

Abstract
A few tumor antigen (TA)-specific monoclonal antibodies (mAb) have been approved by FDA for the treatment of several major malignant diseases and are commercially available. Once in the clinic, mAb have an average success rate of ~30% and are well tolerated. These results have changed the face of cancer therapy, bringing us closer to more specific and more effective biologic therapy of cancer. The challenge facing tumor immunologists at present is represented by the identification of the mechanism(s) underlying patients’ differential clinical response to mAb-based immunotherapy. This information is expected to lead to the development of criteria to select patients to be treated with mAb-based immunotherapy. In the past in vitro and in vivo evidence has shown that TA-specific mAb can mediate their therapeutic effect by inducing tumor cell apoptosis, inhibiting the targeted antigen function, blocking tumor cell signaling and/or mediating complement-or cell-dependent lysis of tumor cells. More recent evidence suggests that TA-specific mAb can induce TA-specific cytotoxic T cell responses by enhancing TA uptake by dendritic cells (DC) and cross-priming of T cells. In this manuscript, we briefly summarize the TA-specific mAb that have received FDA approval. Next we review the potential mechanisms underlying the therapeutic efficacy of TA-specific mAb with emphasis on the induction of TA-specific cellular immune responses and their potential to contribute to the clinical efficacy of TA-specific mAb-based immunotherapy. Lastly, we discuss the potential negative impact of immune escape mechanisms on the clinical efficacy of TA-specific mAb-based immunotherapy.
Keywords: ADCC, antibody, cancer, complement, cytotoxic T lymphocyte, HLA antigen, Immunoglobin Fc receptor, immune escape, immunotherapy, monoclonal antibody, NK cell
I. Introduction
The concept that antibodies could be used for the treatment of malignant disease originated more than a century ago [1]. The first generation of antibody-based therapies were based on the use of tumor antigen (TA)-specific allogeneic, autologous or xenogeneic polyclonal antibodies, which were ill-suited as cancer-specific therapies because of their limited or lack of specificity and reproducibility. It was not until the development of the hybridoma technology [2], that antibody-based immunotherapy of malignant diseases became a practical reality. The hybridoma technology enabled the production of a large number of human TA-specific murine mAb. The clinical application of some of them yielded a handful of promising results that were however overshadowed by disappointing outcomes in early clinical trials implemented with TA-specific mouse mAb in patients with various types of cancer [3]. In hindsight, the inadequate response rates (RR) observed with first generation TA-specific mAb most likely reflected their murine origins resulting in high immunogenicity and poor ability to recruit immune effector mechanisms [4–6]. These hurdles have been recently overcome by the generation of chimeric, humanized and human mAb resulting in reduced or lack of immunogenicity and improved ability to recruit effector cells [7].
Today, TA-specific mAb have been established as highly sensitive and reproducible probes in the diagnostic arena [8–13] as well as in clinically and commercially successful therapies for a variety of malignant diseases [3]. The average clinical success rate of TA-specific mAb-based immunotherapy, which manifests itself as statistically significant disease free interval and survival prolongation as well as reduction of tumor mass in some of the treated patients, is about 30% with ranges from 0 to 60% [3,14]. Only little is known about why merely a limited percentage of the treated patients respond clinically to TA-specific mAb-based immunotherapy. The mechanisms underlying patients’ differential clinical response to TA-specific mAb-based immunotherapy represent a topic of intense research at present, since this information has both basic research and clinical relevance. It will contribute to our understanding of the molecular basis of the clinical efficacy of TA-specific mAb-based immunotherapy and it will lead to the development of criteria to select patients to be treated with TA-specific mAb based immunotherapy. In this review we first summarize the TA-specific mAb that have received FDA approval. Second, we describe the potential mechanisms underlying the therapeutic efficacy of TA-specific mAb with emphasis on the induction of TA-specific cellular immune responses. Finally, we discuss the potential negative impact of tumor immune escape mechanisms on the clinical efficacy of TA-specific mAb-based immunotherapy.
II. TA-specific mAb
In recent years, the regulatory approval and sales of new human medicines indicates an increasing number of biologic therapies, i.e., small molecular inhibitors and mAb specifically designed to target malignant cells [15]a. In fact the global sale of mAb-derived biologic therapies in 2006 was $20.6 billion dollars, indicating a major paradigm shift in industrial R&D from pharmaceutical to biologic therapies [15]a. At the end of 2007, more than thirty years of clinical studies have resulted in the approval of six unconjugated, humanized, or chimeric TA-specific mAb for cancer therapy along with one drug immunoconjugate and two radioisotope immunoconjugates (Table 1) [3,14]. The results of clinical trials in patients treated with TA-specific mAb have demonstrated that the goal of cancer-targeted therapy is realistic and may be superior to older non-specific conventional chemo- and radio-therapy-based regimens, at a minimum enhancing their activity. Once in the clinic, TA-specific mAb are well tolerated with an average success rate of 30% [3,14]. Clinical responses have been found to include complete (CR) and partial (PR) responses as well as statistically significant increases in progression free survival (PFS) and in disease free-interval and -survival in patients with a number of malignant diseases (Table 1). In general, compared with solid tumors, hematologic neoplasms have proven easier to target with TA-specific mAb-based therapies because therapeutic efficacy can be achieved at lower doses and tumor penetration is more readily achieved [3,14]. Moreover, radioimmunotherapy (RIT) has been more successful for hematologic malignancies such as Non Hodgkin’s Lymphoma (NHL). However fewer of these agents are entering clinical trials due to complexities in manufacture and safety concerns compared with unconjugated mAb [3,14]. An additional limitation of RIT relates to delivery of the dose of radioactivity to the tumor compared with normal tissues [3,14]. The poor specificity index is due to slow pharmacokinetics and slow tumor perfusion.
Table 1.
FDA approved TA-specific mAb for human cancers.
| Target | Isotype | FDA approved disease | |
|---|---|---|---|
| Rituximab | CD20 | Chimeric IgG1 | CD20 (+) low grade lymphoma1, diffuse large B cell lymphoma1, follicular lymphoma1 |
| 90Y Ibritumomab + tiuxetan | CD20 | Radiolabeled murine IgG1 | CD20(+) low grade lyphoma2 |
| 131I Tositumomab | CD20 | Radiolabeled murine IgG1 | CD20(+) low grade lyphoma3 |
| Alemtuzumab | CD53 | Humanized IgG1 | Chronic lymphocytic leukemia4 |
| Gemtuzumab + Ozogamacin | CD33 | Rec. humanized IgG4- conjugated to calicheamicin | Acute myelogenous leukemia5 |
| Tastuzumab | HER2/neu | Humanized IgG1 | Her2/neu (+) breast cancer6 |
| Cetuximab | EGFR | Chimeric IgG1 | EGFR(+) Colon cancer7 |
| Panitumumab | EGFR | Fully human IgG2 | EGFR(+) Colon cancer8 |
| Bevacizumab | VEGF | Humanized IgG1 | Colon cancer9, recurrent or advance non-small cell lung cancer, metastatic breast cancer |
Low grade lymphoma: 2nd line monotherapy; diffuse large B cell lymphoma and follicular lymphoma: 1st line chemoimmune therapy as well as maintenance for follicular lymphoma.
2nd line monotherapy.
2nd line monotherapy.
1st and 2nd line monotherapy.
>60 years of age, 2nd line monotherapy.
2nd line monotherapy, adjuvant and 1st line chemoimmunetherapy.
2nd line monotherapy or chemoimmune therapy.
2nd line monotherapy.
1st line chemoimmune therapy.
Compared to traditional chemo- and radio-therapy-based regimens, in general the side effects of immunotherapy with non-conjugated TA-specific mAb are fairly mild [3,14]. Nonetheless, toxicities do occur and may be classified as mechanism-independent and mechanism-dependent (Table 2). Most of the toxicities related to TA-specific mAb are mechanism-independent and are related to allergic or hypersensitivity reactions caused by a protein containing xenogeneic sequences [3,14]. These reactions occur during or just after the first injection and are summarized in Table 2. Moreover, human anti-mouse Ig antibodies, including anti-idiotypic antibodies, can complex with circulating therapeutic mAb, and inhibit their targeting of tumor cells. It should be noted that conjugated antibodies often have a lower therapeutic index than non-conjugated TA-specific mAb and for this reason they often result in more side effects [3,14]. Rare, but more serious side effects of TA-specific mAb-based immunotherapy are often related to mechanism-dependent toxicities and result from the binding of a therapeutic antibody to its target antigen (Table 2) [3,14].
Table 2.
Toxicities related to TA-specific mAb for human cancers.
| Mechanism-dependent | Mechanism-independent | |
|---|---|---|
| Rituximab | Transitory lymphocyte B depletion Reactivation of Hepatitis B/C, CMV and parvovirus B19 Reversible ventricular tachycardia |
Hypersensitivity reactions:1 |
| - urticarial or morbilliform skin eruptions | ||
| - fever, chills, headache | ||
| - gastrointestinal upset | ||
| - anaphylactoid reactions | ||
| 90Y Ibritumomab + tiuxetan | Transitory lymphocyte B depletion Myelosuppression2 |
- hypotension |
| - angioedema | ||
| 131I Tositumomab | Hypothyroidism Hematologic toxicities: -thrombocytopenia, neutropenia and anemia Transitory lymphocyte B depletion |
Development of human anti-mouse Ig antibodies |
| Alemtuzumab | Pancytopenia Autoimmune idiopathic thrombocytopenia & hemolytic anemia Infections3 |
|
| Gemtuzumab + Ozogamacin | Myelosuppression Infection |
|
| Tastuzumab | Cardiac dysfunction4 | |
| Cetuximab | Severe infusion reactions5 Flushing, seborrheic dermatitis, & acneiform eruptions |
|
| Panitumumab | Severe infusion reactions5 Pulmonary fibrosis Hypomagnesemia |
|
| Bevacizumab | hypertension, proteinuria, minor bleeding or thrombosis Bowel perforation6 Wound healing complications and bleeding7 |
Caused by mAb containing xenogeneic sequences. These reactions occur during or just after the first injection
Patients must have > 25% bone marrow involvement to be considered eligible for treatment.
Pneumocystis jiroveci pneumonia and herpes virus prophylaxis is recommended for patients being treated with alemtuzumab
When administered in combination with paclitaxel or an anthracycline and cyclophosphamide.
Rare and usually occur within minutes of the initial infusion.
Alone or in combination with chemotherapy.
In patients treated within 60 days from surgery.
III. Molecular mechanisms underlying the therapeutic efficacy of TA-specific mAb-based immunotherapy
The majority of non-conjugated TA-specific mAb approved for clinical use display intrinsic anti-tumor effects mediated by one or more of the mechanisms outlined in Table 3. They can be broadly divided into those that require immune effector cells and those that do not. It should be noted that these mechanisms do not function independently, but extensively interact with each other. The relative importance of each mechanism varies with the type of tumor and the treatment administered. Moreover, it should be stressed that TA-specific mAb have been clearly shown to be able to inhibit their specific receptor and induce apoptosis in the targeted tumor cell without the influence of immune cells in vitro. Nevertheless, it is still debated whether immune effector cells or receptor blockade is the dominant mode of action in vivo or if both pathways need to cooperate to achieve therapeutic effect.
Table 3.
Molecular mechanisms underlying therapeutic efficacy of TA-specific mAb-based therapy.
| Immune effector cell-inpependent1 | Immune effector cell-dependent |
|---|---|
| Induce apoptosis | Activation of complement mediated: |
| Induce alteration in intracellular signaling | – phagocytosis |
| – complement-dependent cytotoxicity | |
| Inhibit growth factor binding to its cognate receptor | Trigger antibody dependent cellular cytotoxicity |
| Inhibit growth factor receptor activation | Induction of tumor cell necrosis or apopotsis leading to |
| – presentation of TA by APC | |
| – activation of CD(+) T cell mediated kill | |
| – activation of B cells and eosinophils | |
| – activation of TA-specific CTL |
Ultimately results in inhibition of cancer cell proliferation, tumor-induced angiogenesis, cancer cell invasion and metastasis, as well as potentating antitumor activity of cytotoxic drugs and radiotherapy
TA-specific mAb can block activation signals that are needed for continued malignant cell growth and/or viability by blocking the interactions between the ligand and its receptor, inducing modulation of the receptor or interfering with ligand binding and/or dimerization of the receptor [reviewed in 3,14]. The latter mechanisms appear to be particularly important for the epidermal growth factor receptor (EGFR)- [16–21], CD20- [22–26] and vascular endothelial growth factor (VEGF)- [27–30] specific mAb. Alternatively, some TA-specific mAb may exert their effects through Fc-based mechanisms such as antibody-dependent cell-mediated cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC) [31,32]. ADCC and CDC are dependent on interactions of antibody Fc domains with cellular Fcγ receptors (FcγR) expressed on immune accessory cells and with the classical complement activating protein C1q, respectively. The potential to mediate ADCC and CDC is a function of an antibody’s subclass and is also influenced by the nature of its glycosylation [33]. Triggering of both ADCC and CDC not only activates natural killer (NK) cells, neutrophils, mononuclear phagocytes and/or dendritic cells (DC), but also induces secretion of IFN-γ, TNF-α, chemokines and opsonins that recruit immune effector cells. As a result tumor cell proliferation and angiogenesis are inhibited, antigen presentation is increased and tumor cells are lysed [31,32,34].
Several of the clinically approved TA-specific mAb, such as rituximab, cetuximab, trastuzumab, alemtuzumab, panitumumab and 131I–tositumomab, have been shown to activate ADCC and CDC in vitro and when administered to mice transplanted with TA-expressing tumor cells. In many cases it is difficult to unravel whether the therapeutic efficacy of TA-specific mAb depends more on ADCC or CDC; however there has been some work in this area. It is noteworthy that the most convincing evidence for ADCC and CDC in the anti-tumor activity of TA-specific mAb comes from hematologic malignancies, i.e., the CD52- and CD20-specific mAb alemtuzumab and rituximab, respectively [3,14]. Whether this finding reflects the accessibility of tumor cells to both TA-specific mAb and plasma proteins of the complement cascade as well as immune effector cells remains to be determined. Nevertheless the role of CDC in the anti-tumor activity of mAb, which recognize antigens expressed by malignant lymphoid cells, is suggested by experimental and clinical findings. First, alemtuzumab mediates significant CDC of chronic lymphocytic leukemia (CLL) cells in vitro [35]. Second, the ability of rituximab to cure immunocompetent mice challenged with murine lymphoma EL4 cells stably transfected with human CD20 is completely abolished in syngeneic knockout mice lacking C1q [36]. Lastly, complement consumption has been observed after administration of the CD20-specific mAb rituximab to patients with lymphoma [32,37]. It is noteworthy that target antigen density appears to be a critical factor for CDC, since Golay et. al [38] have shown that the success of rituximab in mediating CDC against malignantB cells is highly dependent on CD20 density. Whether the lack of convincing evidence for the role of CDC in the anti-tumor activity of TA-specific mAb utilized for solid tumors reflects inadequate TA density remains to be determined.
The role of ADCC in the anti-tumor activity of TA-specific mAb is also suggested by experimental and clinical findings. First, transgenic mice that lack type I and type III FcγR have provided the conclusive evidence that mAb are capable of targeting immune effector cells to cancer cells in vivo [31,39]. In this regard, FcγR (−) mice, unlike wild-type mice, fail to demonstrate protective immunity against tumor challenge using a number of antigen/antibody systems. Furthermore, the removal of the Fc portion from TA-specific mAb reduces TA-specific mAb related side effects as well as their antitumor activity. In contrast, deletion of the inhibitory type II FcγR (FcγRIIb) results in an increased protective effect suggesting that FcγRIIb modulates TA-specific ADCC activity in vivo [31]. The role of interactions with cellular FcγR in the clinical efficacy of TA-specific mAb-based immunotherapy is further supported by the statistically significant correlation between improved clinical RR to mAb-based immunotherapy and particular “high responder” FcγR polymorphisms in patients with (i) CD20(+) follicular cell lymphoma (FL) treated with rituximab; (ii) metastatic colon cancer (CC) treated with cetuximab, or (iii) metastatic breast cancer (BC) treated with trastuzumab [3]. Nevertheless, it should be stressed that the type of FcγR polymorphism does not appear to be associated with improved clinical RR in every patient with a certain disease and in every disease. For example, Fcγ RIIIa-158F polymorphism is an indicator of improved clinical RR in patients with CC and BC treated with cetuximab and trastuzumab, respectively, but is linked to poor RR in patients with hematologic malignancies treated with rituximab [3]. Moreover, FcγRII and RIII polymorphisms are not associated with improved clinical RR in CLL patients treated with alemtuzumab or rituximab, in diffuse large B cell lymphoma (DLCBCL) patients treated with rituximab and in FL NHL patients treated with sequential CHOP and rituximab [3]. Whether these conflicting findings reflect the effect of other variables, such as characteristics of the TA-specific mAb used, induction of TA-specific cytotoxic T lymphocytes (CTL), tumor sensitivity to apoptosis and host immune cell dysfunction, on the role of FcγR polymorphisms in the patients’ clinical response to antibody-based immunotherapy is not known.
At present, the variables underlying the differential clinical RR of the patients treated with antibody-based immunotherapy have not been identified. The data we have summarized suggest that ADCC and CDC play a part in the clinical efficacy of at least some TA-specific mAb. However, other mechanism(s) are likely to underlie the durable clinical responses observed in some patients treated with TA-specific mAbs, since every tumor cell is not completely eradicated during therapy with TA-specific mAbs. Given the number of interactions that are predicted to occur among different components of the immune system, several investigators have begun to examine the ability of TA-specific mAb to induce TA-specific cytotoxic T cell responses in TA-specific mAb treated patients. In this regard, TA-specific mAb are likely to enhance antigen presenting cell (APC) internalization andantigen presentation of TA as well as cross-priming of T cells via endocytosis and phagocytosis of TA–containing immune complexes and antibody-opsonized tumor target cells, respectively [40,41]. Furthermore, the activation of both ADCC as well as CDC can further augment and focus the generation of TA-specific T cell immunity through the production of cytokines, chemokines and opsonins which ultimately lead to i) amplification of ADCC and CDC; ii) recruitment and activation of immune effector cells; iii) maturation of APC; iv) enhancement of antigen presentation, and iv) generation of TA-specific CD4(+) and CD8(+) T cell immunity (Figure 1). Moreover, generation of cleavage products from components of the complement pathway, e.g., C3b, may activate complement receptor 3 (CR3) on the surface of effector cells and induce CR3-dependent cellular cytotoxicity [42].
Figure 1. Triggering of immune effector mechanisms by TA-specific mAb-based immunotherapy.

TA-specific mAb may participate in host dependent immune effector mechanisms including (A) activation of (i) complement mediated phagocytosis and/or (ii) CDC; (B) induction of ADCC and/or (C) induction of tumor cell necrosis or apoptosis. The latter mechanisms result in the release of TA as well as the production of cytokines and opsonins that lead to (I) TA uptake, APC maturation and presentation of TA by APC; (II) generation of CD8(+) TA-specific CTL through CD4(+) T cell help, and (III) activation of CD4(+) T helper cells which leads to activation of NK cells, granulocytes and macrophage through release of Th1 cytokines, CD4(+) T cell-mediated killing, and activation of B cells and eosinophils through release of Th2 cytokines.
The possibility that TA-specific mAb can enhance the immunogenicity of TA and induce TA-specific cellular immunity is supported by the following lines of preclinical and clinical evidence. First, incubation of ovarian cancer cells with the CA125-specific mouse IgG1 mAb oregovomab in vitro can induce CA125- and autologous ovarian TA-specific CTL [43]. Second, cross-presentation mediated by FcγRs on human DC can enhance the presentation of multiple myeloma antigens to patient-derived T cells, thus suggesting that uptake of antibody-opsonized tumor cells andcellular fragments by APCs could lead to antigen/epitope spreading and induction of immunity to several TA [40]. Third, in vitro incubation of cells with trastuzumab results in augmentation of HER2-specific CTL killing of HER2(+) tumors, via trastuzumab’s ability to mediate HER2 internalization and to enhance HLA class I antigen-restricted presentation of endogenous HER2 via proteasomal processing [44]. Fourth, immunization of mice with DC pulsed with antibody-opsonizedTA acquired via either FcγR-mediated endocytosis [40,45,46] or phagocytosis [43] induces CD4 and CD8 T cell-mediated tumor immunity. Lastly, induction of a TA-specific CTL response has been recently documented in patients treated with TA-specific mAb-based immunotherapy [47]. Six of 10 evaluable breast cancer patients showed augmented HER2-specific CD4 T cell responses during therapy with trastuzumab. Furthermore, the number of patients with detectable HER2-specific antibodies among the 27 treated with trastuzumab increased from 8 (29%) before treatment to 15 (56%) during treatment (P<0.001). Similar results have been obtained by one of us (RLF) in studies in progress. Specifically, HLA class I antigen restricted, TA-specific CTL were detected in 4 of 7 HLA-A*0201+ patients with head and neck squamous cell cancer treated with the EGFR-specific mAb cetuximab.
TA-specific mAb are likely to prime antigen-specific CD4(+) and/or CD8(+) T cells by cross-presentation of TA released by dying cells. In this regard, once TA-specific mAb mediate tumor cell lysis by any of the mechanisms outlined above, they can also participate in cross presentation of antigen to professional APC (Figure 2). The term cross-presentation has been used to describe the uptake and representation of cell-associated antigens primarily in the HLA class I antigen pathway of professional APC [48,49]. Among them, B cells and most prominently DC and macrophages, all of which express FcγR, have been reported to present HLA class I antigen-restricted exogenous antigens [48,49]. An alternative, although not exclusive possibility is represented by the induction of anti-idiotypic antibodies, i.e. antibodies specific for the variable region of the administered TA-specific mAb [50,51]. In some cases, these anti-idiotypic antibodies may mimic the TA recognized by the original TA-specific mAb and function as a surrogate antigen [50,51]. The anti-idiotypic antibody as such or complexed with the TA-specific mAb may be taken up by professional APC and induce a TA-specific T cell immune response [50,51].
Figure 2. Induction of TA-specific CTL responses by TA-specific mAb-based immunotherapy.
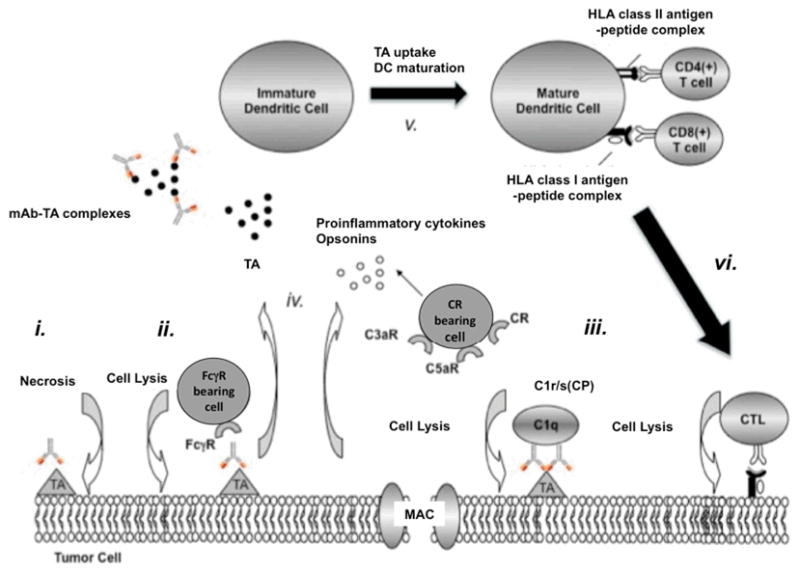
TA-specific mAb may enhance the generation and promote the survival of TA-specific CTL via several mechanisms. TA-specific mAb may (i) induce tumor cell death or activate (ii) ADCC and (iii) CDC. The latter results in 1) the formation of the lytic membrane-attack complex (MAC); 2) the generation of opsonins (C3b), and 3) the release of the anaphylatoxins C3a and C5a. The culmination of the above events (i, ii, iii) leads to the release of Th1 cytokines, (iv) the formation of TA-specific mAb-TA complexes and (v) the uptake of TA and TA-specific mAb-TA complexes by APC. Ultimately, mature DC present processed TA to CD4(+) and CD8(+) T cells, and promote the generation of (vi) TA-specific CTL.
IV. Tumor cell resistance to TA-specific CTL
Despite appropriate TA expression, patients may not have a clinical response to TA-specific mAb-based immunotherapy and/or may develop resistance to therapy during the course of treatment. The multiple mechanisms by which TA-specific mAb may exert their anti-tumor effects, make it difficult to determine which “escape” mechanism(s) is/are most important in patients. Furthermore, the intrinsic genetic instability of tumor cells [52] limits our ability to predict how successful TA-specific mAb-based immunotherapy will be. Nevertheless several mechanisms of tumor cell resistance to TA-specific mAb-based immunotherapy have been suggested (reviewed in 3,14). For the purpose of this manuscript we have focused on tumor cells’ ability to evade immune recognition and destruction by TA-specific CTL.
If T cells do play a role in the clinical efficacy of TA-specific mAb-based immunotherapy, then the escape mechanisms, which have been identified in patients treated with T cell-based immunotherapies, will have relevance. The latter include changes in TA, HLA antigen, and antigen processing machinery (APM) component expression as well as CD4(+)CD25(+) T regulatory (Treg) cells, each of which modulates the interaction between tumor cells and T cells. Specifically, because of their genetic instability [52], tumor cells may change in the expression of molecules such as TA, HLA class I antigens and/or APM components; all of them play a crucial role in the generation of the HLA class I antigen-TA peptide complex, which mediates the recognition of tumor cells by host’s CTL. Approximately 10–30% of tumor cells may fail to express TA and a variable degree of inter- and intra-lesional heterogeneity may also be present in a patient [53]. Furthermore, tumor cells may present TA-derived peptide analogs with antagonist activity resulting in suboptimal T cell activation [54]. These defects have been found to render malignant cells ineffective targets for TA-specific T cells. Moreover, HLA class I antigen downregulation or loss by tumor cells occur with a frequency of about 10–80% depending on the type of malignancy [54]. These abnormalities are caused by distinct molecular mechanisms (reviewed in 55). In some of these cases, HLA class I antigen expression can be restored by cytokines, providing the potential for benefit by combining TA-specific mAb-based immunotherapy with administration of cytokines.
It is noteworthy that the role of HLA class I antigen abnormalities in the clinical course of the disease is highlighted by their increased frequency in recurrent cancers in patients treated with T cell-based immunotherapy [54]. These findings have important implications if the generation of TA-specific CTL underlies, at least in part, the clinical efficacy of TA-specific mAb-based immunotherapy. Specifically, the loss of HLA class I antigens will facilitate the outgrowth of tumor cells that have acquired the ability to escape T cell recognition because of these defects. If this interpretation is correct, the outgrowth of tumor cells with HLA class I antigen abnormalities is likely to reflect escape of tumor cells from immune recognition more than dormancy or ignorance of the patient’s immune system. This possibility is supported by the disease progression frequently observed in patients with malignancy in spite of the development and/or persistence of a TA-specific CTL response [55]. Moreover, given the high rate of mutations in tumor cells, the generation of TA-specific CTL through TA-specific mAb therapy, even when successful, is not likely to be able to completely eradicate a patient’s malignancy, since the eventual outcome is likely to be outgrowth of tumor cells with HLA class I antigen abnormalities.
HLA class I antigen-TA peptide complex loss by tumor cells may not solely be responsible for their immune resistance since in several cases TA-specific CTL coexist with cancer cells expressing the target components required for recognition [56]. Factors such as the type of T cell response induced (i.e. tolerance), lack of CD4+ helper-T-cell activation, host immune cell dysfunction, activating antigen-specific CD4(+)CD25(+) Treg cells, production of inflammatory and lytic molecules by tumor cells and/or high tumor burden may also be responsible for the lack of association between self-TA specific T-cell responses and clinical outcome [56–60].
V. Towards improving immunotherapy
Clearly we are at an early stage in our understanding of the molecular mechanisms by which TA-specific mAb-based immunotherapy mediates effective clinical responses. Although not conclusive, the information we have reviewed has focused attention on the potential role of TA-specific adaptive immunity in patients treated with TA-specific mAb-based immunotherapy. Such responses would be desirable, because they provide a mechanism for long-term protection and immunologic memory. Moreover, the generation of TA-specific adaptive immunity provides a mechanism to explain the improved clinical responses in at least some patients treated with repeated administrations of some TA-specific mAb.
If T cells do play a role in the clinical efficacy of TA-specific mAb-based immunotherapy, one might ask why T cells activated in this setting are effective at controlling tumor cell growth but not when patients are vaccinated with T cell-based activation strategies. Several possibilities can be envisioned to underlie the ability of TA-specific mAb to induce more clinically effective TA-specific T cell immune responses. First, the type of TA targeted by the TA-specific T cell responses generated by TA-specific mAb-based immunotherapy may be very important. In this regard, most of the currently used TA in T cell-based immunotherapy trials represent differentiation and/or shared TA that have been identified from tumor metastases and CTL from patients who, in the majority of cases, have failed to reject their cancer. Moreover, many of these TA are selected on the basis of their immunogenicity and tissue distribution without paying much attention to their function in tumor cell biology. Whether any of the identified TA are tumor-rejection antigens and whether stage IV cancer patients represent the most appropriate source to identify clinically relevant TA is not known at present. Second, TA-specific mAb have the potential to generate adaptive immune responses against the entire TA repertoire of a particular patient’s tumor since they are able to enhance cross-presentation of multiple TA via their direct anti-tumor effects. Third, the adaptive immunity potentially generated by TA-specific mAb provides a means to match therapeutic regimens with changing tumor antigenic profiles which are likely to occur during the course of the disease. Fourth, the potential ability of TA-specific mAb to generate adaptive immunity against multiple TA increases the chances to target also unique TA which have been suggested to be more effective targets of T cell-based immunotherapy than shared TA [61]. Lastly, the polyvalent vaccine-like potential of TA-specific mAb not only eliminates the requirement for patient selection based on HLA type, but may also be able to counteract escape mechanisms caused by selective loss of HLA class I allospecificities and TA. However, it should be noted that one disadvantage of the use of TA-specific mAb is the lack of knowledge about the identity of the TA mediating tumor recognition, that are targeted in adaptive immune responses; the lack of this information hinders the standardization of the assays designed to monitor immune responses in patients treated with TA-specific mAb-based immunotherapy.
The ability of TA-specific mAb to induce more clinically effective TA-specific T cell immune responses may also stem from their ability to activate multiple arms of the host’s immune system including ADCC, CDC, innate immune effector cells, DC, NK, B and T cells. This is in contrast to the narrow view of strictly activating TA-specific CTL that has been attempted in the majority of the T cell-based vaccine strategies to date. In this regard, recent progress into our understanding of the mechanisms underlying activation and proliferation of the adaptive arm of the immune response provides support for the concept that tumor cells may directly and/or indirectly lead to dysfunction and/or death of immune cells in the tumor microenvironment [56]. Therefore, it is unlikely that T cell-based vaccination strategies will be successful in the setting of tumor cell-induced immune suppression. Through their ability to activate ADCC, CDC and APC, TA-specific mAb may promote a Th1 cytokine rich environment thereby enhancing both the number and function of DC, B cells as well as NK cells and ultimately preventing premature death of T cell effectors. It is noteworthy that the potential ability of TA-specific mAb to activate NK cells may prove significant, since there is growing evidence that NK cells are required for the activation of DC as well as the generation of antigen-specific T and B cell response both in vitro and in vivo [62–64].
If it is determined that the induction of such TA-specific T cell immune responses does contribute to the clinical efficacy of TA-specific mAb-based immunotherapy, in vivo induction of ADCC or CDC leading to TA-specific CTL could be viewed as potential biomarkers of clinical response. In this regard, it should be noted that the ability of TA-specific mAb to induce ADCC, CDC and TA-specific T cell immune responses, might have important implications for the development of future TA-specific mAb. As noted above, the potential of a TA-specific mAb to mediate ADCC and CDC is a function of not only its subclass but also its degree and type of glycosylation [33]. Notably, deglycosylated antibodies are unable to bind FcγR and the presence of sialic acid residues confers anti-inflammatory properties to mAb. Therefore, the type and amount of glycosylation of TA-specific mAb is likely to play an important role in balancing the pro- versus anti-inflammatory properties of administered antibodies. It is possible that this vaccine-like property of TA-specific mAb-based immunotherapy could be exploited to selectively amplify or bias the resulting adaptive immune response by promoting antigen presentation, host antibody production and expansion of TA-specific CTL. Nonetheless, it must be kept in mind that if indeed T cells are major players in the clinical efficacy of TA-specific mAb-based immunotherapy, we will continue to face the negative impact on the outcome of immunotherapy of escape mechanisms selected for by immune pressure. As a result, even when successful it is likely that immunotherapy will facilitate the emergence and expansion of tumor cell populations with TA and/or HLA antigen defects and eventually the recurrence of malignant lesions. The latter suggests that immunotherapy for the treatment of cancer may only be successful in a limited number of patients. Even when successful, it is likely that the selective pressure imposed by immunotherapy will facilitate the emergence and expansion of tumor cell populations with TA and/or HLA antigen defects and eventually the recurrence of malignant lesions. Therefore, it will be important to combine immunotherapy with other types of immunological and non-immunological strategies, which utilize distinct mechanisms to control tumor growth. In this regard, the concomitant targeting of other cells in the tumor microenvironment, which are crucial for malignant cell survival and proliferation, may counteract the negative impact of tumor cell genetic instability on immunological and non-immunological therapies.
Acknowledgments
This work was supported by a ASDS Cutting Edge Research Grant (MC) and by PHS grants RO1CA110249(SF), RO1CA113861(SF), RO1CA10494(SF), R01CA105500(SF) and RO3 RFA PA-08-209 (XW) awarded by the National Cancer Institute.
Footnotes
Statement of Translational Relevance
Tumor antigen (TA)-specific monoclonal antibodies (mAb) have been successfully implemented into standard treatment regimens for patients with a variety of malignant diseases. Despite the clinical successes scant information is available regarding the variables underlying patients’ differential clinical response to mAb-based immunotherapy. The scant information in this area has a negative impact on the optimization of the use of TA-specific mAb in therapeutic strategies and represents a major obstacle to the selection of patients to be treated with mAb-based immunotherapy. This paper discusses the potential mechanisms underlying the therapeutic efficacy of mAb-based immunotherapy in patients with malignant disease. This information is expected to contribute to our understanding of the molecular basis of the clinical efficacy of mAb-based immunotherapy and to lead to the development of criteria to select patients with malignant disease to be treated with mAb-based immunotherapy.
VI. References
- 1.Ehrlich P. Collected studies on immunity. New York, NY: J. Wiley & Sons; 1906. [Google Scholar]
- 2.Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256:495–497. doi: 10.1038/256495a0. [DOI] [PubMed] [Google Scholar]
- 3.Campoli M, Ferrone S. Immunotherapy of malignant disease: The coming age of therapeutic monoclonal antibodies. In: DeVita V, Hellman S, Rosenberg S, editors. Cancer: Principles & Practice of Oncology. Vol. 23. Lippincott Williams and Wilkins; New York, NY: 2009. pp. 1–18. [Google Scholar]
- 4.Badger C, Anasetti C, Davis J, Berstein I. Treatment of malignancy with unmodified antibody. Pathol Immunopathol Res. 1987;6:419–434. doi: 10.1159/000157067. [DOI] [PubMed] [Google Scholar]
- 5.Khazaeli MB, Conry RM, LoBuglio AF. Human immune response to monoclonal antibodies. J Immunother. 1994;15:42–52. doi: 10.1097/00002371-199401000-00006. [DOI] [PubMed] [Google Scholar]
- 6.Groner B, Hartmann C, Wels W. Therapeutic antibodies. Curr Mol Med. 2004;4:539–547. doi: 10.2174/1566524043360483. [DOI] [PubMed] [Google Scholar]
- 7.Adams GP, Weiner LM. Monoclonal antibody therapy of cancer. Nat Biotechnol. 2005;23:1147–1157. doi: 10.1038/nbt1137. [DOI] [PubMed] [Google Scholar]
- 8.Martin EW, Jr, Thurston MO. The use of monoclonal antibodies (MAbs) and the development of an intraoperative hand-held probe for cancer detection. Cancer Invest. 1996;14:560–571. doi: 10.3109/07357909609076901. [DOI] [PubMed] [Google Scholar]
- 9.Hoffman EJ, Tornai MP, Janecek M, Patt BE, Iwanczyk JS. Intraoperative probes and 90 imaging probes. Eur J Nucl Med. 1999;26:913–935. doi: 10.1007/s002590050468. [DOI] [PubMed] [Google Scholar]
- 10.Dvorak HF. Vascular permeability factor/vascular endothelial growth factor: a critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J Clin Oncol. 2002;20:4368–4380. doi: 10.1200/JCO.2002.10.088. [DOI] [PubMed] [Google Scholar]
- 11.Kobayashi H, Brechbiel MW. Dendrimer-based nanosized MRI contrast agents. Curr Pharm Biotechnol. 2004;5:539–549. doi: 10.2174/1389201043376571. [DOI] [PubMed] [Google Scholar]
- 12.Mohammed AA, Shergill IS, Vandal M, Gujral SS. ProstaScint and its role in the diagnosis of prostate cancer. Expert Rev Mol Diagn. 2007;7:345–349. doi: 10.1586/14737159.7.4.345. [DOI] [PubMed] [Google Scholar]
- 13.Nie S, Xing Y, Kim GJ, Simons JW. Nanotechnology applications in cancer. Annu Rev Biomed Eng. 2007;9:257–288. doi: 10.1146/annurev.bioeng.9.060906.152025. [DOI] [PubMed] [Google Scholar]
- 14.Reichert JM, Rosensweig CJ, Faden LB, Dewitz MC. Monoclonal antibody successes in the clinic. Nat Biotechnol. 2005;23:1073–1078. doi: 10.1038/nbt0905-1073. [DOI] [PubMed] [Google Scholar]
- 15.Buelow R, Schooten WV. Immunotherapy in 2020. Springer Berlin Heidelberg; Berlin, Germany: 2007. The future of antibody therapy; pp. 83–106. [Google Scholar]
- 16.Sunada H, Magun BE, Mendelsohn J, MacLeod CL. Monoclonal antibody against epidermal growth factor receptor is internalized without stimulating receptor phosphorylation. Proc Natl Acad Sci USA. 1986;83:3825–3829. doi: 10.1073/pnas.83.11.3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang X, Jia XC, Corvalan JR, Wang P, Davis CG, Jakobovits A. Eradication of established tumors by a fully human monoclonal antibody to the epidermal growth factor receptor without concomitant chemotherapy. Cancer Res. 1999;59:1236–1243. [PubMed] [Google Scholar]
- 18.Mendelsohn J, Baselga J. Status of epidermal growth factor receptor antagonists in the biology and treatment of cancer. J Clin Oncol. 2003;21:2787–2799. doi: 10.1200/JCO.2003.01.504. [DOI] [PubMed] [Google Scholar]
- 19.Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ, Kussie P, Ferguson KM. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell. 2005;7:301–311. doi: 10.1016/j.ccr.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 20.Nahta R, Yu D, Hung MC, Hortobagyi GN, Esteva FJ. Mechanisms of disease: understanding resistance to HER2-targeted therapy in human breast cancer. Nat Clin Pract Oncol. 2006;3:269–280. doi: 10.1038/ncponc0509. [DOI] [PubMed] [Google Scholar]
- 21.Hudis CA. Trastuzumab mechanism of action and use in clinical practice. N Engl J Med. 2007;357:39–51. doi: 10.1056/NEJMra043186. [DOI] [PubMed] [Google Scholar]
- 22.Alas S, Bonavida B. Rituximab inactivates signal transducer and activation of transcription 3 (STAT3) activity in B-non-Hodgkin’s lymphoma through inhibition of the interleukin 10 autocrine/paracrine loop and results in down-regulation of Bcl-2 and sensitization to cytotoxic drugs. Cancer Res. 2001;61:5137–5144. [PubMed] [Google Scholar]
- 23.Bezombes C, Grazide S, Garret C, et al. Rituximab antiproliferative effect in B-lymphoma cells is associated with acid-sphingomyelinase activation in raft microdomains. Blood. 2004;104:1166–1173. doi: 10.1182/blood-2004-01-0277. [DOI] [PubMed] [Google Scholar]
- 24.Janas E, Priest R, Wilde JI, White JH, Malhotra R. Rituxan (anti-CD20 antibody)-induced translocation of CD20 into lipid rafts is crucial for calcium influx and apoptosis. Clin Exp Immunol. 2005;139:439–446. doi: 10.1111/j.1365-2249.2005.02720.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bonavida B. Rituximab-induced inhibition of antiapoptotic cell survival pathways: implications in chemo/immunoresistance, rituximab unresponsiveness, prognostic and novel therapeutic interventions. Oncogene. 2007;26:3629–3636. doi: 10.1038/sj.onc.1210365. [DOI] [PubMed] [Google Scholar]
- 26.Ghetie MA, Crank M, Kufert S, Pop I, Vitetta E. Rituximab but not other anti-CD20 antibodies reverses multidrug resistance in 2 B lymphoma cell lines, blocks the activity of P-glycoprotein (P-gp), and induces P-gp to translocate out of lipid rafts. J Immunother. 2006;29:536–544. doi: 10.1097/01.cji.0000211307.05869.6c. [DOI] [PubMed] [Google Scholar]
- 27.Zhu Z, Hattori K, Zhang H, et al. Inhibition of human leukemia in an animal model with human antibodies directed against vascular endothelial growth factor receptor 2. Correlation between antibody affinity and biological activity. Leukemia. 2003;17:604–611. doi: 10.1038/sj.leu.2402831. [DOI] [PubMed] [Google Scholar]
- 28.Lin MI, Sessa WC. Antiangiogenic therapy: creating a unique “window” of opportunity. Cancer Cell. 2004;6:529–531. doi: 10.1016/j.ccr.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 29.Winkler F, Kozin SV, Tong RT, et al. Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: role of oxygenation, angiopoietin-1, and matrix metalloproteinases. Cancer Cell. 2004;6:553–563. doi: 10.1016/j.ccr.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 30.Cao Y. Molecular mechanisms and therapeutic development of angiogenesis inhibitors. Adv Cancer Res. 2008;100:113–131. doi: 10.1016/S0065-230X(08)00004-3. [DOI] [PubMed] [Google Scholar]
- 31.Cassard L, Cohen-Solal J, Camilleri-Broet S, et al. Fc gamma receptors and cancer. Springer Semin Immunopathol. 2006;28:321–328. doi: 10.1007/s00281-006-0058-8. [DOI] [PubMed] [Google Scholar]
- 32.Wang SY, Weiner G. Complement and cellular cytotoxicity in antibody therapy of cancer. Expert Opin Biol Ther. 2008;8:759–768. doi: 10.1517/14712598.8.6.759. [DOI] [PubMed] [Google Scholar]
- 33.Kaneko Y, Nimmerjahn F, Ravetch JV. Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science. 2006;313:670–673. doi: 10.1126/science.1129594. [DOI] [PubMed] [Google Scholar]
- 34.Strome SE, Sausville EA, Mann D. A mechanistic perspective of monoclonal antibodies in cancer therapy beyond target-related effects. Oncologist. 2007;12:1084–1095. doi: 10.1634/theoncologist.12-9-1084. [DOI] [PubMed] [Google Scholar]
- 35.Lopez-Albaitero A, Lee SC, Morgan S, et al. Role of polymorphic Fc gamma receptor IIIa and EGFR expression level in cetuximab mediated, NK cell dependent in vitro cytotoxicity of head and neck squamous cell carcinoma cells. Cancer Immunol Immunother. 2009;58:1855–1864. doi: 10.1007/s00262-009-0697-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zent CS, Secreto CR, LaPlant BR, et al. Direct and complement dependent cytotoxicity in CLL cells from patients with high-risk early-intermediate stage chronic lymphocytic leukemia (CLL) treated with alemtuzumab and rituximab. Leuk Res. 2008;32:1849–1856. doi: 10.1016/j.leukres.2008.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Di Gaetano N, Cittera E, Nota R, Vecchi A, et al. Complement activation determines the therapeutic activity of rituximab in vivo. J Immunol. 2003;171:411581–1587. doi: 10.4049/jimmunol.171.3.1581. [DOI] [PubMed] [Google Scholar]
- 38.Coiffi B. Rituximab therapy in malignant lymphoma. Oncogene. 2007;26:3603–3613. doi: 10.1038/sj.onc.1210376. [DOI] [PubMed] [Google Scholar]
- 39.Golay J, Lazzari M, Facchinetti V, et al. CD20 levels determine the in vitro susceptibility to rituximab and complement of B-cell chronic lymphocytic leukemia: further regulation by CD55 and CD59. Blood. 2001;98:3383–3389. doi: 10.1182/blood.v98.12.3383. [DOI] [PubMed] [Google Scholar]
- 40.Rafiq K, Bergtold A, Clynes R. Immune complex-mediated antigen presentation induces tumor immunity. J Clin Invest. 2002;110:71–79. doi: 10.1172/JCI15640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dhodapkar KM, Krasovsky J, Williamson B, Dhodapkar MV. Antitumor monoclonal antibodies enhance cross-presentation of cellular antigens and the generation of myeloma-specific killer T cells by dendritic cells. J Exp Med. 2002;195:125–133. doi: 10.1084/jem.20011097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kalergis AM, Ravetch JV. Inducing tumor immunity through the selective engagement of activating Fc receptors on dendritic cells. J Exp Med. 2002;195:1653–1659. doi: 10.1084/jem.20020338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gelderman KA, Tomlinson S, Ross GD, Gorter A. Complement function in mAb- mediated cancer immunotherapy. Trends Immunol. 2004;25:158–164. doi: 10.1016/j.it.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 44.Ma J, Samuel J, Kwon GS, Noujaim AA, Madiyalakan R. Induction of anti-idiotypic humoral and cellular immune responses by a murine monoclonal antibody recognizing the ovarian carcinoma antigen CA125 encapsulated in biodegradable microspheres. Cancer Immunol Immunother. 1998;47:13–20. doi: 10.1007/s002620050499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zum Buschenfelde CM, Hermann C, Schmidt B, Peschel C, Bernhard H. Antihuman epidermal growth factor receptor 2 (HER2) monoclonal antibody trastuzumab enhances cytolytic activity of class I-restricted HER2-specific T lymphocytes against HER2-overexpressing tumor cells. Cancer Res. 2002;62:2244–2247. [PubMed] [Google Scholar]
- 46.Kalergis AM, Ravetch JV. Inducing tumor immunity through the selective engagement of activating Fc receptors on dendritic cells. J Exp Med. 2002;195:1653–1659. doi: 10.1084/jem.20020338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Akiyama K, Ebihara S, Yada A, et al. Targeting apoptotic tumor cells to FcR provides efficient and versatile vaccination against tumors by dendritic cells. J Immunol. 2003;170:1641–1648. doi: 10.4049/jimmunol.170.4.1641. [DOI] [PubMed] [Google Scholar]
- 48.Taylor C, Hershman D, Shah N, et al. Augmented HER-2 specific immunity during treatment with trastuzumab and chemotherapy. Clin Cancer Res. 2007;13:5133–5143. doi: 10.1158/1078-0432.CCR-07-0507. [DOI] [PubMed] [Google Scholar]
- 49.Thery C, Amigorena S. The cell biology of antigen presentation in dendritic cells. Curr Opin Immunol. 2001;13:45–51. doi: 10.1016/s0952-7915(00)00180-1. [DOI] [PubMed] [Google Scholar]
- 50.Burgdorf S, Kurts C. Endocytosis mechanisms and the cell biology of antigen presentation. Curr Opin Immunol. 2008;20:89–95. doi: 10.1016/j.coi.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 51.de Cerio AL, Zabalegui N, Rodriguez-Calvillo M, Inoges S, Bendandi M. Anti-idiotype antibodies in cancer treatment. Oncogene. 2007;26:3594–3602. doi: 10.1038/sj.onc.1210371. [DOI] [PubMed] [Google Scholar]
- 52.Pejawar-Gaddy S, Finn OJ. Cancer vaccines: accomplishments and challenges. Crit Rev Oncol Hematol. 2008;67:93–102. doi: 10.1016/j.critrevonc.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 53.Onyango P. Genomics and cancer. Curr Opin Oncol. 2002;14:79–85. doi: 10.1097/00001622-200201000-00014. [DOI] [PubMed] [Google Scholar]
- 54.Neller MA, Lopez JA, Schmidt CW. Antigens for cancer immunotherapy. Semin Immunol. 2008;20:286–295. doi: 10.1016/j.smim.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 55.Chang CC, Campoli M, Ferrone S. Classical and nonclassical HLA class I antigen and NK Cell-activating ligand changes in malignant cells: current challenges and future directions. Adv Cancer Res. 2005;93:189–234. doi: 10.1016/S0065-230X(05)93006-6. [DOI] [PubMed] [Google Scholar]
- 56.Xue SA, Stauss HJ. Enhancing immune responses for cancer therapy. Cell Mol Immunol. 2007;4:173–184. [PubMed] [Google Scholar]
- 57.Whiteside T, Campoli M, Ferrone S. Tumor induced immune suppression mechanisms and possible solutions. In: Nagorsen D, Marincola FM, editors. Analyzing T Cell Responses. How to analyze cellular immune responses against tumor associated antigens. Springer-Verlag; Netherlands: 2005. pp. 43–81. [Google Scholar]
- 58.Nizar S, Copier J, Meyer B, et al. T-regulatory cell modulation: the future of cancer immunotherapy? Br J Cancer. 2009;100:1697–1703. doi: 10.1038/sj.bjc.6605040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ferrone S, editor. Semin Cancer Biol. 2002. Tumour immune escape; p. 12. [DOI] [PubMed] [Google Scholar]
- 60.Rivoltini L, Carrabba M, Huber V, et al. Immunity to cancer: attack and escape in T lymphocyte-tumor cell interaction. Immunol Rev. 2002;188:97–113. doi: 10.1034/j.1600-065x.2002.18809.x. [DOI] [PubMed] [Google Scholar]
- 61.Yu Z, Restifo NP. Cancer vaccines: progress reveals new complexities. J Clin Invest. 2002;110:289–294. doi: 10.1172/JCI16216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Andoniou CE, Coudert JD, Degli-Esposti MA. Killers and beyond: NK-cell-mediated control of immune responses. Eur J Immunol. 2008;38:2938–2942. doi: 10.1002/eji.200838882. [DOI] [PubMed] [Google Scholar]
- 63.Werner H. Tolerance and reactivity of NK cells: Two sides of the same coin? Eur J Immunol. 2008;38:2930–2933. doi: 10.1002/eji.200838755. [DOI] [PubMed] [Google Scholar]
- 64.Zimmer J, Andres E, Hentges F. NK cells and Treg cells: A fascinating dance cheek to cheek. Eur J Immunol. 2008;38:2942–2945. doi: 10.1002/eji.200838813. [DOI] [PubMed] [Google Scholar]