
Figure 1. Phylogenetic tree of bla CTX-M sequences (n = 73) inferred by Bayesian analysis.
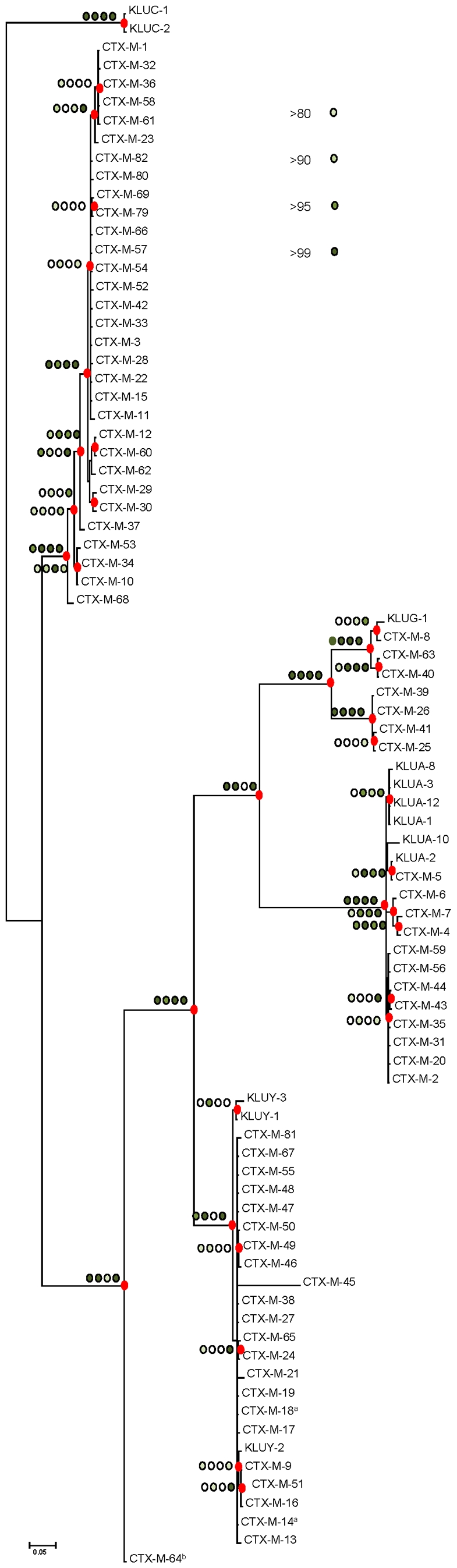
Support for relevant nodes in the tree (indicated by a red dot) was estimated by approximate likelihood ratio, bootstrapping (1000 replicates) with maximum-likelihood reconstruction, bootstrapping (1000 replicates) with neighbor-joining reconstruction, and Bayesian posterior probability, color-coded with increasing density of green with the following equivalences: white <0.80, lightest green ≥0.80, light green ≥0.90, medium green ≥0.95 and dark-green ≥0.99. Kluyvera spp. bla sequences (n = 12) from putative ancestors were included in the analyses and the tree was arbitrarily rooted with Kluyvera cryocrescens. The discrepancy detected between this topology and the one generated by Barlow et al., consisting of the position of the bla CTX-M-11 sequence, might be due to the existence of two different entries in the PubMed corresponding to this bla gene: GenBank accession number AY005110 (this study) and AJ310929 [10], differing in six nucleotides and four amino acid changes, probably sufficient to ascribe these variants to different branches. a bla CTX-M-14 and bla CTX-M-18 genes show identical nucleotide sequences; b bla CTX-M-64 gene corresponds to a hybrid sequence, probably resulting from a homologous recombination event between bla CTX-M-15 and bla CTX-M-14 genes [32].