

Abstract

While β-ketoesters are useful Michael donors, they were previously ineffective in Michael-Michael cascade reactions using α,β-unsaturated aldehydes in conjunction with diphenylprolinol silyl ether organocatalysts. However, through rational modification of substrates and manipulation of the catalytic cycle, we developed an efficient Michael-Michael cascade reaction using β-ketoesters of type 9. In this transformation, highly substituted fused carbocycles are generated in a single step in up to 87% yield and 99% ee.
Organocatalyzed cascade reactions are a powerful synthetic tool in green chemistry, as environmentally-inert catalysts are used in the formation of multiple bonds and stereocenters in a single reaction flask.1 Diphenyl prolinol silyl ethers such as 1a (Scheme 1) have recently emerged as highly effective catalysts for organocascade reactions. This class of catalysts has been employed in cascade processes in which the formation of multiple C-C single bonds generates 3-, 5-, or 6-membered carbocycles. These include: Michael-SN2 alkylation,2 Michael-Aldol,3 Michael-Knoevenagel,4 Michael-Mannich,5 Michael-Henry,6 Michael-Wittig,7 and Michael-Michael8 cascade reactions.
Scheme 1.

1a-catalyzed cascade reactions.
Among the latter class of reactions, of those cascades initiated by activation of an α,β-unsaturated aldehyde through iminium catalysis, a β-dicarbonyl compound was used as a Michael donor in only one example8b (Reaction 1, Scheme 1), despite the fact that this type of Michael donor is commonly employed in organocatalytic conjugate addition reactions.9 However, when the unsaturation was relocated relative to the β-dicarbonyl moiety in an attempt to form substituted cyclohexanes, the desired transformation (i.e., Reaction 4) was surprisingly not observed. When conjugated β-ketoesters with terminal, monosubstituted olefins (5) were used, the initial Michael addition was instead followed by a Morita-Baylis-Hillman reaction (Reaction 2).10 Alternatively, when conjugated β-ketoesters with internal olefins (7) were used, subsequent to the initial Michael addition, acetal formation occurred in lieu of another Michael addition (Reaction 3).11 Presumably, the second Michael addition is kinetically slow and/or thermodynamically unfavorable, as it would disrupt the highly conjugated system.
We reasoned that using conjugated β-ketoesters of type 9, in which the olefin is part of a carbocycle, would modulate the reactivity of these substrates and might enable the desired Michael-Michael cascade reaction. First, disubstitution at the 1-position of the alkene would preclude the undesired Morita-Baylis-Hillman pathway. Additionally, the fact that the alkene is cyclic and is not part of a system with extended conjugation may alter the kinetic and thermodynamic preference, respectively, for the desired Michael addition pathway relative to the undesired acetalization pathway. Moreover, substrates of type 9 would produce highly-substituted fused carbocycles, with a chiral catalyst, as well as the thermodynamic preference for the ring junction, establishing multiple stereocenters.
Using conjugated β-ketoester 9a, the 1a-catalyzed Michael-Michael cascade reaction with 3a in DCE did generate 10 and 11, albeit in very low conversion even after ten days (entry 1, Table 1). While the initial Michael addition was complete within 12 hours, the subsequent Michael addition was exceedingly sluggish. To promote this second step, a preliminary screen of additives known to facilitate catalyst turnover (i.e., benzoic acid) or to enhance enamine formation (i.e., Et3N, NaOAc) was carried out. While basic additives did not accelerate the reaction (data not shown), with benzoic acid, enhanced diastereomeric ratios and excellent ee's of the major diastereomer, 10b, and its epimer, 10a, were acheived (entry 2). Although the conversion was also improved, it was still low after extended reaction times. As suspected, ethanol, a protic solvent that can participate in hydrogen bonding interactions with the β-ketoester moiety, further accelerated the second Michael addition and greatly improved conversion (entry 3).
Table 1.
Catalyst Optimizationa
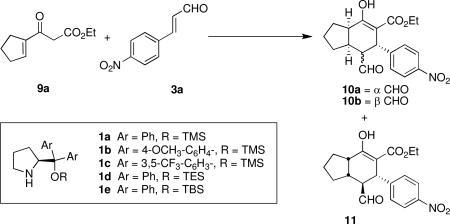 | |||||||
|---|---|---|---|---|---|---|---|
| entry | catalyst | solvent | time (h) | % conversionb | dr (10:11)b % | ee (10a)c | % ee(10b)c |
| 1d | 1a | DCE | 240 | 13 | 78:22 | nd | nd |
| 2 | 1a | DCE | 168 | 25 | 84:16 | 99 | 99 |
| 3 | 1a | EtOH | 168 | 61 | 78:22 | 99 | 99 |
| 4 | 1b | EtOH | 168 | 20 | 85:15 | 87 | 99 |
| 5 | 1c | EtOH | 168 | 0 | nd | nd | nd |
| 6 | 1d | EtOH | 168 | 66 | 79:21 | 99 | 98 |
| 7 | 1e | EtOH | 216 | 65 | 80:20 | 87 | 96 |
Reaction conditions: 3a (1 equiv), 9a (1 equiv), cat. (20 mol %), PhCOOH (20 mol %), solvent (0.3 M), rt.
Determined by 1H NMR of crude reaction mixture.
Determined by chiral HPLC.
Reaction run without PhCOOH.
The use of a more electron-rich catalyst, 1b, drastically slowed both Michael additions in the cascade reaction (entry 4), while the use of a more electron-deficient catalyst, 1c, completely suppressed the second Michael addition (entry 5). Catalysts with different silyl groups did not provide both 10a and 10b in 99% ee, as had catalyst 1a (entries 6-7). In all cases, the ee of the minor diastereomer, 11, was diminished relative to that of 10a and 10b, ranging from 33% (using 1d) to 82% (using 1b).
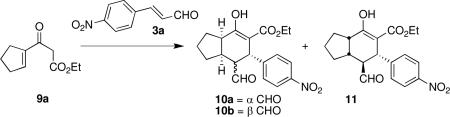
With optimal catalyst 1a in hand, optimization of other reaction conditions ensued. Both non-polar solvents and polar aprotic solvents led to substantially reduced conversions relative to reactions run in ethanol (entries 1-4, Table 2). Switching to trifluoroethanol, a solvent that is a stronger hydrogen bond donor than ethanol, gave surprising results.
Table 2.
Optimization of Reaction Conditionsa
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| entry | mol % 1a | additive | solvent | time (h) | % conversionb | dr (10:11)b | ee (10a)c | ee (10b)c |
| 1 | 20 | PhCO2H | toluene | 168 | 5 | 80:20 | nd | nd |
| 2 | 20 | PhCO2H | Et2O | 168 | <1 | nd | nd | nd |
| 3 | 20 | PhCO2H | THF | 168 | 0 | nd | nd | nd |
| 4 | 20 | PhCO2H | MeCN | 168 | 32 | 93:7 | 99 | 99 |
| 5 | 20 | PhCO2H | CF3CH2OH | 2 | 12 | 89:11 | nd | nd |
| 6 | 20 | PhCO2H | CF3CH2OH | 41 | 17 | 91:9 | 99 | 99 |
| 7 | 20 | -- | CF3CH2OH | 17 | 85 | 90:10 | 99 | 99 |
| 8 | 10 | -- | CF3CH2OH | 17 | 87 | 91:9 | 99 | 99 |
| 9 | 5 | -- | CF3CH2OH | 17 | 80 | 91:9 | 99 | 99 |
| 10 | 1 | -- | CF3CH2OH | 88 | 25 | 88:12 | 99 | 99 |
| 11d | 5 | -- | CF3CH2OH | 46 | 76 | 92 : 8 | 99 | 99 |
Reaction conditions: 3a (1 equiv), 9a (1 equiv), 1a, additive (20 mol %), solvent (0.3 M), rt.
Determined by 1H NMR of crude reaction mixture.
Determined by chiral HPLC.
Reaction run at 0 °C.
After only two hours, product formation was detected (entry 5). Additionally, the ratio of 10a:10b was 1:29, whereas in all previous experiments the ratios of 10a:10b ranged from 1:1.4 to 1.7:1. After 41 hours, there was little improvement in conversion, but the ratio of 10a:10b was 1:5 (entry 6). Moreover, the first Michael addition had not gone to completion under these conditions; the single Michael adduct along with substantial amounts of 3a and 9a were present after 41 hours. As the diastereomeric ratio and ee's were both excellent in this solvent, we sought to improve the conversion. Consideration of these, and other, observations with respect to the proposed mechanism of this transformation (Scheme 2) provided insight as to how to accomplish this.
Scheme 2.

Proposed catalytic cycle.
First, the observation that the initial Michael addition had not gone to completion indicated that either the “catalyst release 1” pathway of iminium 12 competes with the Michael addition to 12, or that 14 can revert back to starting materials. Subjecting single Michael adduct 14 to catalyst 1a and benzoic acid in trifluoroethanol resulted in rapid conversion to 10 only, revealing that the formation of 14 is not reversible. Thus, the “catalyst release 1” pathway appears to be favored in the presence of benzoic acid in trifluoroethanol more so than in other solvents.
When the initial Michael addition does occur to provide 13, if pathway A is followed, the second Michael addition occurs. In trifluoroethanol, this second Michael addition is rapid, as evidenced by the conversion to 10 within two hours. However, in this solvent, as in other solvents, 13 prefers the “catalyst release 2” pathway, which provides single Michael adduct 14. Compound 14 can react with 1a to re-enter the catalytic cycle and go on to product, as rapidly occurs in the absence of 3a. However, for reasons explained above, in trifluoroethanol and benzoic acid, there would be a substantial amount of 3a in solution, which evidently competes for the catalyst. Together, this accounts for the presence of 14 after 41 hours and for the plateau in conversion to 10 (entry 6, Table 2).
Finally, since epimerization of 10a and 10b occurrs in the presence of benzoic acid and 1a, but not benzoic acid alone, we speculated that it proceeds via an enamine intermediate (16), and not via a keto-enol equilibrium. The fact that the conversion to 10a and 10b did not change dramatically over the course of 39 hours under these conditions, while the ratio of 10a to 10b did, suggests that 10b formed first and was slowly converted to, and reaching equilibrium with, 10a. It would therefore appear that in the presence of benzoic acid in trifluoroethanol, “catalyst release 3” predominates over formation of enamine 16 either from 15 prior to catalyst release or from 10b (via 15) after its ejection from the catalytic cycle.
Thus, whereas in other solvents a sluggish second Michael addition hampered conversion, these observations collectively implied that under these conditions, catalyst release (i.e., turnover) was the culprit. Impeding catalyst release from 12 and from 13 should enable a rapid, efficient and highly selective Michael-Michael cascade reaction. We therefore ran the reaction in the absence of benzoic acid, which, as mentioned earlier, is known to facilitate catalyst turnover. Gratifyingly, this led to 85% conversion, 9:1 dr, and 99% ee of 10a and 10b (present in a 1:1.1 ratio) in only 17 hours (entry 7, Table 2)!
Lowering the catalyst loading to 10 mol % slightly improved both conversion and diastereoselectivity (entry 8). A catalyst loading of 5 mol % led to a slight decrease in conversion, but maintained the high selectivity of the reaction (entry 9). Further lowering of the catalyst loading to 1 mol % resulted in reduced conversion and a slight erosion of diastereoselectivity (entry 10). Finally, running the reaction at 0 °C led to a more sluggish reaction with a nominal improvement in diastereoselectivity (entry 11).
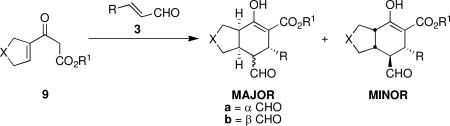
Using 10 mol % 1a and trifluoroethanol as solvent at rt, an investigation of substrate scope was undertaken (Table 3). The 1a-catalyzed Michael—Michael cascade reaction afforded products in high yields and high selectivity using α,β-unsaturated aromatic aldehydes with electron-withdrawing, electron-releasing, and electronically neutral substitution at either the meta- or para- positions (entries 1-4). However, ortho (nitro) substitution was not tolerated. In addition, α,β-unsaturated aldehydes with heteroaromatic and non-aromatic substitutents afforded products in slightly reduced yields, but in 98% ee and 97:3 dr (entries 5-6). Finally, different ester substituents (entry 7) and, notably, heteroatom substitution in the cyclopentane ring (entry 8) were well-tolerated in this reaction, leading to rapid and highly selective product formation. A ketoester with a cyclohexane (i.e., 9 where X = (CH2)2) formed additional diastereomers in this reaction (data not shown).
Table 3.
Substrate Scope.a
 | ||||||
|---|---|---|---|---|---|---|
| entry | Product (major) | time (h) | yield (%)b | dr (maj:min)c | % ee (a)d | % ee (b)d |
| 1 |  |
20 | 82 | 92:8 | 99 | 98 |
| 2 | 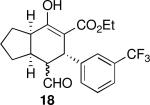 |
17 | 79 | 93:7 | 99 | 99 |
| 3 | 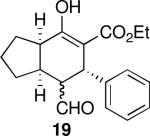 |
40 | 87 | 92:8 | 99 | 98 |
| 4 | 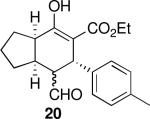 |
168 | 79 | 93:7 | 99 | 99 |
| 5 | 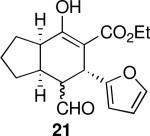 |
142 | 61 | 97:3 | 98 | 98 |
| 6 | 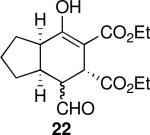 |
22 | 67 | 97:3 | 98 | 98 |
| 7 | 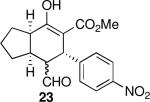 |
21 | 76 | 91:9 | 97 | 98 |
| 8 | 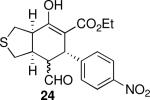 |
28 | 80 | 93:7 | 98 | 96 |
Reaction conditions: 3 (1 equiv), 9 (1 equiv), 1a (10 mol %), CF3CH2OH (0.3 M), rt.
Isolated yield.
Determined by 1H NMR of isolated products.
Determined by chiral HPLC.
A model for the stereochemical outcome of this reaction is depicted in Scheme 2. The stereochemistry at C3 was was assigned by analogy with other 1a-catalyzed conjugate additions and is assumed to arise from the initial Michael addition occurring from the face opposite the bulky group in iminium 12. The remaining stereochemistries were established by X-ray crystallograpy (see supporting information). They are consistent with the intramolecular Michael addition of 13 occurring with the Michael acceptor approaching the enamine from the face opposite, and the proton at C3 projected out towards, the bulky group. Additionally, we established that 10a and 10b were epimers by subjecting pure 10a to benzoic acid in the presence of 1a, which produced a mixture of 10a and 10b.
In conclusion, through rational modification of substrates and manipulation of the catalytic cycle, we developed a new, efficient 1a-catalyzed Michael-Michael cascade reaction. This transformation generates highly substituted, fused carbocycles with dr's ≥ 91:9 and ee's ≥ 96%. Further investigations into this, and other novel, 1a-catalyzed cascade reaction(s) are presently underway.
Supplementary Material
Acknowledgement
This work was supported by a grant from the National Institutes of Health (1SC2GM082360). The authors gratefully acknowledge Dr. Cliff Soll and Dr. Louis Todaro (both at the City University of New York at Hunter College) for their assistance with high-resolution mass spectrometry and X-ray crystallography, respectively. The authors also wish to thank Prof. Gary Molander (University of Pennsylvania) for thoughtful discussions.
Footnotes
SUPPORTING INFORMATION AVAILABLE General experimental conditions and full characterization data for compounds 9b, 9c, 10a, 10b, 11, 17a, 17b, 18a, 18b, 19a, 19b, 20a, 20b, 21a, 21b, 22a, 22b, 23a, 23b, 24a and 24b, including an X-ray crystal structure of 10a. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For a review, see: Enders D, Grondal C, Hüttl MRM. Angew. Chem. Int. Ed. 2007;46:1570–1581. doi: 10.1002/anie.200603129.
- 2.a Xie H, Zu L, Li H, Wang J, Wang W. J. Am. Chem. Soc. 2007;48:5835–5839. [Google Scholar]; b Vesely J, Zhao G-L, Bartoszewicz A, Córdova A. Tetrahedron Lett. 2008;49:4209–4212. [Google Scholar]; c Ibrahem I, Zhao G-L, Rios R, Vesely J, Sundén H, Dziedzic P, Córdova A. Chem. Eur. J. 2008;14:7867–7879. doi: 10.1002/chem.200800442. [DOI] [PubMed] [Google Scholar]
- 3.a Marigo M, Bertelsen S, Landa A, Jørgensen KA. J. Am. Chem. Soc. 2006;128:5475–5479. doi: 10.1021/ja058490o. [DOI] [PubMed] [Google Scholar]; b Enders D, Narine AA, Benninghaus TR, Raabe G. Synlett. 2007:1667–1670. [Google Scholar]; c Wang J, Li H, Xie H, Zu L, Shen X, Wang W. Angew. Chem. Int. Ed. 2007;46:9050–9053. doi: 10.1002/anie.200703163. [DOI] [PubMed] [Google Scholar]; d Hong B-C, Nimje RY, Sadani AA, Liao J-H. Org. Lett. 2008;10:2345–2348. doi: 10.1021/ol8005369. [DOI] [PubMed] [Google Scholar]; e Zhao G-L, Dziedzic P, Ullah F, Eriksson L, Córdova A. Tetrahedron Lett. 2009;50:3458–3462. [Google Scholar]; f Rueping M, Kuenkel A, Tato F, Bats JW. Angew. Chem. Int. Ed. 2009;48:3699–3702. doi: 10.1002/anie.200900754. [DOI] [PubMed] [Google Scholar]; g Enders D, Wang C, Bats JW. Synlett. 2009:1777–1780. [Google Scholar]
- 4.a Hayashi Y, Toyoshima M, Hiroaki G, Ishikawa H. Org. Lett. 2009;11:45–48. doi: 10.1021/ol802330h. [DOI] [PubMed] [Google Scholar]; b Łukaz A, Richter B, Vila C, Krawczyk H, Jørgensen KA. Chem. Eur. J. 2009;15:3093–3102. doi: 10.1002/chem.200802285. [DOI] [PubMed] [Google Scholar]
- 5.Ibrahem I, Rios R, Vesely J, Córdova A. Tetrahedron Lett. 2007;48:6252–6257. [Google Scholar]
- 6.a Hayashi Y, Okano Y, Aratake S, Hazelard D. Angew. Chem. Int. Ed. 2007;46:4922–4925. doi: 10.1002/anie.200700909. [DOI] [PubMed] [Google Scholar]; b Reyes E, Jiang H, Milelli A, Elsner P, Hazell RG, Jørgensen KA. Angew. Chem. Int. Ed. 2007;46:9202–9205. doi: 10.1002/anie.200704454. [DOI] [PubMed] [Google Scholar]
- 7.Liu Y.-k., Ma C, Jiang K, Liu T-Y, Chen Y-C. Org. Lett. 2009;11:2848–2851. doi: 10.1021/ol9010568. [DOI] [PubMed] [Google Scholar]
- 8.a Enders D, Hüettl MRM, Grondal C, Raabe G. Nature. 2006;441:861–863. doi: 10.1038/nature04820. [DOI] [PubMed] [Google Scholar]; B Zu L, Li H, Xie H, Wang J, Jiang W, Tang Y, Wang W. Angew. Chem. Int. Ed. 2007;46:3732–3734. doi: 10.1002/anie.200700485. [DOI] [PubMed] [Google Scholar]; C Carlone A, Cabrera S, Marigo M, Jørgensen KA. Angew. Chem. Int. Ed. 2007;46:1101–1104. doi: 10.1002/anie.200604479. [DOI] [PubMed] [Google Scholar]; D Bor-Cheng H, Nimje RY, Wu M-F, Sadani AA. Eur. J. Org. Chem. 2008:1449–1457. [Google Scholar]; E Penon O, Carlone A, Mazzanti A, Locatelli M, Sambri L, Bartoli G, Melchiorre P. Chem. Eur. J. 2008;14:4788–4791. doi: 10.1002/chem.200800440. [DOI] [PubMed] [Google Scholar]; F Zhao G-L, Ibrahem I, Dziedzic P, Sun J, Bonneau C, Córdova A. Chem. Eur. J. 2008;14:10007–10011. doi: 10.1002/chem.200801082. [DOI] [PubMed] [Google Scholar]; G Bencivenni G, Wu L-Y, Mazzanti A, Giannichi B, Pesciaioli F, Song M-P, Bartoli G, Melchiorre P. Angewandte Chem. Int. Ed. 2009;48:1–5. doi: 10.1002/anie.200903192. [DOI] [PubMed] [Google Scholar]; H Hong B-C, Nimje RY, Liao J-H. Org. Biomol. Chem. 2009:3095–3101. [Google Scholar]
- 9.For a review of organocatalyzed conjugate addition reactions, including those involving β-dicarbonyl compounds as Michael donors, see: Alma i, D., Alonso DA, Nájera C. Tetrahedron Asymm. 2007;18:299–365.
- 10.Cabrera S, Alemán J, Bolze P, Bertelsen S, Jørgensen KA. Angew. Chem. Int. Ed. 2008;47:121–125. doi: 10.1002/anie.200704076. [DOI] [PubMed] [Google Scholar]
- 11.Zhu M-K, Wei Q, Gong L-Z. Adv. Synth. Catal. 2008;350:1281–1285. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.