

Abstract
B cell malignancies, such as human Burkitt’s lymphoma (BL), often harbor translocations that link c-myc or other proto-oncogenes to the immunoglobulin heavy chain locus (IgH)1. The nature of elements that activate oncogenes within such translocations has been a longstanding question. Translocations within IgH involve DNA double strand breaks (DSBs) initiated either by the RAG1/2 endonuclease during V(D)J recombination or by activation induced cytidine deaminase (AID) during class switch recombination (CSR)2-4. V(D)J recombination in progenitor B (pro-B) cells assembles IgH variable region exons upstream of μ constant region (Cμ) exons, which are the first of several sets of CH exons (“CH genes”) within a CH locus that spans several hundred kilobases5,6. In mature B cells, CSR deletes Cμ and replaces it with a downstream CH gene6. An enhancer (iEμ) between the variable region exons and Cμ promotes V(D)J recombination in developing B cells7. In addition, the IgH 3’ regulatory region (IgH3’RR) lies downstream of the CH locus and modulates CSR by long-range transcriptional enhancement of CH genes8-10. Transgenic mice bearing iEμ or IgH3’RR sequences fused to c-myc are predisposed to B lymphomas, demonstrating such elements can confer oncogenic c-myc expression11-16. However, in many B cell lymphomas, IgH/c-myc translocations delete iEμ and place c-myc up to 200kb upstream of the IgH3’RR1. We now address the oncogenic role of the IgH3’RR by inactivating it in two distinct mouse models for B cell lymphoma with IgH/c-myc translocations. The IgH3’RR is dispensable for pro-B lymphomas with V(D)J recombination-initiated translocations, but required for peripheral B cell lymphomas with CSR-associated translocations. As the IgH3’RR is not required for CSR-associated IgH breaks or IgH/c-myc translocations in peripheral B cell lymphoma progenitors, we conclude this regulatory region confers oncogenic activity via long-range and developmental stage-specific activation of translocated c-myc genes.
Individual CH genes are organized into germline transcription units which consist, from 5’ to 3’, of a non-coding “I” exon, a switch (S) region and the CH exons6. CSR to a particular CH gene requires introduction of AID-initiated DSBs into the donor S region upstream of Cμ (Sμ) and into a downstream acceptor S region6. The I exon is preceded by a germline promoter that is up-regulated by particular activation treatments, with transcription targeting AID to specific S regions17. The IgH3’RR contains multiple enhancer elements18-20 and controls germline transcription of certain CH promoters over distances of 100kb or more8. However, the IgH3’RR is not required for V(D)J recombination, expression of rearranged IgH genes, or transcription through Sμ or Sγ110. To test potential roles of the IgH3’RR in B cell lymphomagenesis, we bred an IgH3’RR inactivating mutation, which deletes the key HS3b and HS4 enhancers10 (Supp. Fig. 1), into non-homologous end-joining (NHEJ) and p53 tumor suppressor deficient backgrounds that predispose to either pro-B or peripheral B cell lymphoma.
XRCC4 and DNA ligase 4 (Lig4) form a NHEJ ligation complex required for V(D)J recombination21. Mice with germline inactivation of either Lig4 or Xrcc4 and the p53 tumor suppressor develop pro-B cell lymphomas with complex translocations (“complicons”) involving IgH on chromosome 12 (chr12) and a region downstream of c-myc on chromosome 15 (chr15)22. These translocations arise from joining RAG1/2-induced DSBs in the IgH JH region to DSBs downstream of c-myc, leading to dicentric chr12;15 translocations and c-myc amplification via breakage-fusion-bridge cycles22,23. Analysis of Lig4/p53 double-deficient mice (“LP” mice) that harbored IgH3’RR inactivating mutations on either one or both IgH alleles (referred to as LPR+/- or LPR-/- respectively) revealed that both genotypes succumbed to pro-B cell lymphomas with kinetics similar to those of LP mice22 (Fig. 1a; Supp. Table 1). Likewise, all analyzed LPR+/- and LPR-/- tumors had characteristic chr12 to15 translocations (T(12;15)) and 15;12 complicons (C(15;12)) harboring amplified c-myc (Fig. 1b; Supp. Fig. 2; Supp. Table 1). In addition, Southern blotting with a probe that distinguishes IgH3’RR-deleted from wild-type (wt) IgH alleles (Fig. 1c, top) revealed that IgH/c-myc translocations/amplifications involved the wt allele in some LPR+/- tumors and the IgH3’RR-deleted allele in others (Fig. 1c, bottom). Thus, the IgH3’RR is dispensable for LP pro-B cell lymphomas with IgH/c-myc complicons.
Figure 1.
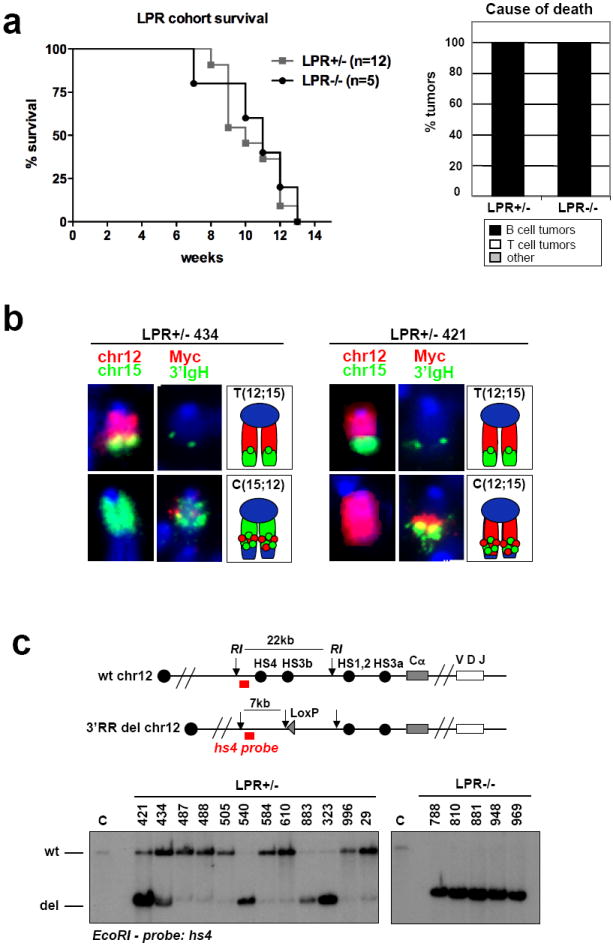
Deletion of the Igh3’RR does not affect development of pro-B-cell lymphomas. a, Left, Kaplan–Meier curve of the LPR1+/− (n=12) and LPR−/− (n=5) cohorts. Curves represent total survival. Right, the percentage of mice in the LPR+/− and LPR−/− cohorts succumbing to B-cell lymphomas, thymic lymphomas or other causes of death. b, Examples of cytological aberrations in representative LPR+/− tumours with translocations to the wild-type (mouse number 434) or the 3’RR-deleted (mouse number 421) Igh alleles. In each set of panels: left, paints specific for chr12 (red) and chr15 (green); middle, FISH analysis on separated metaphases with 3’Igh (green) and c-myc (red) probes; right, graphic representation. Only chromosomes involved in translocations are shown. Whole metaphases are presented in Supplementary Fig. 2. c, Southern blot analysis of LPR+/− (left) and LPR−/− (right) tumour DNA with a probe downstream of hs4, which distinguishes the wild-type (WT) and 3’RR-deleted (del) Igh alleles. A schematic of the wild-type and del Igh locus, with the position of the probe, is on the top. Numbers refer to individual mice in the cohort (see Supplementary Table 1). C, control, total spleen DNA from wild-type mouse; RI, EcoRI.
Specific inactivation of Xrcc4 by a loxP/Cre approach in peripheral B cells of p53-deficient mice (referred to as “CXP” mice) leads to surface Ig-negative peripheral B cell lymphomas24,25. CXP B cell lymphomas arise from progenitors that delete or aberrantly rearrange their Igκ and Igλ light chain loci and routinely harbor a T(12;15) that fuses IgH S regions to sequences just upstream of c-myc, leading to high level c-myc expression from a translocated single copy c-myc gene25,26. Such T(12;15)s lack iEμ as they occur downstream of this element24. To test potential roles of the IgH3’RR in CXP tumorigenesis, we followed tumor development in cohorts of CXPR+/- and CXPR-/- mice. CXPR+/- mice succumbed to the same tumor spectrum as CXP mice25, with 40% developing surface Ig-negative B cell lymphomas that appeared identical to CXP B cell lymphomas (Fig. 2a and Supp. Table 2). The remaining CXPR+/- mice succumbed to thymic lymphomas or other tumors associated with germline p53 deficiency. In contrast, none of 17 analyzed CXPR-/- mice developed B cell lymphoma; instead, most died from thymic lymphoma (Fig. 2a and Supp. Table 2). Thus, homozygous IgH3’RR inactivation abrogates CXP B cell lymphomas.
Figure 2. Deletion of the IgH3’RR abrogates development of peripheral B cell lymphomas.
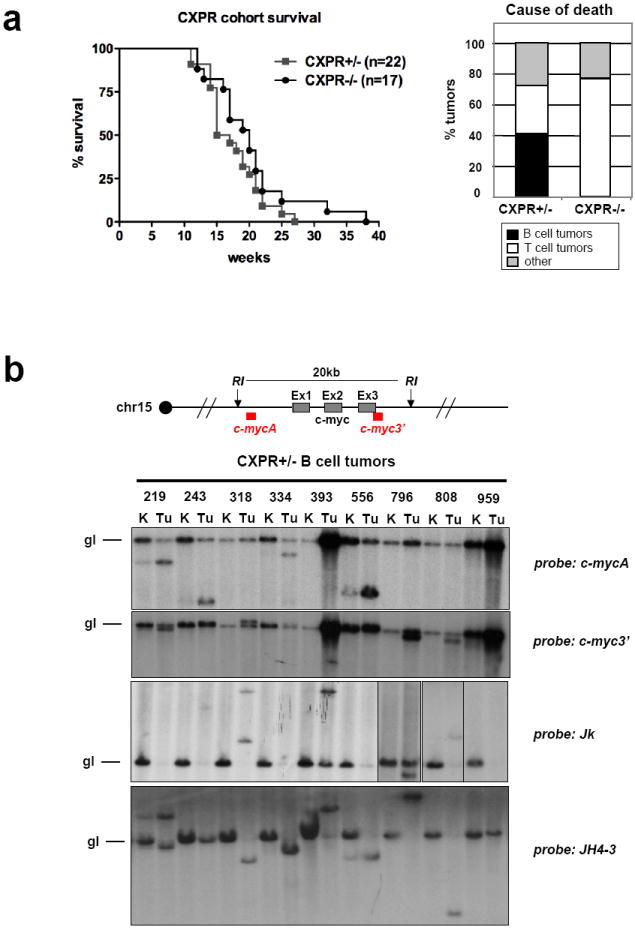
a. Left: Kaplan-Meier curve of the CXPR+/- (n=22) and CXPR-/- (n=17) cohorts. Curves represent total survival. Right: Percentage of mice in the CXPR+/- and CXPR-/- cohorts succumbing to B cell lymphomas, thymic lymphomas or to other cause of death. b. Southern blot analysis of CXPR+/- tumor DNA with probes indicated on the right of each panel. Numbers refer to individual mice in the cohort (see Supp. Table 2). K, kidney, used as control; Tu, tumor. A schematic of the c-myc locus, indicating the 5’ and 3’ probes used to detect c-myc rearrangements is on the top.
All analyzed CXPR+/- B cell tumors had a clonal T(12;15), based on spectral karyotyping (Fig. 3c, Supp. Fig. 3). As in CXP B cell lymphomas25, most T(12;15) from CXPR+/- B cell lymphomas split c-myc, as evidenced by Southern blotting with 5’ and 3’ c-myc locus probes (Fig. 2b, top panels). However, two tumors (393 and 959) had c-myc amplification without detectable c-myc rearrangements (Fig. 2b), and tumor 796, while harboring a clonal T(12;15) that split c-myc, also contained metaphases with a translocation that fused this T(12;15) to chromosome 16, resulting in low level IgH/c-myc amplification (Supp. Fig.3; data not shown). Northern blotting revealed elevated c-myc expression in all CXPR+/- B cell tumors, with tumors 393, 796 and 959 showing highest levels (Supp. Fig. 4), suggesting that amplification may sometimes be selected secondary to ectopic activation to achieve maximal expression, which may be relevant to certain human lymphomas that acquire c-myc amplification during tumor progression12.
Figure 3. T(12;15) in CXPR+/- B cell tumors always involves the wt IgH allele.
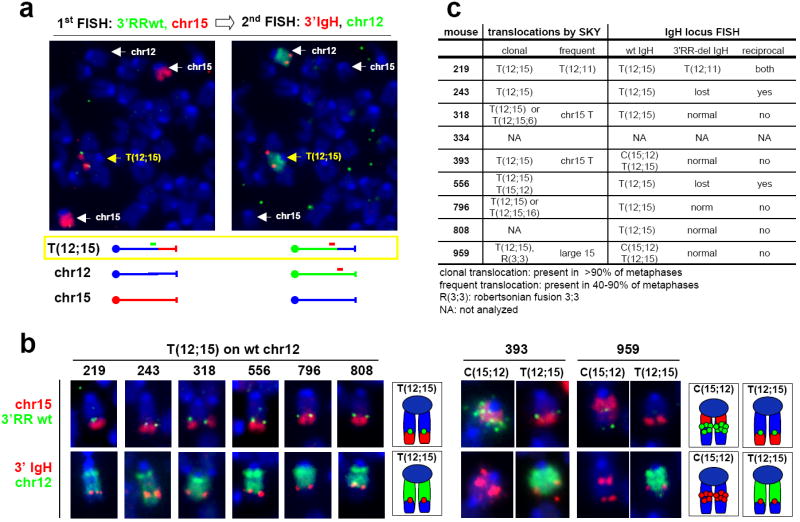
a. Example of FISH and chromosome paints on CXPR+/- tumor metaphases. The same metaphase was sequentially analyzed with the different set of probes indicated at the top of each panel. A schematic of the different chromosomal species detected is shown at the bottom. b. Summary of FISH and chromosome paint analyses on all analyzed CXPR+/- tumors. Numbers refer to individual mice in the cohort. Sequential hybridization with the set of probes indicated on the left was performed. Only chromosomes involved in translocations are shown, along with a graphic representation. The whole metaphases are presented in Supp. Fig. 4. c. Table summarizing SKY and FISH data for CXPR+/- B cell tumors.
To determine which IgH allele was involved in CXPR+/- lymphoma T(12;15)s, we analyzed tumor metaphases by fluorescence in situ hybridization (FISH) with a probe specific for the deleted portion of the IgH3’RR (3’RR wt probe, green) and a chr15 paint (red). In this assay, the translocated portion of chr15 (red paint) co-localizes with a green signal if the wt IgH allele is translocated, but not if the IgH3’RR-deleted allele is translocated (Fig. 3a, left panel). Sequential re-probing of these metaphases with a green chr12 paint (FISH 2, green) plus a 3’ IgH BAC probe (FISH 2, red) (Fig. 3a, right panel) revealed that all CXPR+/- tumor IgH/c-myc translocations involved the wt IgH allele (Fig. 3b, c and Supp. Fig. 5). These results, coupled with the absence of B cell tumors in CXPR-/- mice, demonstrate that the IgH3’RR is required for peripheral CXP B cell lymphomas via a role in oncogenic IgH/c-myc translocations.
The IgH3’RR might influence appearance of oncogenic IgH/c-myc translocations by mechanistically promoting them through induction of CSR at certain S regions and/or by long-range activation of translocated c-myc expression. To distinguish between these potential mechanisms, we asked whether the IgH3’RR-deleted allele is a substrate for AID-induced DSBs. As one measure, we activated CXPR+/- and CXPR-/- B cells for 4 days with αCD40/IL4 and found that both genotypes switched to IgG1, as expected by the fact that the 3’IgHRR is not required for CSR to this IgH isotype8,10 (Supp. Fig. 6a). We also analyzed metaphases from αCD40/IL4-stimulated CXPR+/-and CXPR-/- B cells via two-color FISH27 and found that the increased level of IgH breaks in the absence of XRCC424 was not markedly affected by the IgH3’RR mutation (Fig. 4a). CSR to IgG3 is severely impaired in mice homozygous for IgH3’RR inactivating mutations due to inhibition of Iγ3 transcription8,10. However, LPS/αIgD-dextran stimulation to induce IgG3 CSR led to similarly increased IgH breaks in CXPR+/- and CXPR-/- B cells (Supp. Fig. 6b; Fig. 4a), likely reflecting unimpeded AID activity on Sμ, which is transcribed independently of the IgH3’RR10. Southern blotting further revealed that most CXPR+/- B cell tumors had Sμ rearrangements or deletions on both alleles, again indicative of substantial AID activity on IgH3’RR-deleted alleles (Fig. 4b). We conclude that introduction of AID-induced IgH lesions is not markedly impaired by the IgH3’RR deletion.
Figure 4. Deletion of the IgH3’RR does not affect IgH locus breaks and IgH/c-myc translocations.
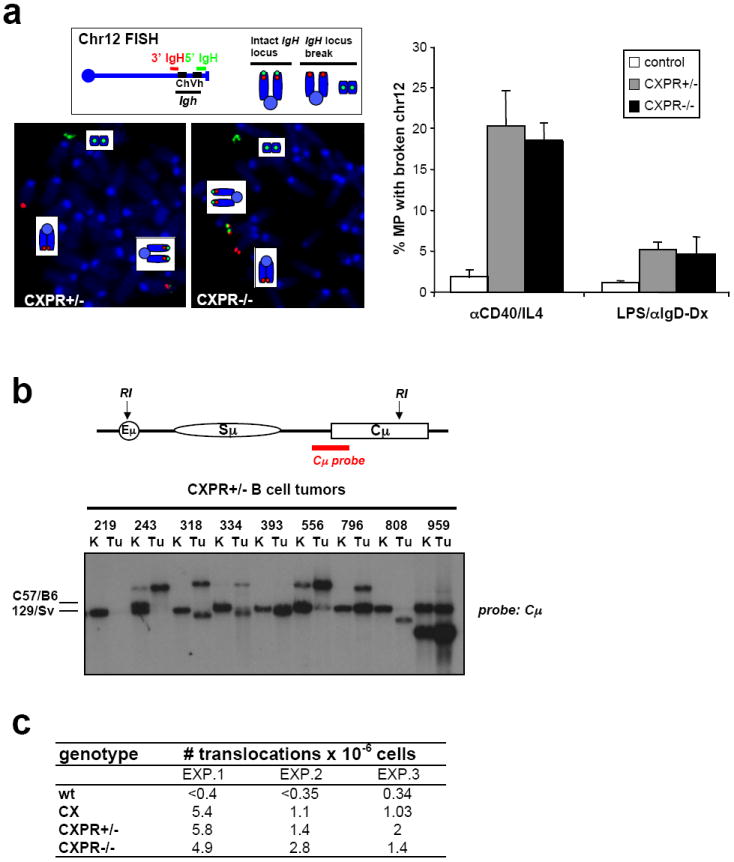
a. Left, top: Diagram showing location of 3’ IgH (red) and 5’ IgH (green) BAC probes used for FISH. An intact IgH locus on chr12 shows colocalized red and green signals, a broken locus shows split red and green signals. Left, bottom: Examples of metaphases from CXPR+/-and CXPR-/- αCD40/IL4-stimulated splenic B cells showing IgH breaks. Right: Quantification of IgH locus breaks in day 4 αCD40/IL4-activated or day 5 LPS/αIgD-dextran-activated B cells from control, CXPR+/-, and CXPR-/- mice. At least three mice per each genotype and at least 60 metaphases per mouse were analyzed; data are presented as average ± sd. b. Southern blot analysis of CXPR+/- tumor DNA with a Cμ probe that detects Sμ rearrangements. A schematic of the IgH locus, with position of the probe is on the top. Position of germline bands in C57/B6 and 129Sv backgrounds is indicated on the left; the 3’RR-deleted allele is from 129Sv background. Numbers refer to individual mice in the cohort. K, kidney, used as control; Tu, tumor. Note that in some cases kidneys contained infiltration of tumor cells, as judged by tumor-specific rearranged JH and c-myc bands (Fig. 2b). c. Frequency of IgH/c-myc translocations was measured by PCR assays using Sμ and c-myc primers. DNA samples were isolated from day4 αCD40/IL4-activated wt, CX, CXPR+/- and CXPR-/- splenic B cells.
While AID-induced IgH breaks occur at high frequency on IgH3’RR-deleted alleles, IgH/c-myc translocations still might be inhibited, for example by a different distribution of IgH DSBs or by effects on proximity of the two loci26. Therefore, we employed a PCR assay28 to directly evaluate potential effects of the IgH3’RR deletion on IgH/c-myc translocation frequency and found IgH/c-myc translocations, indeed, occurred at similar frequencies in αCD40/IL4-stimulated CXPR+/- and CXPR-/- B cells (Fig. 4c). Moreover, one CXPR+/- B cell tumor had a T(12;11) involving the IgH3’RR-deleted allele in addition to its T(12;15), again indicating the IgH3’RR-deleted allele is a translocation target (Fig. 3c, Supp. Fig. 3). We conclude that the IgH3’RR is dispensable for generation of IgH/c-myc translocations in XRCC4-deficient B cells.
The IgH locus has long been speculated to have cis-acting elements that activate c-myc or other oncogenes in the context of translocations. We now demonstrate that the IgH3’RR is required for oncogenicity of IgH/c-myc translocations that ectopically activate c-myc in mouse CXP B cell lymphomas. As a substantial proportion of CXPR+/- (Supp. Fig. 7) and CXP lymphomas25 have translocations that fuse c-myc to Sμ, oncogenic IgH3’RR activity extends at least 200kb upstream. Thus, our findings define a major oncogenic role for the IgH3’RR in activating c-myc subsequent to IgH/c-myc translocations; although we do not rule out an additional role in promoting translocations by enhancing AID-mediated lesions in certain S regions regulated by this element. Although high-copy iEμ transgenes predispose to pro-B cell lymphoma in mice12, the role of iEμ in IgH/oncogene translocations remains to be determined. In this context, knock-in of c-myc into the IgH JH region led to peripheral B cell lymphomas, as opposed to pro-B cell lymphomas12, suggesting iEμ alone may not always be sufficient to activate c-myc in the endogenous setting. In this regard, our finding that the IgH3’RR is dispensable for LP pro-B cell lymphomas may explain why c-myc is amplified in these tumors and ectopically activated in peripheral CXP B cell tumors. Specifically, the IgH3’RR is not active in pro-B cells9 and would not activate IgH translocations upstream of a single-copy c-myc, favoring selection for translocations downstream of c-myc that promote gene amplification. Given the similar organization of mouse and human IgH9, our findings suggest that the IgH3’RR also supports activated oncogene expression in human B cell tumors with IgH S region translocations that eliminate iEμ (e.g. sporadic BL29) and, potentially, even in some with IgH JH region translocations that leave iEμ intact (e.g. endemic BL29) In this regard, targeted inhibition of the B cell-specific IgH3’RR could theoretically provide a therapeutic strategy for such human B lymphomas.
METHODS SUMMARY
Mouse strains
IgH3’RR-deleted mice were generated previously10 and crossed into Lig4+/-/p53+/-30 or CD21-Cre/XRCC4c/c/p53+/-25 mice to obtain triple or quadruple heterozygous animals, which were appropriately crossed to obtain the experimental cohorts. Mice were analyzed as outlined in the text at 8–30 weeks of age. The Institutional Animal Care and Use Committee of Children’s Hospital (Boston, Massachusetts) approved all animal work.
Splenic B-cell purification, activation in culture, and CSR assays
CD43− B cells were isolated and cultured as previously described27. Cells were processed at day 4 of stimulation with αCD40/IL4 or at day 5 of stimulation with LPS/αIgD-dextran for DNA isolation, metaphase preparation and flow cytometry analysis (see Methods).
Two-color FISH
Metaphase spreads were prepared and FISH experiments performed according to standard protocols27. FISH probes are detailed in Methods. Whole chromosome paints specific for mouse chromosome 12 and 15 were used according to the manufacturer’s instructions (Applied Spectral Imaging).
PCR assay to detect IgH/c-myc translocations
IgH/c-myc translocation junctions were amplified by PCR from genomic DNA prepared from splenic B cells activated for 4 days with αCD40/IL4, using primers and conditions previously described28 (see Methods). DNA corresponding to 50000 or 100000 cells was analyzed in separate reactions. PCR products were run on agarose gel, blotted and hybridized with an internal oligonucleotide probe in the c-myc locus.
Supplementary Material
Acknowledgments
We thank Lynn G. Jiang for help with cloning translocation breakpoints. This work was supported by NIH grant CA92625 and the Leukemia and Lymphoma Society of America (LLS) SCORE grant to F.W.A. M.G. was a Special Fellow of LLS and C.T.Y. was supported by an NCI training grant. M.G. and C.T.Y were also supported by the de Villiers International Achievement Award of the LLS to F.W.A. E.P. was a Fellow of the Fondation pour la Recherche Medicale. F.W.A. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature
Author contributions. F.W.A. and M.G. planned studies and interpreted data. M.G. performed experiments, with technical help from J.M.B. C.T.Y generated CXP mice. E.P and M.C. generated IgH3’RR-deleted mice and helped interpret data. F.W.A. and M.G. wrote the paper.
Author information. Reprints and permissions information is available at www.nature.com/reprints. The authors declare no competing financial interests.
References
- 1.Kuppers R, Dalla-Favera R. Mechanisms of chromosomal translocations in B cell lymphomas. Oncogene. 2001;20:5580–5594. doi: 10.1038/sj.onc.1204640. [DOI] [PubMed] [Google Scholar]
- 2.Lieber MR, Yu K, Raghavan SC. Roles of nonhomologous DNA end joining, V(D)J recombination, and class switch recombination in chromosomal translocations. DNA Repair (Amst) 2006;5:1234–1245. doi: 10.1016/j.dnarep.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 3.Ramiro A, et al. The role of activation-induced deaminase in antibody diversification and chromosome translocations. Adv Immunol. 2007;94:75–107. doi: 10.1016/S0065-2776(06)94003-6. [DOI] [PubMed] [Google Scholar]
- 4.Robbiani DF, et al. AID is required for the chromosomal breaks in c-myc that lead to c-myc/IgH translocations. Cell. 2008;135:1028–1038. doi: 10.1016/j.cell.2008.09.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bassing CH, Swat W, Alt FW. The mechanism and regulation of chromosomal V(D)J recombination. Cell. 2002;109(Suppl):S45–55. doi: 10.1016/s0092-8674(02)00675-x. [DOI] [PubMed] [Google Scholar]
- 6.Chaudhuri J, et al. Evolution of the immunoglobulin heavy chain class switch recombination mechanism. Adv Immunol. 2007;94:157–214. doi: 10.1016/S0065-2776(06)94006-1. [DOI] [PubMed] [Google Scholar]
- 7.Henderson A, Calame K. Transcriptional regulation during B cell development. Annu Rev Immunol. 1998;16:163–200. doi: 10.1146/annurev.immunol.16.1.163. [DOI] [PubMed] [Google Scholar]
- 8.Cogne M, et al. A class switch control region at the 3’ end of the immunoglobulin heavy chain locus. Cell. 1994;77:737–747. doi: 10.1016/0092-8674(94)90057-4. [DOI] [PubMed] [Google Scholar]
- 9.Khamlichi AA, Pinaud E, Decourt C, Chauveau C, Cogne M. The 3’ IgH regulatory region: a complex structure in a search for a function. Adv Immunol. 2000;75:317–345. doi: 10.1016/s0065-2776(00)75008-5. [DOI] [PubMed] [Google Scholar]
- 10.Pinaud E, et al. Localization of the 3’ IgH locus elements that effect long-distance regulation of class switch recombination. Immunity. 2001;15:187–199. doi: 10.1016/s1074-7613(01)00181-9. [DOI] [PubMed] [Google Scholar]
- 11.Adams JM, et al. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature. 1985;318:533–538. doi: 10.1038/318533a0. [DOI] [PubMed] [Google Scholar]
- 12.Janz S. Myc translocations in B cell and plasma cell neoplasms. DNA Repair (Amst) 2006;5:1213–1224. doi: 10.1016/j.dnarep.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 13.Schmidt EV, Pattengale PK, Weir L, Leder P. Transgenic mice bearing the human c-myc gene activated by an immunoglobulin enhancer: a pre-B-cell lymphoma model. Proc Natl Acad Sci USA. 1988;85:6047–6051. doi: 10.1073/pnas.85.16.6047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Truffinet V, et al. The 3’ IgH locus control region is sufficient to deregulate a c-myc transgene and promote mature B cell malignancies with a predominant Burkitt-like phenotype. J Immunol. 2007;179:6033–6042. doi: 10.4049/jimmunol.179.9.6033. [DOI] [PubMed] [Google Scholar]
- 15.Wang J, Boxer LM. Regulatory elements in the immunoglobulin heavy chain gene 3’-enhancers induce c-myc deregulation and lymphomagenesis in murine B cells. J Biol Chem. 2005;280:12766–12773. doi: 10.1074/jbc.M412446200. [DOI] [PubMed] [Google Scholar]
- 16.Yan Y, Park SS, Janz S, Eckhardt LA. In a model of immunoglobulin heavy-chain (IGH)/MYC translocation, the Igh 3’ regulatory region induces MYC expression at the immature stage of B cell development. Genes Chromosomes Cancer. 2007;46:950–959. doi: 10.1002/gcc.20480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Manis JP, Tian M, Alt FW. Mechanism and control of class-switch recombination. Trends Immunol. 2002;23:31–39. doi: 10.1016/s1471-4906(01)02111-1. [DOI] [PubMed] [Google Scholar]
- 18.Madisen L, Groudine M. Identification of a locus control region in the immunoglobulin heavy-chain locus that deregulates c-myc expression in plasmacytoma and Burkitt’s lymphoma cells. Genes Dev. 1994;8:2212–2226. doi: 10.1101/gad.8.18.2212. [DOI] [PubMed] [Google Scholar]
- 19.Dariavach P, Williams GT, Campbell K, Pettersson S, Neuberger MS. The mouse IgH 3’-enhancer. Eur J Immunol. 1991;21:1499–1504. doi: 10.1002/eji.1830210625. [DOI] [PubMed] [Google Scholar]
- 20.Lieberson R, Giannini SL, Birshtein BK, Eckhardt LA. An enhancer at the 3’ end of the mouse immunoglobulin heavy chain locus. Nucleic Acids Res. 1991;19:933–937. doi: 10.1093/nar/19.4.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lieber MR. The mechanism of human nonhomologous DNA end joining. J Biol Chem. 2008;283:1–5. doi: 10.1074/jbc.R700039200. [DOI] [PubMed] [Google Scholar]
- 22.Zhu C, et al. Unrepaired DNA breaks in p53-deficient cells lead to oncogenic gene amplification subsequent to translocations. Cell. 2002;109:811–821. doi: 10.1016/s0092-8674(02)00770-5. [DOI] [PubMed] [Google Scholar]
- 23.Difilippantonio MJ, et al. Evidence for replicative repair of DNA double-strand breaks leading to oncogenic translocation and gene amplification. J Exp Med. 2002;196 doi: 10.1084/jem.20020851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yan CT, et al. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature. 2007;449:478–482. doi: 10.1038/nature06020. [DOI] [PubMed] [Google Scholar]
- 25.Wang JH, et al. Oncogenic transformation in the absence of Xrcc4 targets peripheral B cells that have undergone editing and switching. J Exp Med. 2008;205:3079–3090. doi: 10.1084/jem.20082271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang JH, et al. Mechanisms promoting translocations in editing and switching peripheral B cells. Nature. 2009;460:231–236. doi: 10.1038/nature08159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Franco S, et al. H2AX prevents DNA breaks from progressing to chromosome breaks and translocations. Mol Cell. 2006;21:201–214. doi: 10.1016/j.molcel.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 28.Ramiro AR, et al. Role of genomic instability and p53 in AID-induced c-myc-Igh translocations. Nature. 2006;440:105–109. doi: 10.1038/nature04495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Neri A, Barriga F, Knowles DM, Magrath IT, Dalla-Favera R. Different regions of the immunoglobulin heavy-chain locus are involved in chromosomal translocations in distinct pathogenetic forms of Burkitt lymphoma. Proc Natl Acad Sci U S A. 1988;85:2748–2752. doi: 10.1073/pnas.85.8.2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frank KM, et al. DNA ligase IV deficiency in mice leads to defective neurogenesis and embryonic lethality via the p53 pathway. Mol Cell. 2000;5:993–1002. doi: 10.1016/s1097-2765(00)80264-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.