

Abstract
Glycoprotein B (gB) enables the fusion of viral and cell membranes during entry of herpesviruses. However, gB alone is insufficient for membrane fusion; the gH/gL heterodimer is also required. The crystal structure of the herpes simplex virus type 1 (HSV-1) gB ectodomain, gB730, has demonstrated similarities between gB and other viral fusion proteins, leading to the hypothesis that gB is a fusogen, presumably directly involved in bringing the membranes together by refolding from its initial or prefusion form to its final or postfusion form. The only available crystal structure likely represents the postfusion form of gB; the prefusion form has not yet been determined. Previously, a panel of HSV-1 gB mutants was generated by using random 5-amino-acid-linker insertion mutagenesis. Several mutants were unable to mediate cell-cell fusion despite being expressed on the cell surface. Mapping of the insertion sites onto the crystal structure of gB730 suggested that several insertions might not be accommodated in the postfusion form. Thus, we hypothesized that some insertion mutants were nonfunctional due to being “trapped” in a prefusion form. Here, we generated five insertion mutants as soluble ectodomains and characterized them biochemically. We show that the ectodomains of all five mutants assume conformations similar to that of the wild-type gB730. Four mutants have biochemical properties and overall structures that are indistinguishable from those of the wild-type gB730. We conclude that these mutants undergo only minor local conformational changes to relieve the steric strain resulting from the presence of 5 extra amino acids. Interestingly, one mutant, while able to adopt the overall postfusion structure, displays significant conformational differences in the vicinity of fusion loops, relative to wild-type gB730. Moreover, this mutant has a diminished ability to associate with liposomes, suggesting that the fusion loops in this mutant have decreased functional activity. We propose that these insertions cause a fusion-deficient phenotype not by preventing conversion of gB to a postfusion-like conformation but rather by interfering with other gB functions.
Herpes simplex virus type 1 (HSV-1) is the prototype of the diverse herpesvirus family that includes many notable human pathogens (26). In addition to the icosahedral capsid and the tegument that surround its double-stranded DNA genome, herpesviruses have an envelope—an outer lipid bilayer—bearing a number of surface glycoproteins. During infection, HSV-1 must fuse its envelope with a cellular membrane in order to deliver the capsid into a target host cell. Among its viral glycoproteins, only glycoprotein C (gC), gB, gD, gH, and gL participate in this entry process, and only the last four are required for fusion (28). Although gD is found only in alphaherpesviruses, all herpesviruses encode gB, gH, and gL, which constitute their core fusion machinery. Of these three proteins, gB is the most highly conserved.
We recently determined the crystal structure of a nearly full-length ectodomain of HSV-1 gB, gB730 (18). The crystal structure of the ectodomain of gB from Epstein-Barr virus, another herpesvirus, has also been subsequently determined (4). The two structures showed similarities between gB and other viral fusion proteins, in particular, G from an unrelated vesicular stomatitis virus (VSV) (25), leading to the hypothesis that gB is a fusogen, presumably directly involved in bringing the viral and host cell membranes together to enable their fusion. However, gB alone is known to be insufficient for membrane fusion; the gH/gL heterodimer is also required. This insufficiency raises the question of exactly how gB functions during viral entry. Answering this question is critical for understanding the complex mechanism that herpesviruses use to enter their host cells.
In acting as a viral fusogen, gB must undergo dramatic conformational changes, refolding through a series of conformational intermediates from its initial, or prefusion form, to its final, or postfusion form (15). These conformational changes are not only necessary to bring the two membranes into proximity; they are also thought to provide the energy for the fusion process. The prefusion form corresponds to the protein present on the viral surface prior to initiation of fusion. The postfusion form represents the protein after fusion of the viral and host cell membranes. The available gB structure likely represents its postfusion form, since it shares more in common with the postfusion rather than the prefusion structure of vesicular stomatitis virus (VSV) G (3, 17). However, the prefusion form has not yet been characterized.
Recently, a panel of gB mutants was generated by using random linker-insertion mutagenesis (21). Of these mutants, 16 were particularly interesting because they were nonfunctional in cell-cell fusion assays despite being expressed on the cell surface at levels that indicate proper folding for transport. These observations suggested that each insertion somehow interfered with gB function. Insertions in 12 of these mutants are located within the available structure of the gB ectodomain, which allowed Lin and Spear to analyze their locations (21).
The most prominent examples of such nonfunctional mutants are two mutants with insertions after residues I185 or E187, henceforth referred to as “cavity mutants” because both I185 and E187 point into a cavity inside the gB trimer (Fig. 1B and D). Although this cavity might accommodate a single 5-amino-acid insertion, it “is not large enough to accommodate three 5-amino-acid insertions” (21) that would be present in the trimer (one insertion per protomer).
FIG. 1.

Location of the insertion sites in the sequence of gB and the structure of the postfusion form of its ectodomain. (A) Linear diagram of the full-length gB with functional domains highlighted (as in reference 18). Domain I is shown in cyan, domain II in green, domain III in yellow, domain IV in orange, domain V in red, and the disordered region between domains II and III in purple. Regions absent from the crystal structure of gB730 are shown in gray. Sequences in the region of 5-amino-acid insertions (residues 181 to 190 and residues 661 to 680) are shown in black. Arrows mark the locations of 5-amino-acid insertions, shown as red text. (B) Crystal structure of gB730 (18). Residues preceding the 5-amino-acid insertions in mutants studied here are shown as spheres colored by domain, consistent with panel A. Boxes delineate the hinge region, enlarged in panel C, and the cavity region, enlarged in panel D. (C) Close-up view of the hinge region shown in molecular surface representation, with residues 663 to 675 displayed as sticks. Hydrophobic residues are colored orange. Residues preceding the 5-amino-acid insertions in mutants studied here are labeled with asterisks; remaining labels correspond to additional hydrophobic residues in the 663-675 region. (D) Enlarged view of the cavity region. Residues that line the cavity and are not solvent exposed are colored magenta. Residue E187 of each protomer is colored teal and shown as spheres. Fusion loops for two protomers are marked with asterisks; the third pair of fusion loops lies behind the crystal structure and is not visible. Panels B, C, and D were made by using Pymol (http://www.pymol.org/).
Five other nonfunctional mutants have insertions after residues D663, T665, V667, I671, or L673, respectively. We refer to them as “hinge mutants.” These residues lie in the region located between domains IV and V, which has been termed the hinge region because it may play an important role during the conformational transition from the prefusion to the postfusion form (17). Lin and Spear proposed that insertions following these residues “would likely affect hinge regions” (21), with the implication that they may prevent gB from refolding into the postfusion conformation. Our analysis suggested that insertions after these residues could, perhaps, be sterically accommodated in the structure but would probably be energetically unfavorable by causing several buried hydrophobic side chains in the 665-673 region, such as F670, I671, and L673, to become exposed (Fig. 1B and C).
In light of these observations, we hypothesized that the insertion mutants are “trapped” in a prefusion form. We decided to test this hypothesis by determining whether the ectodomain of gB containing one of these insertion mutations is able to assume the conformation seen in the crystal structure of the wild-type gB ectodomain, which we are referring to as the likely postfusion conformation. For this purpose, we chose one cavity mutant, containing an insertion after E187, and four hinge mutants, containing insertions after T665, V667, I671, or L673, respectively. We chose to test four hinge mutants because structure analysis suggested to us that insertions following the respective residues might not affect the structure in precisely the same way. We expressed the soluble ectodomain of each mutant by using a baculovirus expression system and characterized the purified proteins by using biochemical and biophysical methods. Surprisingly, we found that the ectodomains of all five mutants assume a conformation similar to that of the wild-type gB ectodomain. The four hinge mutants had biochemical properties and overall three-dimensional structures that were indistinguishable from those of the wild-type gB ectodomain. We conclude that these mutants undergo only minor local conformational changes to relieve the steric strain resulting from the presence of 5 extra amino acids. Interestingly, the cavity mutant, while able to adopt the overall postfusion structure, still displayed significant conformational differences relative to wild-type gB. Because these conformational differences are in the vicinity of fusion loops, we conclude that the fusion loops in this mutant have decreased functional activity.
MATERIALS AND METHODS
Construction of gB mutants.
Five mutants were constructed: E187i5, T665i5, V667i5, I671i5, and L673i5. Each mutant is designated using the amino acid residue preceding the insertion; “i5” indicates insertion of five residues. Mutant gB constructs were generated by “splicing by overlap extension” PCR (SOE-PCR) method (16). The E187i5 mutant contains a 5-amino-acid insertion DSLNK following residue E187, the T665i5 mutant contains a 5- amino-acid insertion MFKHT following residue T665, the V667i5 mutant contains a 5-amino-acid insertion MFKHV following residue V667, the I671i5 mutant contains a 5-amino-acid insertion DLLNI following residue I671, and the L673i5 mutant contains a 5-amino-acid insertion NSLNI following residue L673. When introducing insertions, we strived to keep the same amino acid sequence of the inserted residues as in the original mutants obtained by random linker insertion mutagenesis (21). Nevertheless, the I671i5 mutant contains the DLLNI insertion instead of the original DCLNI insertion due to a cloning error. The E187i5 mutant has DSLNK sequence instead of the original DCLNK, and the L673i5 mutant has NSLNI sequence instead of the original NCLNI to avoid introducing an unpaired cysteine. Resulting PCR products were subcloned into the pKH3 plasmid, which encodes gB730 in a pFastBac1 vector. In this plasmid, the endogenous signal sequence, which targets gB to the cell surface, is replaced with the signal sequence from honeybee mellitin for improved secretion in insect cells. The resulting plasmids are pKH28 (E187i5), pKH38 (T665i5), pKH39 (V667i5), pKH26 (I671i5), and pKH40 (L673i5). For fusion assays, E187i5 and I671i5 mutations were introduced into the pPEP98 plasmid, encoding the full-length HSV-1 gB in pCAGGS background. The resulting plasmids are pKH32 (E187i5) and pKH33 (I671i5). The presence of insertions, as well as the absence of mutations elsewhere within the amplified region, was verified by sequencing.
Plasmids.
Plasmids pT7EMCLuc (encoding the firefly luciferase gene) and pCAGT7 (encoding T7 polymerase), as well as pPEP98, pPEP99, pPEP100, and pPEP101 (encoding the genes for HSV-1 gB, gD, gH, and gL, respectively) were gifts of P. G. Spear (23, 24). Plasmid pSC386 encodes the gene for the HSV receptor HVEM (10).
Cells.
Mouse melanoma cells expressing nectin-1 (designated C10) were grown in 10% fetal bovine serum (FBS)-Dulbecco modified Eagle medium (DMEM) containing 500 mg of G418/ml (22). CHO-K1 cells, a gift from P. Spear, were grown in Ham F-12 medium containing 10% FBS. Sf9 cells were grown in Sf-900 II SFM (Invitrogen).
Fusion assay.
The luciferase reporter gene activation assay was used as previously described (9, 23, 24) with some modifications (1). CHO-K1 cells were transfected according to the Lipofectamine-2000 protocol. Cells growing in 24-well plates that were transfected with plasmids encoding HSV glycoproteins and the T7 RNA polymerase (0.2 μg/well of each) were designated as effector cells. Cells growing in six-well plates that were transfected with plasmids encoding the HSV receptor HVEM (pSC386) and luciferase under the control of the T7 promoter (0.8 μg/well of each) were designated as target cells. At 5 h posttransfection, cells were refed with 10% FBS-F12 medium. One hour later, target cells were treated with trypsin and cocultivated with the effector cells growing in the 24-well plates. At 18 to 20 h postcocultivation, cells were washed once with phosphate-buffered saline (PBS) and then lysed in 200 μl of 1× reporter lysis buffer (Luciferase Assay System, Promega) per well. Cell lysates were incubated at −20°C overnight. Finally, 190 μl of lysate from each sample was mixed with 100 μl of luciferase substrate (Promega) and immediately assayed for light output by luminometry.
Surface expression.
Surface expression was assayed by using fluorescence-activated cell sorting (FACS) according to a procedure adapted from Krummenacher et al. (20). Six-well plates were seeded with C10 cells at a density 0.5 × 106 cells/well. Transfection was done by using the GenePorter method (3 μg of DNA plus 15 μl of GenePorter in 1 ml of serum-free DMEM). At 24 h posttransfection, cells were removed with EDTA (Gibco-BRL). A total of 106 cells were transferred into a 5-ml snap-cap tube, and 1 ml of FACS medium (3% fetal calf serum, 0.01% sodium azide in PBS) was added. Cells were next pelleted at low speed and incubated with 50 μl of staining solution (R68 IgG, 10 μg/ml in FACS medium) for 60 min at 4°C. After being washed with FACS medium, cells were incubated with 50 μl of secondary antibody (1:500 goat anti-rabbit phycoerythrin-conjugated polyclonal antibody [PAb]) for 30 min at 4°C. The cells were washed again, resuspended in 500 μl of fix solution (3% paraformaldehyde, 3% FBS in PBS), and subjected to FACS analysis.
Protein expression and purification.
Recombinant baculoviruses were generated by using Bac-to-Bac technology (Invitrogen). Sf9 cells were grown to a density of 2 × 106 cells/ml and infected with the recombinant baculoviruses. The amount of viral stock was optimized to obtain the maximal protein yield. Typically, 5 ml of the viral stock from the third passage (P3) was added per 2 × 109 cells. Supernatant was harvested 72 h postinfection. I671i5, T665i5, V667i5, and L673i5 mutant proteins were purified from culture supernatant by immunoaffinity chromatography using immobilized monoclonal antibody DL16 according to the protocol developed earlier for the purification of the wild-type gB730 (18). After extensive washing with 10 mM Tris-HCl (pH 8.0) and 500 mM NaCl, bound protein was eluted with 3 M KSCN and concentrated by using Millipore Ultra-15 (molecular mass cutoff, 30 kDa). During this concentration step, the buffer was exchanged for 10 mM Tris·HCl (pH 8.0) and 500 mM NaCl. The protein was further purified by size exclusion chromatography in 20 mM Tris-HCl (pH 8.0), 100 mM NaCl, and 1 mM EDTA (Superdex S200; GE Healthcare) and concentrated by using Millipore Ultra-4 (molecular mass cutoff, 50 kDa) to 3 to 4 mg/ml. E187i5 was purified in a similar way, except that the monoclonal antibody C226 was used for immunoaffinity purification.
Size exclusion chromatography.
Size exclusion chromatography was performed on a Biologic Duoflow chromatography system (Bio-Rad) equipped with a Superdex S200 column (GE Healthcare) equilibrated with 20 mM Tris-HCl (pH 8.0), 100 mM NaCl, and 1 mM EDTA. The Superdex S200 column was calibrated by using thyroglobulin (670 kDa), ferritin (440 kDa), catalase (232 kDa), aldolase (158 kDa), and ovalbumin (44 kDa); blue dextran was used to determine the void volume (GE Healthcare).
Antibodies.
Hybridoma cell line expressing anti-HSV-1-gB mouse monoclonal antibody DL16 was a gift from R. J. Eisenberg and G. H. Cohen (University of Pennsylvania). The corresponding IgG was purified from supernatants of hybridoma cells using protein A resin at the GRASP facility at Tufts Medical Center. IgGs for the mouse monoclonal antibodies C226, SS10, SS144, and SS55 were also a gift from R. J. Eisenberg and G. H. Cohen. Monoclonal antibody (MAb) DL16 is a non-neutralizing antibody that recognizes a discontinuous, trimer-dependent epitope (5). C226 recognizes an epitope located in domain II (5). SS10 recognizes a pseudocontinuous epitope encompassing residues 640 to 670, within domain IV (5). SS55 recognizes a discontinuous epitope within domain I (5). SS144 recognizes a pseudocontinuous epitope within domain V (5). MAbs C226, SS10, SS144, and SS55 are virus neutralizers (5).
Western blots and immunoprecipitation.
For Western blot analysis of the mutant and the wild-type ectodomains, unless noted otherwise, all samples were prepared under mildly denaturing conditions (nonreducing, unboiled samples containing 0.1% sodium dodecyl sulfate [SDS]) to preserve the trimeric species of gB730 (8). For Western blot analysis with MAbs C226, SS55, or SS144, 150 ng of protein was used per lane. For Western blot analysis with MAb DL16, 250 ng of protein was used per lane, with the exception of gB730, for which 300 ng of protein was used. For Western blot analysis with MAb SS10, 100 ng of protein was used per lane, and samples were partially denatured by boiling in nonreducing buffer containing 1% SDS. Samples were then resolved by 4 to 15% Tris SDS-PAGE (Bio-Rad) and transferred to a nitrocellulose membrane. Membranes were blocked with 5% skimmed milk in Tris-buffered saline with 0.05% Tween and then probed with MAbs C226, SS10, SS55, or SS144 at a 1:5,000 dilution or with MAb DL16 at a 1:1,000 dilution. This was followed by incubation with anti-mouse IgG-horseradish peroxidase conjugate at a 1:10,000 dilution. The blots were developed by using a Pierce Western Pico chemiluminescence kit. Wild-type ectodomain, gB730, was used as a positive control.
For Western blot analysis of the mutant and wild-type full-length proteins, 20 μl of each lysate (1/10 of one well of a six-well plate) was loaded onto a 10% SDS-polyacrylamide gel and separated under mildly denaturing conditions (8). After transfer to nitrocellulose, proteins were probed with MAb DL16 or PAb R68 (1 μg/ml).
For immunoprecipitation, transfected C10 cells expressing mutant or wild-type full-length gB proteins were lysed in lysis buffer (10 mM Tris [pH 8.0], 150 mM NaCl, 10 mM EDTA, 1% NP-40, 0.5% sodium deoxycholate). Next, 25 μl of each lysate (1/8 of one well of a six-well plate) was diluted in 70 μl of immunoprecipitation (IP) binding buffer (10 mM Tris [pH 8.0], 100 mM NaCl, 0.01% NP-40, 0.05% bovine serum albumin), followed by incubation with 5 μg of either anti-gB MAb or anti-c-myc (negative control) overnight at 4°C. The proteins were precipitated with protein A-agarose beads (Invitrogen) for 2 h at 4°C, and the beads were washed with IP binding buffer, followed by a high-salt buffer wash (10 mM Tris [pH 8.0], 0.5 M NaCl, 0.01% NP-40, 0.05% bovine serum albumin) and a final wash in IP binding buffer. Samples were then subjected to electrophoresis on a 10% SDS-polyacrylamide gel under reducing, denaturing conditions, and the mutant or the wild-type gB proteins were detected by Western blotting with the anti-gB PAb R68. Immunoprecipitation of purified, truncated gB proteins was performed as described above with the exception that proteins were diluted in IP binding buffer for the antibody incubation step.
Electron microscopy.
Protein was incubated on electron microscopy (EM) grids and stained with 0.75% solution of uranyl formate. EM images were obtained at room temperature by using a Tecnai G2 Spirit BioTWIN equipped with an AMT 2k charge-coupled device camera microscope at the Harvard Medical School EM core facility.
Limited proteolysis.
Mutants E187i5, T665i5, V667i5, I671i5, and L673i5 or the wild-type gB730 (3 μg) in 8 μl of 20 mM Tris-HCl (pH 8.0), 100 mM NaCl, and 1 mM EDTA were incubated with 30, 60, 120, or 300 ng of TLCK (Nα-p-tosyl-l-lysine chloromethyl ketone)-treated chymotrypsin (Worthington) at room temperature for 1 h. This corresponds to substrate/protease ratios of 100:1, 50:1, 25:1, and 10:1, respectively. The digestion was stopped by adding denaturing, reducing SDS-PAGE sample buffer and heating the samples for 5 min at 95°C. The results of the proteolysis were analyzed on 4 to 12% NuPAGE gels (Invitrogen) under denaturing and reducing conditions. For N-terminal sequencing of the protein bands, the digests were separated on 4 to 12% NuPAGE gels and then transferred to a polyvinylidene difluoride membrane. The membranes were stained with Coomassie R-250. Bands of interest were cut out and submitted for sequencing by Edman degradation at the Tufts University core facility.
Liposome flotation assay.
Conditions for liposome flotation experiments were adapted from previous methods (11, 19) and modified as described previously (13). Liposomes were purchased from Encapsula Nanosciences (Nashville, TN) at a size of 400 nm, containing a 1.7:1 molar ratio of soy phosphatidylcholine to cholesterol. Liposomes were stored at 4°C and used for up to 1 month according to the manufacturer's instructions. Purified soluble proteins (1 μg), liposomes (25 μg), and 15 μl of 200 mM sodium citrate were combined, and the final reaction volume was adjusted to 50 μl with PBS. Protein-liposome mixtures were then incubated at 37°C for 1 h. To eliminate unwanted electrostatic protein-lipid associations, mixtures were incubated with 1 M KCl for 15 min at 37°C (11) before being loaded onto a sucrose gradient. Mixtures were adjusted to 40% sucrose in a final volume of 500 μl and overlaid with 4 ml of 25% sucrose-PBS and 500 ml of 5% sucrose-PBS. The gradients were centrifuged for 3 h in a Beckman SW55Ti rotor at 246,000 × g and 4°C. Seven equal fractions (∼700 μl each) were collected starting from the top of the gradient. For dot blots, 200 μl of each fraction was spotted onto nitrocellulose filters using a vacuum manifold (Schleicher & Schuell) and then probed using the anti-gB rabbit PAb R68. Blots were then incubated with horseradish peroxidase-conjugated goat anti-rabbit antibodies and visualized by using enhanced chemiluminescence (Pierce).
RESULTS
Choice of mutated residues.
We recreated five insertion mutants, including one cavity (E187i5) and four hinge (T665i5, V667i5, I671i5, and L673i5) mutants, as soluble proteins ending at amino acid 730 (Fig. 1). An insertion following residue E187 was ostensibly the most likely to destabilize the postfusion conformation. Residue E187 has a side chain pointing into the cavity located inside the gB trimer in domain I (Fig. 1). Although this cavity may be spacious enough to accommodate a single 5-amino-acid insertion, it is unquestionably too small to accommodate the three 5-amino-acid insertions that would occur in the trimer, one per protomer. We also focused on the hinge region, residues 665 to 673 (Fig. 1), because 5-amino-acid insertions after residues T665, V667, I671, or L673 also cause the fusion-deficient phenotype despite adequate surface expression.
Cell fusion activity.
The insertions in three of our mutants (E187i5, I671i5, and L673i5) differ by one amino acid from the insertion mutants reported previously (21). To ensure that our mutants and those of Lin and Spear had the same phenotypes, we introduced our insertions into the full-length gB and tested the fusion activity of two mutant gB proteins (E187i5 and I671i5) by using a luciferase reporter assay system (9, 23, 24). Effector cells were prepared by cotransfection of CHO cells with plasmids containing the genes for T7 polymerase, gD, gH, gL, and either wild-type gB or one of the mutants. Target CHO cells were transfected with plasmids encoding the luciferase protein under the control of the T7 promoter and the gD receptor HVEM. Target and effector cells were mixed, incubated for 20 h, lysed, and then assayed for light production (i.e., fusion). Neither E187i5 (0% wild-type levels of fusion) nor I671i5 (7%) were able to substitute for wild-type gB in the cell-cell fusion assay (Fig. 2A). Thus, the fusion assay demonstrated that our E187i5 and I671i5 mutants had phenotypes similar to those previously studied (21). The mutant gB proteins were expressed on the cell surface as well as the wild-type gB protein, as determined by FACS (Fig. 2B).
FIG. 2.
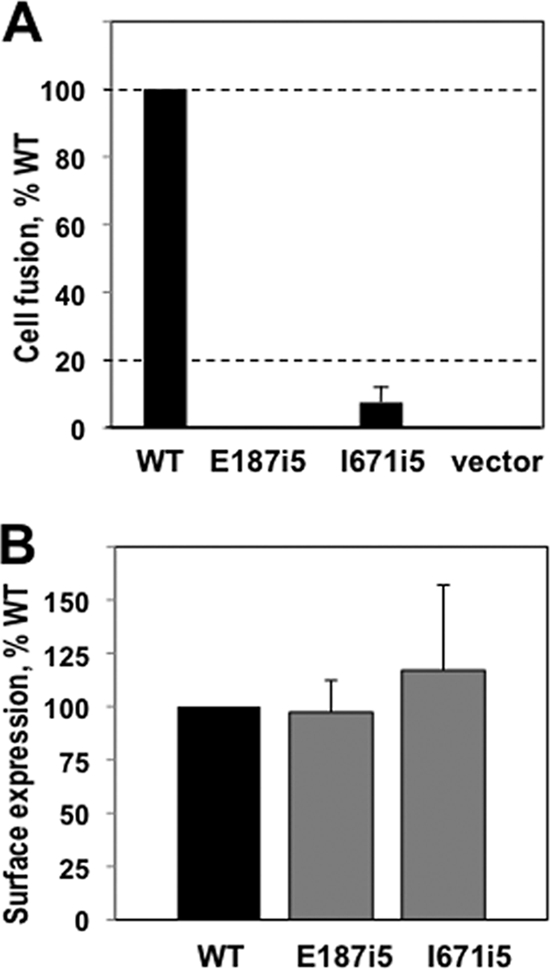
Characterization of the fusion phenotype of the insertion mutants. (A) Quantitative cell-cell fusion assay. Target CHO-K1 cells, expressing the luciferase protein and the HSV receptor HVEM, were cocultivated with effector cells expressing T7 polymerase, gD, gH, gL, and either wild-type (WT) gB, mutant, or the empty vector, and assayed for light production 20 h later. Extent of fusion is expressed as percentage WT. (B) FACS assay to determine surface expression of full-length WT and mutant gB proteins. Mouse melanoma C10 cells were transfected with either WT gB, or mutant, or the empty vector. Cells were stained with a primary antibody R68, a secondary antibody goat anti-rabbit-phycoerythrin, fixed with paraformaldehyde, and subjected to FACS analysis. In the above graph, the signal from C10 cells that were transfected with empty vector DNA was subtracted out. Error bars represent the standard error. The mutant gB proteins I671i5 and E187i5 were expressed on the cell surface, as well as the wild-type gB protein.
MAb reactivity.
To reveal the mechanism underlying the fusion-deficient phenotype of the insertion mutants, we wanted to characterize the biochemical properties of the mutants in detail. To do so, we expressed the mutants as soluble ectodomains, residues 31 to 730, in insect cells using recombinant baculovirus technology. We first compared the MAb reactivity of the purified mutants and the wild-type gB730 (Fig. 3A) by using Western blot analysis under mildly denaturing conditions. We used the well-characterized MAbs DL16, C226, SS10, SS55, and SS144, all of which recognize epitopes in different structural regions within the gB ectodomain (5), as described in more detail in Materials and Methods. Thus, these antibodies are particularly well suited for probing the conformation of different regions in mutant gB proteins.
FIG. 3.

gB insertion mutant proteins are similarly recognized by different antibodies, with the exception of E187i5. (A) Purified protein samples were resolved on mildly denaturing 4 to 15% Tris gradient gels and visualized by Western blot analysis using different anti-gB MAbs, as indicated under each blot. Wild-type gB ectodomain, gB730, was used as a positive control in all blots. For each blot and gel in this figure, molecular mass markers are indicated on the left in kilodaltons. “T” stands for trimeric and “M” stands for monomeric gB species. (B) WT gB and the E187i5 and I671i5 mutants were expressed as full-length proteins in C10 cells and immunoprecipitated (IP) with different anti-gB MAbs. IP samples were then resolved on 10% polyacrylamide gels and visualized by Western blot analysis with a polyclonal anti-gB antibody, R68. IP results for anti-myc antibody are shown as a negative control. (C) Recognition of full-length gB versus soluble gB ectodomains by MAb DL16 was tested by IP. IP samples of full-length proteins were prepared as in panel B. Purified soluble ectodomains were diluted in IP binding buffer and immunoprecipitated with DL16. The results of the IP were analyzed by Western blot analysis with R68, as in panel B. IP results for anti-myc antibody are shown as a negative control. (D) Recognition of full-length gB by MAb DL16 was tested by Western blotting. WT gB and the E187i5 and I671i5 mutants were expressed as full-length proteins in C10 cells, and whole lysates were resolved on a 10% polyacrylamide gel under mildly denaturing conditions, blotted, and probed with MAb DL16. Equivalent expression of full-length gB proteins in C10 cells was verified by Western blot analysis with PAb R68 using SDS-10% PAGE under reducing, denaturing conditions. (E) Approximate locations of the epitopes of four of the MAbs used in the present study in the crystal structure of gB730. The location of the epitope for the trimer-specific antibody DL16 is unknown.
We first tested DL16 reactivity. DL16 recognizes a conformational epitope on the gB730 trimer; it does not react with monomeric gB730, be it fully or partially denatured (5). We found that all of the hinge mutants—T665i5, V667i5, I671i5, and L673i5—were recognized by DL16 in Western blots, which suggested that they were trimeric (Fig. 3A). These four mutants were also recognized by antibodies C226, SS10, SS55, and SS144. Thus, the 5-amino-acid insertions within the hinge region did not noticeably influence the antigenic characteristics of the gB mutants expressed as soluble ectodomains, as judged by the results of Western blot analysis with antibodies C226, SS10, SS55, and SS144. In contrast, mutant E187i5 was not recognized by DL16 in Western blots. The lack of DL16 reactivity was not due to the absence of the trimer because MAbs C226, SS10, SS55, and SS144, which recognize both the trimeric and the monomeric forms of gB by SDS-PAGE, reacted with the trimeric form of the E187i5 mutant (Fig. 3A). The fact that DL16 does not recognize the E187i5 mutant indicated, therefore, that the DL16 epitope was disrupted as a consequence of the 5-amino-acid insertion.
We next tested whether the insertions had the same effect on the antibody reactivity of the full-length mutants. Using IP, we tested the reactivity of our antibodies against the two full-length mutants I671i5 and E187i5 (Fig. 3B). We found that all antibodies, including C226, SS10, SS55, SS144, and DL16, recognized both mutants and the wild-type gB (Fig. 3B). The fact that DL16 was able to immunoprecipitate the E187i5 full-length protein was surprising. Therefore, we set out to determine whether the difference in reactivity of DL16 antibody observed for the E187i5 mutant was indeed due to the different antigenic properties of the full-length and truncated mutant protein or due to different assays, i.e., Western blotting with the truncated protein versus IP with the full-length protein. To do so, we tested the reactivity of DL16 antibody against the truncated and the full-length gB mutant proteins by both Western blotting and IP (Fig. 3C and D). Our results showed that the full-length and the truncated E187i5 mutants had the same reactivity with DL16 antibody, but the two assays yielded different results. DL16 did not recognize either the full-length or the truncated E187i5 mutant by Western blotting but was able to precipitate them in the IP assay. The cause of the discrepancy between the two assays probably lies in the conditions used in each assay. It is possible that the DL16 epitope in the E187i5 mutant does not disappear completely and is, instead, rendered unstable such that it is not recognized by the DL16 antibody under somewhat more stringent conditions of a Western blot assay. Nevertheless, the inability of the DL16 antibody to recognize either truncated or full-length E187i5 mutant by Western blotting suggests that the DL16 epitope in this mutant was affected by the 5-amino-acid insertion. Further, the full-length proteins react with antibodies in the same way as the soluble, truncated proteins do, which justifies our use of soluble ectodomains in place of the full-length proteins.
Oligomeric state.
The oligomeric state of the mutant ectodomains was assessed by size exclusion chromatography (SEC). SEC analysis of purified mutant proteins revealed that the T665i5, V667i5, I671i5, and L673i5 proteins had the same elution profiles as the wild-type gB730, suggesting that they all form trimers (Fig. 4). A small fraction of these mutant proteins eluted in the void, which is occasionally observed with the wild-type gB730. In contrast, all of E187i5 eluted very close to the void volume on the Superdex 200 column (Fig. 4). This behavior suggested that E187i5 forms aggregates with a size of >670 kDa, possibly, dimers of trimers or even larger species. Therefore, the presence of the 5-amino-acid insertion following residue E187 appeared to have changed the protein conformation to make the E187i5 mutant much more prone to aggregation. We studied these conformational changes further in the experiments described below.
FIG. 4.

E187i5 mutant protein elutes completely in the void volume of the size exclusion column. An overlay of the size exclusion chromatograms for the mutant and the wild-type ectodomain proteins is shown. Elution volumes of size exclusion standards are indicated with arrows above the traces.
Electron microscopy.
To further assess the conformation of the mutant proteins, we used electron microscopy. The gB730 ectodomain has a characteristic shape that can be easily discerned in the EM images of negative-stained protein particles. In particular, the two ends of the rod-like molecule can be readily distinguished (Fig. 5A). We refer to these ends as the “crown” and the “base,” respectively (Fig. 5A and B). In the structure of gB730, the “crown” corresponds to domain IV, whereas the “base,” at the opposite end, contains domain I (18). We measured the length of gB730 particles in EM images to be 16.0 ± 1.2 nm long (n = 5). This number correlates well with the length of the gB ectodomain calculated from its atomic coordinates, 16.7 nm (18).
FIG. 5.

Negative-stain EM images of wild-type gB730, T665i5 mutant, and E187i5 mutant proteins. (A) Representative particles of WT gB730 and an insertion mutant, T665i5, are shown. Two distinct “crown” and “base” ends of particles are labeled with red arrows in one representative particle. The scale bar for these images is the same as for panel C. (B) The structure of gB730 in surface representation is colored by domain as in Fig. 1A. The crown and base regions are labeled. The approximate location of residue E187 is marked with a white star. (C) Representative particles of two types are shown for the E187i5 mutant protein. Crown regions of individual trimers are labeled with red arrows. (D) Model of how higher-order oligomers are formed from gB730 trimers in the E187i5 mutant, as depicted in panel C. Dimers of trimers and trimers of trimers are shown. The approximate locations of residue E187 in all structures are marked with a white star, as in panel B.
We obtained images of negative-stained samples of all five mutants. Not surprisingly, given their reactivity with DL16 MAb, T665i5, V667i5, I671i5, and L673i5 look the same as the wild-type gB730. Images of a representative mutant, T665i5, are shown in Fig. 5A. The overall shape of the T665i5 particles is very similar to that of the wild-type gB730 particles. In particular, the “crown” and the “base” ends can be seen. Particles of T665i5 mutant protein were measured to be 16.2 ± 0.2 nm long (n = 5). Particles of V667i5, I671i5, and L673i5 were of similar size (data not shown). Therefore, the 5-amino-acid insertions after residues T665, V667, I671, or L673 do not affect the structure of the soluble ectodomain of gB globally. We conclude that the T665i5, V667i5, I671i5, and L673i5 mutant ectodomains adopt the overall wild-type postfusion form. In contrast, E187i5 preferentially formed two kinds of particles: three-armed particles and long rods (Fig. 5C). The length of each arm in the three-armed particle was 16.3 ± 0.5 nm (n = 9), indicating that each arm consists of a gB trimer. We believe that these are trimers of trimers, joined in a “base-to-base” way because crowns can be clearly seen at the tips of several arms (Fig. 5C). We measured the length of the long rods to be 33.0 ± 0.6 nm (n = 5), which is about twice the length of a single trimer. This suggests that each long rod is composed of a dimer of gB trimers. These dimers of trimers also appear to be joined “base-to-base” because crowns can be seen at the opposite ends of the long rods (Fig. 5C and D). So, in the E187i5 samples, two observed types of particles consist of either two or three trimers associating by their “base” ends.
Given that wild-type gB730 does not normally associate into higher-order oligomers, the presence of higher-order oligomers of E187i5 suggests that the insertion of 5 amino acid residues after residue E187 alters the protein conformation to allow or, perhaps, force the mutant protein to associate into dimers of trimers and trimers of trimers. These conformational changes have no apparent effect on the gross architecture of individual trimers because the length of the “arms” is similar to the length of the postfusion trimer of gB730. Moreover, the characteristic features of the postfusion trimers, e.g., the “crown” end, are preserved in the E187i5 mutant molecules. We conclude that whereas the E187i5 mutant adopts the overall postfusion form, it has significant conformational changes at the “base” end of the trimer, which contains domain I.
Limited proteolysis.
To better understand the nature of the conformational changes that caused the E187i5 mutant molecules to associate into dimers of trimers and trimers of trimers, we used limited proteolysis with chymotrypsin to compare the conformation of the mutants to that of the wild-type gB730 protein. The pattern of proteolytic fragments of wild-type gB730, as seen on reducing SDS-PAGE, has been previously reported (5, 18). Briefly, chymotrypsin cleaves gB730 between residues 97 and 98; residues 472 and 473; and, to a lesser extent, residues 489 and 490. These cleavage events generate fragment 98-472 (53 kDa), fragment 473-730 (38 kDa), and a small amount of fragment 490-730 (36 kDa) (Fig. 6). The intact N-terminal fragment 28-97 is never observed on gels, probably because this highly unstructured region is further cleaved into very small pieces that are undetectable (18).
FIG. 6.

Limited proteolysis reveals a new chymotrypsin site in the E187i5 mutant. Wild-type gB730 and five insertion mutants were digested with chymotrypsin at decreasing mass ratios of substrate to protease, indicated above each lane. ND, nondigested. For each gel in this figure, molecular mass markers are indicated on the left in kilodaltons. (A) Digestion pattern of wild-type gB730, the I671i5 mutant, and the E187i5 mutant. (B) Digestion pattern of the T665i5 mutant. (C) Digestion pattern of the V667i5 and L673i5 mutants. Asterisks indicate proteolytic products that are seen in the wild-type gB730 and all of the mutants. Arrows in the E187i5 digestion pattern indicate new bands absent from the digests of the wild-type gB730 and the rest of the mutants. Weak bands corresponding to chymotrypsin autoproteolysis products are observed at 15 and 10 kDa in some of the digests at 25:1 and 10:1 substrate/protease ratios.
Figure 6 shows representative proteolytic patterns generated by limited digestion of gB730, T665i5, V667i5, I671i5, L673i5, and E187i5 with chymotrypsin. T665i5, V667i5, I671i5, and L673i5 were digested into fragments similar to those seen for the wild-type gB730 (Fig. 6). Surprisingly, treatment of E187i5 with chymotrypsin resulted in two additional bands migrating at ∼35 and ∼18 kDa (Fig. 6A). Although the electrophoretic mobility of the 35-kDa fragment observed in E187i5 resembled that of the 36-kDa fragment, observed in wild-type gB730 and containing residues 490 to 730, this fragment represents a different part of the polypeptide chain, as determined by N-terminal sequencing. Specifically, its N-terminal residue is 224, indicating that the 35-kDa fragment most likely corresponds to residues 224 to 472. The 18-kDa proteolytic product, also a new band, was found by N-terminal sequencing to begin with residue 98. The appearance of the 35- and the 18-kDa bands in the digest coincided with the disappearance of the 53-kDa 98-472 fragment (Fig. 6A), supporting our hypothesis that the 35-kDa fragment was composed of residues 224 to 472 and the 18-kDa fragment was composed of residues 98 to 223.
Residues F223 and H224 are located in domain I at the base of the short β-hairpin (Fig. 7). Inspection of the crystal structure clearly shows that residues 223 to 224 are not solvent exposed (Fig. 7) and, therefore, are not accessible to protease. Not surprisingly, neither trypsin nor chymotrypsin cleaves wild-type gB ectodomain, gB730, in the vicinity of these residues (5, 18). However, in the E187i5 mutant protein, chymotrypsin can clearly access these residues and cleave the peptide bond between them. We conclude that the 5-amino-acid insertion after residue E187 causes a conformational change that exposes residues 223 to 224 and makes them accessible to protease.
FIG. 7.

Newly identified proteolytic cleavage site in E187i5 mutant is not exposed in the wild-type gB730. (A) Crystal structure of gB730 is shown as a beige ribbon with residues E187 in all three protomers shown as orange spheres. Residues F223 and H224, the site of chymotrypsin cleavage, are shown as pink sticks. The box delineates the region enlarged in panel B. (B) Close-up view of the region boxed in panel A, rotated 90° clockwise relative to the view in panel A and shown in molecular surface representation. Residues E187 for two protomers are shown as orange spheres and labeled with arrows; the third E187 residue is situated behind the molecule and is not visible. Residues F223 and H224 for one protomer are shown as magenta sticks. Only the tip of the F223 side chain is exposed, shown as magenta surface. The figure was prepared by using Pymol (http://www.pymol.org/).
gB association with liposomes.
It has been recently shown that soluble gB730 associates with liposomes through the fusion loops in domain I (13). To test whether the E187i5 mutant, which contains an insertion in the vicinity of the fusion loops, could be impaired in liposome binding, we incubated purified, soluble wild-type or mutant gB proteins with liposomes at 37°C for 1 h. We then added 1 M KCl to eliminate any nonspecific electrostatic interactions between the proteins and liposomes (11). Next, each protein-liposome mixture was adjusted to 40% sucrose, layered beneath a sucrose step gradient, and centrifuged for 3 h to allow the liposomes and any associated proteins to float to the top of the gradient. Under these conditions, proteins that fail to bind liposomes remain at the bottom of the gradient. Seven equal fractions were collected starting from the top of each gradient and analyzed by immuno-dot blot using anti-gB rabbit polyclonal antibody R69 (Fig. 8). E187i5 was drastically impaired in its ability to bind liposomes, whereas the four hinge mutants (T665i5, V667i5, L673i5, and I671i5) cofloated with liposomes similar to wild-type gB730. Thus, it appears that insertions in E187i5 affected the conformation of the fusion loops rendering them suboptimal for proper insertion into the lipid membranes. Alternatively, the smaller amount of E187i5 associated with liposomes could be due to its aggregation, that is, the formation of dimers of trimers and trimers of trimers, as seen by EM. In these aggregates, the fusion loops are not exposed and thus may be unavailable for lipid interaction.
FIG. 8.
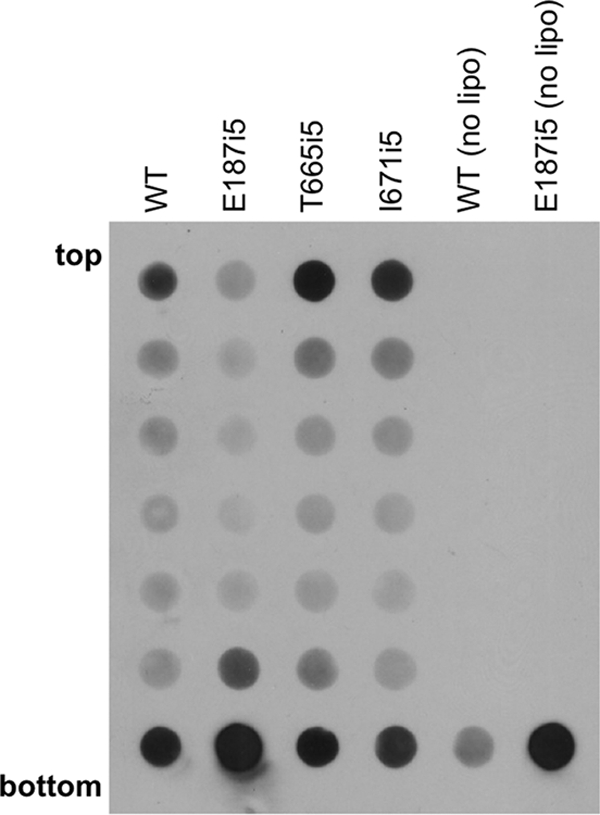
Binding of insertion mutants to liposomes. Purified soluble wild-type (WT) or mutant proteins were incubated with phosphatidylcholine/cholesterol liposomes at 37°C for 1 h. Each sample was then incubated in 1 M KCl for 15 min, adjusted to 40% sucrose, and layered beneath a sucrose step gradient. Gradients were centrifuged for 3 h, fractionated, and analyzed by immuno-dot blot using anti-gB rabbit polyclonal antibody R69. The E187i5 mutant has impaired liposome binding, whereas the two hinge mutants (T665i5 and I671i5) cofloat with liposomes as efficiently as wild-type gB730.
DISCUSSION
Previous work established that 5-amino-acid insertions in HSV-1 gB after residues E187, T665, V667, I671, or L673 result in mutant gB proteins that can be expressed on the cell surface but are unable to mediate cell-cell fusion when coexpressed with gD and gH/gL (21). These mutants were also unable to complement a gB-deficient virus, the assay that serves as a proxy for viral cell entry (21). The results of the study suggested to us that the mutants were fusion deficient due to their inability to adopt the postfusion conformation. Here, we found that despite the presence of the 5-amino-acid insertions, the cavity mutant E187i5 and the hinge mutants T665i5, V667i5, I671i5, and L673i5 were clearly able to adopt the postfusion structure, at least when expressed as soluble ectodomains in the gB730 background. These results were unexpected because inspection of the structure of gB730 along with previous analysis (21) suggested to us that 5-amino-acid insertions at position E187 and also at positions T665, V667, I671, and L673 could not be sterically accommodated in the postfusion structure of wild-type gB730.
Cavity mutant E187i5.
A priori, we hypothesized that the presence of three 5-amino-acid insertions within the threefold central cavity would cause severe steric strain; simply put, these extra 15 residues, 5 per protomer, cannot fit into the small cavity (21). Therefore, we expected that this steric strain would prevent gB from folding into the postfusion conformation. Instead, we found that the E187i5 mutant adopts the overall postfusion conformation but that there are significant localized conformational changes to accommodate the insertions. We hypothesize that the significant steric strain within the cavity, created by the presence of the extra residues in the E187i5 mutant, is relieved by domains I being pushed away from one another. This hypothesis is supported by the destabilization of a MAb epitope, the appearance of a new proteolytic site, and the exposure of buried hydrophobic residues.
First, the separation of domains I weakens the DL16 epitope, as evidenced by a greatly diminished DL16 reactivity in the E187i5 mutant. Although the exact location of the DL16 epitope is unknown, judging by MAb competition data, it is located within domain I (5). Importantly, DL16 only binds trimeric gB, suggesting that its epitope is composed of adjacent residues on two protomers. The destabilized DL16 epitope in the E187i5 mutant strongly suggests that within the E187i5 trimer, domains I in different protomers are not packed as closely together as they are in the wild-type gB730 trimer.
The second consequence of the “spreading out” of domains I is the exposure of residues that are normally buried. In particular, we found that chymotrypsin cleaves the E187i5 mutant between residues F223 and H224, in addition to cleaving at the same sites as in the wild-type gB730. In the wild-type gB730, residues F223 and H224 are not accessible to protease, which means that the E187i5 mutant undergoes conformational changes that result in the exposure of these residues. We hypothesize that the extra 15 residues within the cavity push on the β-sheets lining the cavity causing them to move apart. Finally, exposure of buried hydrophobic residues as the result of this conformational change likely accounts for the self-association (aggregation) of E187i5 mutant molecules into dimers of trimers and trimers of trimers, as seen by EM. Such oligomers probably do not occur in the full-length, membrane-associated mutant protein on the viral or cell surface due to the presence of transmembrane regions. However, the hydrophobic regions that become exposed as a result of insertion would probably render even the full-length E187i5 mutant protein more “sticky” and therefore could interfere with its function.
Hinge mutants T665i5, V667i5, I671i5, and L673i5.
The insertions after residues T665, V667, I671, or L673 would be predicted to lead to local steric strain. These residues lie within the linker region that connects domains IV and V and that descends from the “crown” along the hydrophobic bundle of domain III helices. Such steric strain could be avoided by displacement of several residues preceding and following the insertion site. For example, the insertion after residue I671 would be predicted to displace not only residue I671 itself but also neighboring F670 and D672 and, possibly, T669 and L673, as well (Fig. 1C). Given that T665i5, V667i5, I671i5, and L673i5 mutants have biochemical and biophysical characteristics—antibody reactivity, overall shape, proteolytic sensitivity, and oligomeric state—very similar to those of the wild-type gB730, we hypothesize that the mutants undergo only minor local conformational changes to relieve the steric strain resulting from the presence of five extra amino acids. Inserted residues probably bulge out, while residues immediately preceding and following the insertion change their conformation, but the overall structure of the trimer is maintained. The conformational changes in the hinge mutants are much less pronounced than those in the E187i5 mutant. Nevertheless, we speculate that they are probably energetically unfavorable due to the disruption of hydrophobic packing and exposure of several hydrophobic residues to water. For example, to accommodate a five-residue insertion after I671, several buried hydrophobic side chains—F670, I671, and L673—would need to be removed from the hydrophobic core.
Thus, our results suggest that the postfusion conformation of gB is very stable and highly energetically favorable. gB can assume this conformation even in the presence of mutations that would be predicted to prevent the formation of the postfusion form. The energetic penalty of having extra residues is outweighed by the energetic benefits of forming the low-energy postfusion form. This highly stable postfusion conformation is characteristic of other viral fusion proteins, e.g., influenza hemagglutinin and paramyxovirus (PIV3) F proteins (7, 29). Such stability is probably very important in the fusion process—a significant amount of energy is released during conversion of the prefusion form into the very stable, energetically favorable postfusion form. This energy is thought to be required to overcome the high activation energy of merging the two lipid bilayers (15).
Possible causes of the fusion-deficient phenotype.
Why do the five mutants studied here fail to mediate cell-cell fusion? Our results strongly suggest that the fusion-deficient phenotype of all five mutants is not due to the inability of the 5-amino-acid insertions to be accommodated in the trimeric postfusion conformation. Although we cannot completely eliminate the possibility that, in the context of the full-length membrane-associated gB, these insertions may somehow prevent the formation of the postfusion form, we do not think this possibility is likely because of the stability of the postfusion form of gB. The ectodomain represents the bulk of the full-length gB molecule and is an independently folding unit. We find it unlikely that the mutant ectodomain, when expressed alone, can assume the postfusion conformation, whereas the ectodomain in the context of the full-length molecule cannot. Although additional work is necessary to determine the precise mechanism, we envision several possible mechanisms.
First, the E187i5 mutant might have poorly functional fusion loops, which are located at the tips of the fingerlike β-sheet projections in domain I (Fig. 1D) (13, 14). We hypothesize that, as the result of the insertion after residue E187, spreading apart of domains I pushes apart the fusion loops thereby preventing them from inserting properly into the lipid bilayer. This spatial separation of fusion loops possibly prevents them from adopting the appropriate conformation for membrane insertion. The mutant protein would then be unable to mediate fusion despite being able to refold into the postfusion conformation. The drastically impaired ability of the E187i5 mutant ectodomain to cofloat with liposomes supports this idea. Nevertheless, we cannot exclude the possibility that the impaired liposome binding is due to the presence of higher-molecular-weight aggregates, dimers of trimers and trimers of trimers, in which the fusion loops are sequestered at the oligomeric interface and can no longer bind to liposomes. Importantly, the inability of the E187i5 mutant to function suggests that the three fusion domains (one per protomer) in their entirety are critical for proper gB function, that is, that the overall structure of the fusion domains is critical for function and not just the specific residues within the fusion loops.
Second, mutants may be defective in binding gH or gD. The components of the HSV fusion machinery, gB, gD, and gH/gL, have long been thought to interact to enable membrane fusion during viral entry or cell-cell fusion. Recent studies suggest that gB can directly interact with both gD and gH during the fusion process, although the precise nature of these interactions is yet to be determined (1, 2, 12). Given the importance of coordinated work by these different components, it is reasonable to speculate that mutations could interfere with fusion due to their inability to interact with other viral entry proteins in an appropriate way.
Third, hinge mutants may be deficient in binding a gB receptor. gB730 can block HSV entry into cells that are deficient in heparan sulfate proteoglycans (6). This suggests that it may compete for binding to a cell receptor (6) such as, possibly, the recently identified paired immunoglobulinlike type 2 receptor alpha (27). The MAb SS10 can block entry into cells that are deficient in heparan sulfate proteoglycans (6), possibly, by preventing binding of gB to its cell receptor. Because the SS10 epitope is located in the vicinity of the hinge region (5), it is tempting to speculate that the insertions in the hinge region may interfere with receptor binding. Future work is necessary to test this possibility.
Finally, the T665i5, V667i5, I671i5, and L673i5 mutants, although able to fold into the postfusion conformation, may do so more slowly than the wild-type gB. This slower rate could, perhaps, prevent fusion within the timeline of the fusion assay.
In summary, although the mechanism by which several insertion mutations within gB inhibit membrane fusion is yet unknown, we propose that inability to refold into the trimeric postfusion form is not the reason behind their fusion-deficient phenotype.
Acknowledgments
This study was funded by the NIH grant 1DP20D001996 and by the Pew Scholar Program in Biomedical Sciences (E.E.H.). T.M.C. acknowledges support by the NIH grant AI-076231 to Roselyn J. Eisenberg.
We thank Mike Berne at the Tufts University Core facility for N-terminal protein sequencing and Katie Stiles at the University of Pennsylvania for FACS analysis. We are grateful to Roselyn Eisenberg and Gary Cohen at the University of Pennsylvania for helpful discussions, critical reading of the manuscript, and providing MAbs used in this work.
E.E.H. designed research; J.S., S.S., T.M.C., and E.E.H. performed research; J.S., S.S., and E.E.H. contributed new reagents; J.S., S.S., T.M.C., and E.E.H. analyzed data; and E.E.H. wrote the paper.
Footnotes
Published ahead of print on 25 November 2009.
REFERENCES
- 1.Atanasiu, D., J. C. Whitbeck, T. M. Cairns, B. Reilly, G. H. Cohen, and R. J. Eisenberg. 2007. Bimolecular complementation reveals that glycoproteins gB and gH/gL of herpes simplex virus interact with each other during cell fusion. Proc. Natl. Acad. Sci. U. S. A. 104:18718-18723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Avitabile, E., C. Forghieri, and G. Campadelli-Fiume. 2007. Complexes between herpes simplex virus glycoproteins gD, gB, and gH detected in cells by complementation of split enhanced green fluorescent protein. J. Virol. 81:11532-11537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Backovic, M., and T. S. Jardetzky. 2009. Class III viral membrane fusion proteins. Curr. Opin. Struct. Biol. 19:189-196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Backovic, M., R. Longnecker, and T. S. Jardetzky. 2009. Structure of a trimeric variant of the Epstein-Barr virus glycoprotein B. Proc. Natl. Acad. Sci. U. S. A. 106:2880-2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bender, F. C., M. Samanta, E. E. Heldwein, M. P. de Leon, E. Bilman, H. Lou, J. C. Whitbeck, R. J. Eisenberg, and G. H. Cohen. 2007. Antigenic and mutational analyses of herpes simplex virus glycoprotein B reveal four functional regions. J. Virol. 81:3827-3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bender, F. C., J. C. Whitbeck, H. Lou, G. H. Cohen, and R. J. Eisenberg. 2005. Herpes simplex virus glycoprotein B binds to cell surfaces independently of heparan sulfate and blocks virus entry. J. Virol. 79:11588-11597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen, J., S. A. Wharton, W. Weissenhorn, L. J. Calder, F. M. Hughson, J. J. Skehel, and D. C. Wiley. 1995. A soluble domain of the membrane-anchoring chain of influenza virus hemagglutinin (HA2) folds in Escherichia coli into the low-pH-induced conformation. Proc. Natl. Acad. Sci. U. S. A. 92:12205-12209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cohen, G. H., V. J. Isola, J. Kuhns, P. W. Berman, and R. J. Eisenberg. 1986. Localization of discontinuous epitopes of herpes simplex virus glycoprotein D: use of a nondenaturing (“native” gel) system of polyacrylamide gel electrophoresis coupled with Western blotting. J. Virol. 60:157-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Connolly, S. A., D. J. Landsburg, A. Carfi, D. C. Wiley, G. H. Cohen, and R. J. Eisenberg. 2003. Structure-based mutagenesis of herpes simplex virus glycoprotein D defines three critical regions at the gD/HveA interface. J. Virol. 77:8127-8140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Connolly, S. A., D. J. Landsburg, A. Carfi, D. C. Wiley, R. J. Eisenberg, and G. H. Cohen. 2002. Structure-based analysis of the herpes simplex virus glycoprotein D binding site present on herpesvirus entry mediator HveA(HVEM). J. Virol. 76:10894-10904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Doms, R. W., A. Helenius, and J. White. 1985. Membrane fusion activity of the influenza virus hemagglutinin. J. Biol. Chem. 260:2973-2981. [PubMed] [Google Scholar]
- 12.Gianni, T., M. Amasio, and G. Campadelli-Fiume. 2009. Herpes simplex virus gD forms distinct complexes with fusion executors gB and gH/gL in part through the C-terminal profusion domain. J. Biol. Chem. 284:17370-17382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hannah, B. P., T. M. Cairns, F. C. Bender, J. C. Whitbeck, H. Lou, R. J. Eisenberg, and G. H. Cohen. 2009. Herpes simplex virus glycoprotein B associates with target membranes via its fusion loops. J. Virol. 83:6825-6836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hannah, B. P., E. E. Heldwein, F. C. Bender, G. H. Cohen, and R. J. Eisenberg. 2007. Mutational evidence of internal fusion loops in herpes simplex virus glycoprotein B. J. Virol. 81:4858-4865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harrison, S. C. 2008. Viral membrane fusion. Nat. Struct. Mol. Biol. 15:690-698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heckman, K. L., and L. R. Pease. 2007. Gene splicing and mutagenesis by PCR-driven overlap extension. Nat. Protoc. 2:924-932. [DOI] [PubMed] [Google Scholar]
- 17.Heldwein, E. E., and C. Krummenacher. 2008. Entry of herpesviruses into mammalian cells. Cell Mol. Life Sci. 65:1653-1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heldwein, E. E., H. Lou, F. C. Bender, G. H. Cohen, R. J. Eisenberg, and S. C. Harrison. 2006. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 313:217-220. [DOI] [PubMed] [Google Scholar]
- 19.Kielian, M., M. R. Klimjack, S. Ghosh, and W. A. Duffus. 1996. Mechanisms of mutations inhibiting fusion and infection by Semliki Forest virus. J. Cell Biol. 134:863-872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krummenacher, C., F. Baribaud, M. Ponce De Leon, I. Baribaud, J. C. Whitbeck, R. Xu, G. H. Cohen, and R. J. Eisenberg. 2004. Comparative usage of herpesvirus entry mediator A and nectin-1 by laboratory strains and clinical isolates of herpes simplex virus. Virology 322:286-299. [DOI] [PubMed] [Google Scholar]
- 21.Lin, E., and P. G. Spear. 2007. Random linker-insertion mutagenesis to identify functional domains of herpes simplex virus type 1 glycoprotein B. Proc. Natl. Acad. Sci. U. S. A. 104:13140-13145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller, C. G., C. Krummenacher, R. J. Eisenberg, G. H. Cohen, and N. W. Fraser. 2001. Development of a syngenic murine B16 cell line-derived melanoma susceptible to destruction by neuroattenuated HSV-1. Mol. Ther. 3:160-168. [DOI] [PubMed] [Google Scholar]
- 23.Okuma, K., M. Nakamura, S. Nakano, Y. Niho, and Y. Matsuura. 1999. Host range of human T-cell leukemia virus type I analyzed by a cell fusion-dependent reporter gene activation assay. Virology 254:235-244. [DOI] [PubMed] [Google Scholar]
- 24.Pertel, P. E., A. Fridberg, M. L. Parish, and P. G. Spear. 2001. Cell fusion induced by herpes simplex virus glycoproteins gB, gD, and gH-gL requires a gD receptor but not necessarily heparan sulfate. Virology 279:313-324. [DOI] [PubMed] [Google Scholar]
- 25.Roche, S., S. Bressanelli, F. A. Rey, and Y. Gaudin. 2006. Crystal structure of the low-pH form of the vesicular stomatitis virus glycoprotein G. Science 313:187-191. [DOI] [PubMed] [Google Scholar]
- 26.Roizman, B., and P. E. Pellett. 2001. The family Herpesviridae: a brief introduction, p. 2381-2398. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 4th ed. Lippincott/The Williams and Wilkins Co., Philadelphia, PA.
- 27.Satoh, T., J. Arii, T. Suenaga, J. Wang, A. Kogure, J. Uehori, N. Arase, I. Shiratori, S. Tanaka, Y. Kawaguchi, P. G. Spear, L. L. Lanier, and H. Arase. 2008. PILRα is a herpes simplex virus-1 entry coreceptor that associates with glycoprotein B. Cell 132:935-944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Turner, A., B. Bruun, T. Minson, and H. Browne. 1998. Glycoproteins gB, gD, and gHgL of herpes simplex virus type 1 are necessary and sufficient to mediate membrane fusion in a Cos cell transfection system. J. Virol. 72:873-875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yin, H. S., R. G. Paterson, X. Wen, R. A. Lamb, and T. S. Jardetzky. 2005. Structure of the uncleaved ectodomain of the paramyxovirus (hPIV3) fusion protein. Proc. Natl. Acad. Sci. U. S. A. 102:9288-9293. [DOI] [PMC free article] [PubMed] [Google Scholar]