Google Scholar]
- 68.Brand MD. Uncoupling to survive? The role of mitochondrial inefficiency in ageing. Exp Gerontol. 2000;35:811–820. doi: 10.1016/s0531-5565(00)00135-2. [DOI] [PubMed] [Google Scholar]
- 69.Lemire BD, Behrendt M, DeCorby A, Gaskova D. C. elegans longevity pathways converge to decrease mitochondrial membrane potential. Mech Ageing Dev. 2009;130:461–465. doi: 10.1016/j.mad.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 70.Gates AC, Bernal-Mizrachi C, Chinault SL, Feng C, Schneider JG, Coleman T, Malone JP, Townsend RR, Chakravarthy MV, Semenkovich CF. Respiratory uncoupling in skeletal muscle delays death and diminishes age-related disease. Cell Metab. 2007;6:497–505. doi: 10.1016/j.cmet.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 71.Speakman JR, Talbot DA, Selman C, Snart S, McLaren JS, Redman P, Krol E, Jackson DM, Johnson MS, Brand MD. Uncoupled and surviving: individual mice with high metabolism have greater mitochondrial uncoupling and live longer. Aging Cell. 2004;3:87–95. doi: 10.1111/j.1474-9728.2004.00097.x. [DOI] [PubMed] [Google Scholar]
- 72.Perez VI, Buffenstein R, Masamsetti V, Leonard S, Salmon AB, Mele J, Andziak B, Yang T, Edrey Y, Friguet B, Ward W, Richardson A, Chaudhuri A. Protein stability and resistance to oxidative stress are determinants of longevity in the longest-living rodent, the naked mole-rat. Proc Natl Acad Sci U S A. 2009;106:3059–3064. doi: 10.1073/pnas.0809620106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 74.Ott M, Zhivotovsky B, Orrenius S. Role of cardiolipin in cytochrome c release from mitochondria. Cell Death Differ. 2007;14:1243–1247. doi: 10.1038/sj.cdd.4402135. [DOI] [PubMed] [Google Scholar]
- 75.Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 76.Ow YP, Green DR, Hao Z, Mak TW. Cytochrome c: functions beyond respiration. Nat Rev Mol Cell Biol. 2008;9:532–542. doi: 10.1038/nrm2434. [DOI] [PubMed] [Google Scholar]
- 77.Lanza IR, Nair KS. Functional assessment of isolated mitochondria in vitro. Methods Enzymol. 2009;457:349–372. doi: 10.1016/S0076-6879(09)05020-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cande C, Cohen I, Daugas E, Ravagnan L, Larochette N, Zamzami N, Kroemer G. Apoptosis-inducing factor (AIF): a novel caspase-independent death effector released from mitochondria. Biochimie. 2002;84:215–222. doi: 10.1016/s0300-9084(02)01374-3. [DOI] [PubMed] [Google Scholar]
- 79.Veatch JR, McMurray MA, Nelson ZW, Gottschling DE. Mitochondrial dysfunction leads to nuclear genome instability via an iron–sulfur cluster defect. Cell. 2009;137:1247–1258. doi: 10.1016/j.cell.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mattson MP, Magnus T. Ageing and neuronal vulnerability. Nat Rev Neurosci. 2006;7:278–294. doi: 10.1038/nrn1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Narula J, Haider N, Virmani R, DiSalvo TG, Kolodgie FD, Hajjar RJ, Schmidt U, Semigran MJ, Dec GW, Khaw BA. Apoptosis in myocytes in end-stage heart failure. N Engl J Med. 1996;335:1182–1189. doi: 10.1056/NEJM199610173351603. [DOI] [PubMed] [Google Scholar]
- 82.Miro O, Lopez S, Fernandez-Sola J, Garrabou G, Pedrol E, Badia E, Martinez E, Cardellach F, Gatell JM, Casademont J. Short communication: HIV infection, antiretrovirals, and apoptosis: studies on skeletal muscle. AIDS Res Hum Retroviruses. 2005;21:702–705. doi: 10.1089/aid.2005.21.702. [DOI] [PubMed] [Google Scholar]
- 83.Sandri M, Carraro U, Podhorska-Okolov M, Rizzi C, Arslan P, Monti D, Franceschi C. Apoptosis, DNA damage and ubiquitin expression in normal and mdx muscle fibers after exercise. FEBS Lett. 1995;373:291–295. doi: 10.1016/0014-5793(95)00908-r. [DOI] [PubMed] [Google Scholar]
- 84.Dirks AJ, Leeuwenburgh C. Aging and lifelong calorie restriction result in adaptations of skeletal muscle apoptosis repressor, apoptosis-inducing factor, X-linked inhibitor of apoptosis, caspase-3, and caspase-12. Free Radic Biol Med. 2004;36:27–39. doi: 10.1016/j.freeradbiomed.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 85.Allen DL, Linderman JK, Roy RR, Bigbee AJ, Grindeland RE, Mukku V, Edgerton VR. Apoptosis: a mechanism contributing to remodeling of skeletal muscle in response to hindlimb unweighting. Am J Physiol. 1997;273:C579–C587. doi: 10.1152/ajpcell.1997.273.2.C579. [DOI] [PubMed] [Google Scholar]
- 86.Alway SE, Degens H, Krishnamurthy G, Smith CA. Potential role for Id myogenic repressors in apoptosis and attenuation of hypertrophy in muscles of aged rats. Am J Physiol Cell Physiol. 2002;283:C66–C76. doi: 10.1152/ajpcell.00598.2001. [DOI] [PubMed] [Google Scholar]
- 87.Veis DJ, Sorenson CM, Shutter JR, Korsmeyer SJ. Bcl-2-deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell. 1993;75:229–240. doi: 10.1016/0092-8674(93)80065-m. [DOI] [PubMed] [Google Scholar]
- 88.Holloszy JO. Biochemical adaptations in muscle. Effects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle. J Biol Chem. 1967;242:2278–2282. [PubMed] [Google Scholar]
- 89.Baldwin KM, Klinkerfuss GH, Terjung RL, Mole PA, Holloszy JO. Respiratory capacity of white, red, and intermediate muscle: adaptative response to exercise. Am J Physiol. 1972;222:373–378. doi: 10.1152/ajplegacy.1972.222.2.373. [DOI] [PubMed] [Google Scholar]
- 90.Bizeau ME, Willis WT, Hazel JR. Differential responses to endurance training in subsarcolemmal and intermyofibrillar mitochondria. J Appl Physiol. 1998;85:1279–1284. doi: 10.1152/jappl.1998.85.4.1279. [DOI] [PubMed] [Google Scholar]
- 91.Chow LS, Greenlund LJ, Asmann YW, Short KR, McCrady SK, Levine JA, Nair KS. Impact of endurance training on murine spontaneous activity, muscle mitochondrial DNA abundance, gene transcripts, and function. J Appl Physiol. 2007;102:1078–1089. doi: 10.1152/japplphysiol.00791.2006. [DOI] [PubMed] [Google Scholar]
- 92.Davies KJ, Packer L, Brooks GA. Biochemical adaptation of mitochondria, muscle, and whole-animal respiration to endurance training. Arch Biochem Biophys. 1981;209:539–554. doi: 10.1016/0003-9861(81)90312-x. [DOI] [PubMed] [Google Scholar]
- 93.Demirel HA, Powers SK, Naito H, Hughes M, Coombes JS. Exercise-induced alterations in skeletal muscle myosin heavy chain phenotype: dose–response relationship. J Appl Physiol. 1999;86:1002–1008. doi: 10.1152/jappl.1999.86.3.1002. [DOI] [PubMed] [Google Scholar]
- 94.Dudley GA, Abraham WM, Terjung RL. Influence of exercise intensity and duration on biochemical adaptations in skeletal muscle. J Appl Physiol. 1982;53:844–850. doi: 10.1152/jappl.1982.53.4.844. [DOI] [PubMed] [Google Scholar]
- 95.Gollnick PD, King DW. Effect of exercise and training on mitochondria of rat skeletal muscle. Am J Physiol. 1969;216:1502–1509. doi: 10.1152/ajplegacy.1969.216.6.1502. [DOI] [PubMed] [Google Scholar]
- 96.Howald H, Hoppeler H, Claassen H, Mathieu O, Straub R. Influences of endurance training on the ultrastructural composition of the different muscle fiber types in humans. Pflugers Arch. 1985;403:369–376. doi: 10.1007/BF00589248. [DOI] [PubMed] [Google Scholar]
- 97.Kent-Braun JA, McCully KK, Chance B. Metabolic effects of training in humans: a 31P-MRS study. J Appl Physiol. 1990;69:1165–1170. doi: 10.1152/jappl.1990.69.3.1165. [DOI] [PubMed] [Google Scholar]
- 98.Short KR, Vittone JL, Bigelow ML, Proctor DN, Rizza RA, Coenen-Schimke JM, Nair KS. Impact of aerobic exercise training on age-related changes in insulin sensitivity and muscle oxidative capacity. Diabetes. 2003;52:1888–1896. doi: 10.2337/diabetes.52.8.1888. [DOI] [PubMed] [Google Scholar]
- 99.Young IG, Anderson S. The genetic code in bovine mitochondria: sequence of genes for the cytochrome oxidase subunit II and two tRNAs. Gene. 1980;12:257–265. doi: 10.1016/0378-1119(80)90108-0. [DOI] [PubMed] [Google Scholar]
- 100.Zamora AJ, Tessier F, Marconnet P, Margaritis I, Marini JF. Mitochondria changes in human muscle after prolonged exercise, endurance training and selenium supplementation. Eur J Appl Physiol Occup Physiol. 1995;71:505–511. doi: 10.1007/BF00238552. [DOI] [PubMed] [Google Scholar]
- 101.Evans MJ, Scarpulla RC. Interaction of nuclear factors with multiple sites in the somatic cytochrome c promoter. Characterization of upstream NRF-1, ATF, and intron Sp1 recognition sequences. J Biol Chem. 1989;264:14361–14368. [PubMed] [Google Scholar]
- 102.Fisher RP, Topper JN, Clayton DA. Promoter selection in human mitochondria involves binding of a transcription factor to orientation-independent upstream regulatory elements. Cell. 1987;50:247–258. doi: 10.1016/0092-8674(87)90220-0. [DOI] [PubMed] [Google Scholar]
- 103.Jager S, Handschin C, St Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci U S A. 2007;104:12017–12022. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Holloszy JO. Regulation by exercise of skeletal muscle content of mitochondria and GLUT4. J Physiol Pharmacol. 2008;59(Suppl 7):5–18. [PubMed] [Google Scholar]
- 105.Lin J, Handschin C, Spiegelman BM. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005;1:361–370. doi: 10.1016/j.cmet.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 106.Scarpulla RC. Nuclear activators and coactivators in mammalian mitochondrial biogenesis. Biochim Biophys Acta. 2002;1576:1–14. doi: 10.1016/s0167-4781(02)00343-3. [DOI] [PubMed] [Google Scholar]
- 107.Adhihetty PJ, Taivassalo T, Haller RG, Walkinshaw DR, Hood DA. The effect of training on the expression of mitochondrial biogenesis- and apoptosis-related proteins in skeletal muscle of patients with mtDNA defects. Am J Physiol Endocrinol Metab. 2007;293:E672–E680. doi: 10.1152/ajpendo.00043.2007. [DOI] [PubMed] [Google Scholar]
- 108.Radak Z, Taylor AW, Sasvari M, Ohno H, Horkay B, Furesz J, Gaal D, Kanel T. Telomerase activity is not altered by regular strenuous exercise in skeletal muscle or by sarcoma in liver of rats. Redox Rep. 2001;6:99–103. doi: 10.1179/135100001101536102. [DOI] [PubMed] [Google Scholar]
- 109.Coggan AR, Spina RJ, King DS, Rogers MA, Brown M, Nemeth PM, Holloszy JO. Skeletal muscle adaptations to endurance training in 60- to 70-yr-old men and women. J Appl Physiol. 1992;72:1780–1786. doi: 10.1152/jappl.1992.72.5.1780. [DOI] [PubMed] [Google Scholar]
- 110.Jubrias SA, Esselman PC, Price LB, Cress ME, Conley KE. Large energetic adaptations of elderly muscle to resistance and endurance training. J Appl Physiol. 2001;90:1663–1670. doi: 10.1152/jappl.2001.90.5.1663. [DOI] [PubMed] [Google Scholar]
- 111.Balagopal P, Schimke JC, Ades P, Adey D, Nair KS. Age effect on transcript levels and synthesis rate of muscle MHC and response to resistance exercise. Am J Physiol Endocrinol Metab. 2001;280:E203–E208. doi: 10.1152/ajpendo.2001.280.2.E203. [DOI] [PubMed] [Google Scholar]
- 112.Judge S, Jang YM, Smith A, Selman C, Phillips T, Speakman JR, Hagen T, Leeuwenburgh C. Exercise by lifelong voluntary wheel running reduces subsarcolemmal and interfibrillar mitochondrial hydrogen peroxide production in the heart. Am J Physiol Regul Integr Comp Physiol. 2005;289:R1564–R1572. doi: 10.1152/ajpregu.00396.2005. [DOI] [PubMed] [Google Scholar]
- 113.Radak Z, Naito H, Kaneko T, Tahara S, Nakamoto H, Takahashi R, Cardozo-Pelaez F, Goto S. Exercise training decreases DNA damage and increases DNA repair and resistance against oxidative stress of proteins in aged rat skeletal muscle. Pflugers Arch. 2002;445:273–278. doi: 10.1007/s00424-002-0918-6. [DOI] [PubMed] [Google Scholar]
- 114.Radak Z, Kaneko T, Tahara S, Nakamoto H, Ohno H, Sasvari M, Nyakas C, Goto S. The effect of exercise training on oxidative damage of lipids, proteins, and DNA in rat skeletal muscle: evidence for beneficial outcomes. Free Radic Biol Med. 1999;27:69–74. doi: 10.1016/s0891-5849(99)00038-6. [DOI] [PubMed] [Google Scholar]
- 115.Aniansson A, Grimby G, Rundgren A. Isometric and isokinetic quadriceps muscle strength in 70-year-old men and women. Scand J Rehabil Med. 1980;12:161–168. [PubMed] [Google Scholar]
- 116.Ehsani AA, Spina RJ, Peterson LR, Rinder MR, Glover KL, Villareal DT, Binder EF, Holloszy JO. Attenuation of cardiovascular adaptations to exercise in frail octogenarians. J Appl Physiol. 2003;95:1781–1788. doi: 10.1152/japplphysiol.00194.2003. [DOI] [PubMed] [Google Scholar]
- 117.Brierley EJ, Johnson MA, James OF, Turnbull DM. Effects of physical activity and age on mitochondrial function. QJM. 1996;89:251–258. doi: 10.1093/qjmed/89.4.251. [DOI] [PubMed] [Google Scholar]
- 118.Proctor DN, Sinning WE, Walro JM, Sieck GC, Lemon PW. Oxidative capacity of human muscle fiber types: effects of age and training status. J Appl Physiol. 1995;78:2033–2038. doi: 10.1152/jappl.1995.78.6.2033. [DOI] [PubMed] [Google Scholar]
- 119.Russ DW, Kent-Braun JA. Is skeletal muscle oxidative capacity decreased in old age? Sports Med. 2004;34:221–229. doi: 10.2165/00007256-200434040-00002. [DOI] [PubMed] [Google Scholar]
- 120.Nair KS. Aging muscle: what causes it? E.V. McCollum Lecture 2004. Am J Clin Nutr. 2005;81:953–963. doi: 10.1093/ajcn/81.5.953. [DOI] [PubMed] [Google Scholar]
- 121.Atchley DW, Loeb RF, Richards DW, Jr, ElM B, Driscoll ME. On diabetic acidosis—a detailed study of electrolyte balances following the withdrawal and reestablishment of insulin therapy. J Clin Invest. 1933;12:297–326. doi: 10.1172/JCI100504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Holloszy JO Mortality rate and longevity of food-restricted exercising male rats: a reevaluation [DOI] [PubMed]
- 123.Holloszy JO. Exercise increases average longevity of female rats despite increased food intake and no growth retardation. J Gerontol. 1993;48:B97–B100. doi: 10.1093/geronj/48.3.b97. [DOI] [PubMed] [Google Scholar]
- 124.Holloszy JO, Smith EK, Vining M, Adams S. Effect of voluntary exercise on longevity of rats. J Appl Physiol. 1985;59:826–831. doi: 10.1152/jappl.1985.59.3.826. [DOI] [PubMed] [Google Scholar]
- 125.Walford RL, Harris SB, Weindruch R. Dietary restriction and aging: historical phases, mechanisms and current directions. J Nutr. 1987;117:1650–1654. doi: 10.1093/jn/117.10.1650. [DOI] [PubMed] [Google Scholar]
- 126.Weindruch R, Naylor PH, Goldstein AL, Walford RL. Influences of aging and dietary restriction on serum thymosin alpha 1 levels in mice. J Gerontol. 1988;43:B40–B42. doi: 10.1093/geronj/43.2.b40. [DOI] [PubMed] [Google Scholar]
- 127.Yu BP, Masoro EJ, McMahan CA. Nutritional influences on aging of Fischer 344 rats: 1. physical, metabolic, and longevity characteristics. J Gerontol. 1985;40:657–670. doi: 10.1093/geronj/40.6.657. [DOI] [PubMed] [Google Scholar]
- 128.Nisoli E, Tonello C, Cardile A, Cozzi V, Bracale R, Tedesco L, Falcone S, Valerio A, Cantoni O, Clementi E, Moncada S, Carruba MO. Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science. 2005;310:314–317. doi: 10.1126/science.1117728. [DOI] [PubMed] [Google Scholar]
- 129.Lin SJ, Kaeberlein M, Andalis AA, Sturtz LA, Defossez PA, Culotta VC, Fink GR, Guarente L. Calorie restriction extends Saccharomyces cerevisiae lifespan by increasing respiration. Nature. 2002;418:344–348. doi: 10.1038/nature00829. [DOI] [PubMed] [Google Scholar]
- 130.Bishop NA, Guarente L. Genetic links between diet and lifespan: shared mechanisms from yeast to humans. Nat Rev Genet. 2007;8:835–844. doi: 10.1038/nrg2188. [DOI] [PubMed] [Google Scholar]
- 131.Panowski SH, Wolff S, Aguilaniu H, Durieux J, Dillin A. PHA-4/Foxa mediates diet-restriction-induced longevity of C. elegans. Nature. 2007;447:550–555. doi: 10.1038/nature05837. [DOI] [PubMed] [Google Scholar]
- 132.Bevilacqua L, Ramsey JJ, Hagopian K, Weindruch R, Harper ME. Long-term caloric restriction increases UCP3 content but decreases proton leak and reactive oxygen species production in rat skeletal muscle mitochondria. Am J Physiol Endocrinol Metab. 2005;289:E429–E438. doi: 10.1152/ajpendo.00435.2004. [DOI] [PubMed] [Google Scholar]
- 133.Lambert AJ, Merry BJ. Effect of caloric restriction on mitochondrial reactive oxygen species production and bioenergetics: reversal by insulin. Am J Physiol Regul Integr Comp Physiol. 2004;286:R71–R79. doi: 10.1152/ajpregu.00341.2003. [DOI] [PubMed] [Google Scholar]
- 134.Jia K, Levine B. Autophagy is required for dietary restriction-mediated life span extension in C. elegans. Autophagy. 2007;3:597–599. doi: 10.4161/auto.4989. [DOI] [PubMed] [Google Scholar]
- 135.Wohlgemuth SE, Julian D, Akin DE, Fried J, Toscano K, Leeuwenburgh C, Dunn WA., Jr Autophagy in the heart and liver during normal aging and calorie restriction. Rejuvenation Res. 2007;10:281–292. doi: 10.1089/rej.2006.0535. [DOI] [PubMed] [Google Scholar]
- 136.Lin SJ, Ford E, Haigis M, Liszt G, Guarente L. Calorie restriction extends yeast life span by lowering the level of NADH. Genes Dev. 2004;18:12–16. doi: 10.1101/gad.1164804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bordone L, Guarente L. Calorie restriction, SIRT1 and metabolism: understanding longevity. Nat Rev Mol Cell Biol. 2005;6:298–305. doi: 10.1038/nrm1616. [DOI] [PubMed] [Google Scholar]
- 138.Gerhart-Hines Z, Rodgers JT, Bare O, Lerin C, Kim SH, Mostoslavsky R, Alt FW, Wu Z, Puigserver P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. EMBO J. 2007;26:1913–1923. doi: 10.1038/sj.emboj.7601633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 140.Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE, Tsokos M, Alt FW, Finkel T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci U S A. 2008;105:3374–3379. doi: 10.1073/pnas.0712145105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Feige JN, Lagouge M, Canto C, Strehle A, Houten SM, Milne JC, Lambert PD, Mataki C, Elliott PJ, Auwerx J. Specific SIRT1 activation mimics low energy levels and protects against diet-induced metabolic disorders by enhancing fat oxidation. Cell Metab. 2008;8:347–358. doi: 10.1016/j.cmet.2008.08.017. [DOI] [PubMed] [Google Scholar]
- 142.Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang LL, Scherer B, Sinclair DA. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425:191–196. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- 143.Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, Jin L, Boss O, Perni RB, Vu CB, Bemis JE, Xie R, Disch JS, Ng PY, Nunes JJ, Lynch AV, Yang H, Galonek H, Israelian K, Choy W, Iffland A, Lavu S, Medvedik O, Sinclair DA, Olefsky JM, Jirousek MR, Elliott PJ, Westphal CH. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature. 2007;450:712–716. doi: 10.1038/nature06261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Pearson KJ, Baur JA, Lewis KN, Peshkin L, Price NL, Labinskyy N, Swindell WR, Kamara D, Minor RK, Perez E, Jamieson HA, Zhang Y, Dunn SR, Sharma K, Pleshko N, Woollett LA, Csiszar A, Ikeno Y, Le Couteur D, Elliott PJ, Becker KG, Navas P, Ingram DK, Wolf NS, Ungvari Z, Sinclair DA, de Cabo R. Resveratrol delays age-related deterioration and mimics transcriptional aspects of dietary restriction without extending life span. Cell Metab. 2008;8:157–168. doi: 10.1016/j.cmet.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewis K, Pistell PJ, Poosala S, Becker KG, Boss O, Gwinn D, Wang M, Ramaswamy S, Fishbein KW, Spencer RG, Lakatta EG, Le Couteur D, Shaw RJ, Navas P, Puigserver P, Ingram DK, de Cabo R, Sinclair DA. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444:337–342. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, Geny B, Laakso M, Puigserver P, Auwerx J. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 147.Civitarese AE, Carling S, Heilbronn LK, Hulver MH, Ukropcova B, Deutsch WA, Smith SR, Ravussin E. Calorie restriction increases muscle mitochondrial biogenesis in healthy humans. PLoS Med. 2007;4:e76. doi: 10.1371/journal.pmed.0040076. [DOI] [PMC free article] [PubMed] [Google Scholar]