

Abstract
The 5-halopyrimidine nucleotides damage DNA upon UV-irradiation or exposure to γ-radiolysis via the formation of the 2'-deoxyuridin-5-yl σ-radical. The bromo and iodo derivatives of these molecules are useful tools for probing DNA structure and as therapeutically useful radiosensitizing agents. A series of aryl iodide C-nucleotides were incorporated into synthetic oligonucleotides and exposed to UV-irradiation and γ-radiolysis. The strand damage produced upon irradiation of DNA containing these molecules is consistent with the generation of highly reactive σ-radicals. Direct stand breaks and alkali-labile lesions are formed at the nucleotide analogue and flanking nucleotides. The distribution of lesion type and location varies depending upon the position of the aryl ring that is iodinated. Unlike 5-halopyrimidine nucleotides, the aryl iodides produce interstrand cross-links in duplex regions of DNA when exposed to γ-radiolysis or UV-irradiation. Quenching studies suggest that cross-links are produced by γ-radiolysis via capture of a solvated electron, and subsequent fragmentation to the σ-radical. Cross-link formation is reduced slightly in the presence of O2. These observations suggest that aryl iodide C-nucleotide analogues may be useful as probes for excess electron transfer and radiosensitizing agents.
Keywords: nucleic acid oxidation, DNA damage, nucleotide analogues, interstrand cross-links, radiosensitization
Introduction
The 5-halopyrimidines, most notably 5-iodo-2'-deoxyuridine (IdU) and 5-bromo-2'-deoxyuridine (BrdU), are useful probes for studying excess electron transfer in duplex DNA and nucleic acid structure.1-4 The weak carbon-halogen bond and their structural similarity to thymidine provide the foundation for their utility in these applications. UV-irradiation of BrdU and IdU produces a σ-radical, 2'-deoxyuridin-5-yl (5YL, Scheme 1).5-12 The σ-radical is also produced upon γ-irradiation of BrdU and IdU, possibly by reaction of the 5-halopyrimidines with solvated electrons, followed by cleavage of the radical anion. The σ-radical (5YL) produces alkali-labile lesions by abstracting hydrogen atoms from 5'-adjacent nucleotides. The significantly higher reactivity of 5YL compared to π-carbon radicals is the source for the formation of alkali-labile lesions without the involvement of O2, which is often required for “fixing” damage involving DNA radicals. The ability to produce DNA damage under O2 deficient conditions has resulted in the use of BrdU and IdU as radiosensitizing agents in hypoxic tumors.13-15 Recently, BrdU was shown to produce interstrand cross-links upon γ-irradiation in unpaired (“bubble”) regions of duplex DNA, albeit not in hybridized regions.16-18 Interstrand cross-links are an important family of DNA lesions because they are absolute blocks to replication and transcription.19-21 The biological significance of one particular ICL was recently increased by the observation that it gives rise to even more toxic double-strand breaks upon nucleotide excision repair.22 Molecules that produce ICLs in hybridized regions of DNA upon γ-radiolysis and/or UV-irradiation might be useful as new radiosensitizing agents, as well as mechanistic probes of nucleic acid chemistry. We wish to report on aryl iodide nucleotide analogues that exhibit these desirable properties. 
Scheme 1.

Results and Discussion
Our choices of aryl iodides were based upon two disparate bodies of literature. Aryl iodides are photochemical precursors of aryl radicals.23,24 σ-Radicals are also generated from their respective radical anions.25-27 The aryl radicals are highly reactive and readily add to π-bonds and abstract hydrogen atoms that are bonded to tetrahedral carbons. In addition, nucleotide analogues containing aryl halides as substitutes for pyrimidine nucleobases are useful probes of DNA structure, replication, and nucleic acid-protein interactions.28-32 In particular, IT has been used to explore the flexibility of protein binding sites.33-36 This molecule provided the starting point for our investigation, and led to the examination of the 3 monoiodinated nucleotide analogues (I2, I3, I4). It was assumed that irradiation of IT (Scheme 2) and the monoiodides would produce the respective aryl radicals.
Scheme 2.

Synthesis of iodo substituted aryl nucleotide analogues and their incorporation into oligonucleotides
Oligonucleotides containing IT and I3 were prepared via solid phase oligonucleotide synthesis using procedures previously reported by Kool and Sekine respectively.37,38 The syntheses of oligonucleotides containing I2 and I4 were carried out using a similar strategy. Phosphoramidite 4 was synthesized from the previously reported bromide (1, Scheme 3).39 Bromide (1) was displaced by iodide using a mixture of NaI/CuI and trans-N,N′-dimethyl-1,2-cyclohexanediamine (5) in a pressure bottle in a manner similar to that previously described by Kool.38,39 The iodide (2) was carried on to 4 via standard methods. The phosphoramidite (10) used for synthesizing oligonucleotides containing I4 was prepared from 1,4-diiodobenzene and the silylated 2-deoxyribonolactone (6) employed in the syntheses of related molecules (Scheme 4).40
Scheme 3a.

Scheme 4a.

The phosphoramidites of the iodo nucleotide analogues were incorporated into oligonucleotides via automated solid phase synthesis using an extended (5 min) coupling time. Oligonucleotides containing Kool's diiodo molecule (IT) were prepared using commercially available (Glen Research) fast-deprotecting phosphoramidites, and t-BuOOH (1 M) was used as the oxidizing agent in place of I2. These oligonucleotides were deprotected with concentrated NH4OH (9 h, 25 °C). Oligonucleotides containing I2, I3, or I4 were prepared using standard I2 oxidation and were deprotected using “AMA” conditions (1:1 aqueous methylamine and concentrated NH4OH).41 The oligonucleotides were purified by 20% denaturing polyacrylamide  gel electrophoresis and characterized by ESI-MS following desalting.42 All experiments described herein were carried out using duplexes (11-18) containing these modified oligonucleotides. With the exception of hydroxyl radical digestion experiments to determine the site(s) of cross-linking on the opposing strand, the oligonucleotide containing the nucleotide analogue was radiolabeled at either its 5'- or 3'-terminus.
gel electrophoresis and characterized by ESI-MS following desalting.42 All experiments described herein were carried out using duplexes (11-18) containing these modified oligonucleotides. With the exception of hydroxyl radical digestion experiments to determine the site(s) of cross-linking on the opposing strand, the oligonucleotide containing the nucleotide analogue was radiolabeled at either its 5'- or 3'-terminus.
Strand damage produced upon UV-irradiation of duplex DNA containing the 2,4-diiodo-5-methylbenzene nucleotide analogue (IT)
Photolyses (30 min) were carried out in Pyrex tubes using lamps with maximum emission at 300 nm. When 5'-32P-11 (50 nM) containing IT was photolyzed under anaerobic conditions the nucleotide analogue (X11) and 5'- (T10) and 3'- (T12) flanking nucleotides were the major sites of strand damage (Figure 1A). Direct strand breaks (dsb) and smaller amounts of NaOH labile cleavage were produced. Treatment with piperidine resulted in a slight increase in cleavage at most nucleotide positions. Although detailed product analysis was not carried out, previous studies on oxidative DNA damage suggest that the formation of direct strand breaks and NaOH labile sites are consistent with hydrogen atom abstraction from the deoxyribose component.43 Positions that are only labile to piperidine are often associated with nucleobase lesions, which in this study could result from aryl radical addition to adjacent nucleobases. The data (Figure 1) suggest that all three types of lesions are produced upon photolysis of IT containing DNA. Lesser amounts of strand damage are detected at A8 and T9. Damage at these sites may be attributed to a diffusible species, particularly under aerobic conditions where the relative amounts of damage increases. However, we cannot rule out direct reaction between the aryl radical and these nucleotides due to disruption of the duplex structure by the diiodo precursor. Duplex melting temperatures are reduced considerably when IT is substituted for thymidine opposite dA.44 In addition, reactions of a DNA radical two or more nucleotides away, particularly in single stranded regions is precedented.45 The amount and pattern of strand damage at T10-T12 produced from 5'-32P-11 in the presence of O2 (Figure 1B) is similar to that observed under degassed conditions. However, as mentioned above cleavage at A8 and T9 is greater under these conditions and could be due to the production of reactive oxygen species. The general cleavage pattern is similar when 3'-32P-11 is photolyzed under the respective aerobic or anaerobic conditions.42 Under most conditions cleavage at T10 and X11 is slightly favored over T12. In addition, the cleavage products produced under all conditions from 3'- and 5'-32P-11 comigrate in denaturing PAGE with the products of Maxam-Gilbert sequencing reactions, indicating that they contain phosphate termini.42 The similarity in cleavage patterns in 3'- and 5'-32P-11 is in contrast to what is observed when nucleobase radicals resulting from radical addition to native pyrimidines are generated by γ-irradiation. In those instances, the resulting peroxyl radicals generate tandem lesions by reacting with flanking nucleotides in duplex DNA.43,46 The position of the original nucleobase radical and the other nucleotide in the tandem lesion are alkali-labile. Consequently, the strand scission pattern is different depending upon which oligonucleotide terminus is labeled because only the shortest labeled fragment is observed. In contrast, when radicals are generated from IT in 11, formation of an alkali-labile lesion at this site need not accompany damage at adjacent nucleotides.
Figure 1.

Direct strand scission and alkali-labile lesion formation when duplex DNA (11) containing IT is photolyzed. A. Anaerobic photolysis of 5'-32P-11. B. Aerobic photolysis of 5'-32P-11. dsb, direct strand breaks; NaOH, photolysate treated with 0.1 M NaOH at 37°C; Pip., photolysate treated with 1.0 M piperidine at 90°C.
Another difference between the strand damage derived from IT and nucleobase radicals resulting from radical addition is the effect of O2 on the level and pattern of strand damage. The nucleobase radicals require O2 for strand damage.47 In contrast, strand damage derived from IT is at most weakly dependent on O2. Comparing the reactivity of 2'-deoxyuridin-5-yl (5YL) produced from the 5-halopyrimidines to the presumptive radical(s) derived from IT is perhaps a more fair comparison, as both are σ-radicals. An O2 effect is observed in the reactions where 5YL is produced photochemically, but this may be complicated by the involvement of photoinduced electron transfer in their generation.11 Alternatively, the lack of an O2 effect involving irradiation of IT (with the exception of a small increase in damage at T8 and A9 in the presence of O2) may suggest that this radical trap ineffectively competes with intrastrand reactions of the aryl radical(s). This is consistent with available kinetic data on aryl radicals. Assuming that O2 is present at 0.2 mM, the effective first order rate constant for its reaction with the aryl radical (kO2 = 2 × 109 M−1s−1) is ~4 × 105 s−1. In contrast, an aryl radical abstracts hydrogen atoms from tetrahydrofuran, which should be less reactive than 2-deoxyribose, with a bimolecular rate constant, kTHF = 3.7 × 106 M−1s−1.23 Using this rate constant as a guide we speculate that oxidation of the DNA backbone will compete with O2 trapping of the aryl radical if the effective molarity of the sugar is 0.1 M. Rate constants for intermolecular aryl radical addition to alkenes (107 – 1010 M−1s−1) are also rapid enough to effectively compete with reactions with O2, because the former are effectively intramolecular reactions in this case.25,26 Overall, the available rate constants support the observation that O2 does not compete very effectively with the oligonucleotide for the putative aryl radical.
Dioxygen is also involved in the steps leading to strand scission and alkali-labile damage that follow intranucleotidyl or internucleotidyl reaction of the aryl radical. Consequently, in the absence of an exogenous quenching agent, such as a thiol, traces of O2 could give rise to the observed products. For instance, if 99.9% of the O2 (0.2 mM) is removed from a sample, the remaining concentration (0.2 μM) is approximately an order of magnitude greater than that of 11. Hence, in the absence of thiol it is possible that the same products are formed in degassed and aerobic reactions.
Additional information was gleaned from examining the effects of thiol on strand damage. The ability of β-mercaptoethanol (BME) to prevent direct strand breaks (Figure 2) and alkali-labile lesion formation (Figure 3) was studied under aerobic and anaerobic conditions. Low (1 mM) BME concentration had little if any effect under aerobic conditions on direct strand scission (Figure 2A) and even less of one on piperidine labile lesions (Figure 3A). Although bimolecular rate constants for thiol trapping of aryl radicals (kRSH) are not available, reaction of an aryl radical with Bu3SnH (kSnH = 7.8 × 108 M−1s−1) has been reported.23 Since thiols typically react slightly more rapidly than Bu3SnH with alkyl radicals, the rate constant for an aryl radical reacting with Bu3SnH could be considered a lower limit for reaction of BME with the radical(s) generated from IT (Scheme 2).48 Based upon these assumptions the rate constant for aryl radical trapping by the thiol should be comparable to that of O2 and 1 mM BME will compete with O2 for the aryl radical(s). Therefore, since O2 (0.2 mM) does not compete with inter- and intranucleotidyl reactions of the aryl radical(s), we do not expect 1 mM BME to either. The competition between O2 and BME for reaction with alkyl (π) radicals produced in DNA is expected to be very different from the reaction(s) of the putative aryl (σ) radicals because the rate constants with O2 with these radicals are considerably greater than that with BME.48 Consequently, BME (1 mM) will not compete with O2 (0.2 mM) for any subsequently formed alkyl radicals. These facts are consistent with the lack of an effect of 1 mM BME on strand damage from photolysis of DNA containing IT under aerobic conditions.
Figure 2.

The effect of β-mercaptoethanol on direct strand incision in UV-irradiated 5'-32P-11. A. Aerobic conditions. B. Anaerobic conditions.
Figure 3.

The effect of β-mercaptoethanol on alkali-labile lesion formation (piperidine, 1 M, 90 °C) in UV-irradiated 5'-32P-11. A. Aerobic conditions. B. Anaerobic conditions.
In contrast, 1 mM BME significantly reduced the amount of strand damage under anaerobic conditions (Figures 2B and 3B). These observations are attributed to reactions between thiols and alkyl radicals that are produced via reactions of the initially formed aryl radical(s) with the 2'-deoxyribose rings and/or adjacent nucleobases. β-Fragmentation of sugar radicals produce strand breaks in duplex DNA with rate constants ~2 × 102 s−1 at pH 7.0.49 These reactions and alkyl radical trapping by residual O2 are too slow to compete with quenching (kRSH ≤ 1 × 107 M−1s−1) by 1 mM BME.48 At higher BME concentration (100 mM) the levels of direct strand breaks and alkali-labile lesions at A8 – T12 are reduced to very low levels (<1%). This concentration of BME efficiently competes with O2 for the radicals. Moreover, using the above rate constants as a guide, it is reasonable to assume that the 0.1 M thiol competes with the inter- and intranucleotidyl reactions of the aryl radical(s).
Strand damage produced upon UV-irradiation of duplex DNA containing monoiodinated aryl nucleotide analogues
Irradiation of the 2,4-diiodo-5-methylbenzene nucleotide analogue (IT) could give rise to two different aryl radicals, which may react differently from one another due to conformational constraints in the duplex. Consequently, the photochemistry of duplexes (12, 14) containing ortho-iodoaryl (I2) or para-iodoaryl (I4) nucleotide analogues was examined. Strand damage was produced in lower yields than from IT (11) when otherwise identical duplexes containing these compounds were photolyzed (Figures 4 and 5).42 One explanation for this behavior is that photolysis of IT generates radicals more efficiently. Although the aryl iodide nucleotide analogues all absorb weakly at 300 nm, IT absorbs slightly more strongly than the mono-iodoaryl molecules in the region not blocked by the Pyrex glass and has the longest λmax (242 nm in MeOH).42 However, we cannot determine from these experiments whether the aryl iodides exhibit comparable quantum efficiency for photochemical conversion.
Figure 4.

Direct strand scission and alkali-labile lesion formation when duplex DNA (12) containing I2 is photolyzed. A. Anaerobic photolysis of 5'-32P-12. B. Aerobic photolysis of 5'-32P-12. dsb, direct strand breaks; NaOH, photolysate treated with 0.1 M NaOH at 37°C; Pip., photolysate treated with 1.0 M piperidine at 90°C.
Figure 5.

Direct strand scission and alkali-labile lesion formation when duplex DNA (14) containing I4 is photolyzed. A. Anaerobic photolysis of 5'-32P-14. B. Aerobic photolysis of 5'-32P-14. dsb, direct strand breaks; NaOH, photolysate treated with 0.1 M NaOH at 37°C; Pip., photolysate treated with 1.0 M piperidine at 90°C.
Although UV-irradiation of the ortho-iodoaryl (12, Figure 4) and para-iodoaryl (14, Figure 5) nucleotide analogues produced lower yields of strand damage products than Kool's compound, these molecules generated distinctive cleavage patterns. These observations suggest that diffusible species are not responsible for the majority of strand damage. When the ortho-iodoaryl analogue (I2) is photolyzed (Figure 4), piperidine labile lesion formation is favored at the 3'-adjacent nucleotide (T12) under all conditions.42 Direct strand breaks and NaOH labile lesions were produced in <1% yield at T12 regardless of whether O2 was present or not and independent of which oligonucleotide terminus was labeled. Intranucleotidyl cleavage (X11) was the second most prominent cleavage site, but the majority of lesions at this position in the oligonucleotide were direct strand breaks and they were produced in low yield. There was little if any enhancement in cleavage attributable to NaOH or piperidine treatment. These observations suggest that the aryl radical produced from I2 preferentially reacts with the nucleobase of the 3'-adjacent nucleotide (T12), less so with its own 2'-deoxyribose ring, and little if at all with the 5'-adjacent nucleotide (T10).
Photolysis of para-iodoaryl containing duplex (5'-32P-14) produces low levels of DNA damage, with modest preference for the 5'-flanking nucleotides (T9 and T10) (Figure 5). Cleavage is enhanced 2-fold or more when the DNA is treated with piperidine and there is little if any direct strand scission. In contrast to the ortho-iodoaryl molecule, I4 exhibits little if any specific damage at the 3'-flanking nucleotide (T12) or at its own position (X11).
The damage produced by photolysis of the ortho-iodoaryl and para-iodoaryl containing DNA is somewhat reflective of the proximity of the presumably formed aryl radicals to potential reactive sites as suggested by simple molecular models of a trinucleotide containing IT, which take into account only two conformations of the nucleotide analogue (Scheme 5, Figure 6). For instance, when IT is in the “anti” like conformation, the carbon of the incipient radical ortho to the carbon bonded to the sugar is ~3.9 Å from the C5-carbon of the 3'-adjacent nucleobase (Figure 6A), which could help explain the formation of piperidine labile lesions at T12 (Figures 1 and 3). Adoption of the “syn” like conformational isomer (Figure 6B) places the same radical in a better position to react with the C2'-hydrogen atom of its own sugar ring, which is ~2.6 Å away from the pro-radical center and a comparable distance from the C2'-hydrogen of the 5'-adjacent nucleotide. Although the C1'- and C2'-hydrogen atoms of the 5'-adjacent thymidine are within 3.3 Å and 2.6 Å respectively of the ortho-carbon in IT, there is little evidence for reaction at these positions in 12 even though there is in the former (Figure 4). In contrast, the alkene carbons of the 5'-adjacent thymine ring are ≥ 4.5 Å from the aryl ring carbon and as expected we do not detect damage (piperidine labile) consistent with reaction at this site. Assuming that piperidine labile lesions result from reaction with nucleobases and that direct strand breaks and NaOH labile lesions involve hydrogen atom abstraction from the 2'-deoxyribose rings, with the exception of evidence for direct oxidation of the 5'-adjacent 2'-deoxyribose ring, these models reflect the observed DNA damage overall fairly well (Figure 4).
Scheme 5.

Figure 6.

Molecular modeling (Spartan '02) of 5'-T•IT•T (top to bottom in figure) with IT in the (A.) anti-conformation or (B.) syn-conformation. The opposing strand is omitted for clarity.
Molecular models also indicate that radicals derived from the para-iodoaryl molecule (I4) are not well positioned to react with any of the 2'-deoxyribose rings (Figure 6), which is consistent with the dearth of direct strand breaks or NaOH labile lesions from 5'-32P-14 (Figure 5). Furthermore, the helical nature of the duplex provides better overlap between the incipient aryl radical(s) and the π-bond of the 5'-flanking nucleotide than it does the 3'-adjacent nucleobase (Figure 6). As with the photoreactivity of IT, we cannot rule out the formation of a diffusible species or the disruption of the duplex structure as a source for the piperidine labile lesions at T9 when 5'-32P-14 is photolyzed. Disruption of the duplex structure by IT or I4 is more likely than by I2. The former should experience steric clash with the N6-amino of the opposing dA; whereas an iodide at the ortho-position can occupy the minor groove. Overall, the composite reactivity of ortho-iodoaryl and para-iodoaryl nucleotide analogues is consistent with much of the strand damage produced by IT. The most apparent difference is the absence of significant fractions of direct strand breaks and NaOH labile lesions at T10.
The reactivity of the ortho- and para-aryl radicals led us to investigate the third regioisomeric aryl iodide nucleotide analogue, I3. Strand damage upon photolysis of 5'-32P-13 is more efficient than the other monoiodides but less so than IT (Figure 7).42 Piperidine labile damage is the major form of lesions produced under all conditions, indicating that little if any reactions involve hydrogen atom abstraction from the 2'-deoxyribose rings. In addition, the cleavage pattern for I3 is different from the other molecules examined. Intranucleotidyl cleavage is minimal under most conditions. The majority of alkali-labile lesions are produced at the 3'-and 5'-flanking nucleotides, as well as at T9. The reactivity of the meta-aryl iodide molecule is consistent with observations made in studies on the reactivity of the above aryl iodides. The meta-aryl radical was expected to be inefficient at producing intranucleotidyl direct strand breaks or NaOH labile lesions because of its poor orbital overlap with the hydrogen atoms of its own deoxyribose. The absence of such lesions at the 5'-adjacent thymidine is a little surprising because the distances between the C1'- and C2'-hydrogens at this position are not much greater than they are from the ortho-carbon (Figure 8). However, the lack of reactivity could be due to the fact that the sp2 like orbital of the incipient radical is not directed towards the 2'-deoxyribose hydrogen atoms. In contrast, the aryl radical is well positioned to add into the π-bond of the 5'- or 3'-adjacent pyrimidine when it is present in the “syn” or “anti” conformation, respectively. Finally, the anticipated disruption of base pairing with the opposing dA by the meta-iodo substituent may explain why such a significant level of damage is observed at T9.
Figure 7.

Direct strand scission and alkali-labile lesion formation when duplex DNA (13) containing I3 is photolyzed. A. Anaerobic photolysis of 5'-32P-13. B. Aerobic photolysis of 5'-32P-13. dsb, direct strand breaks; NaOH, photolysate treated with 0.1 M NaOH at 37°C; Pip., photolysate treated with 1.0 M piperidine at 90°C.
Figure 8.

Molecular modeling (Spartan '02) of 5'-T•I3•T (top to bottom in figure) with IT in the anti-conformation (A.) or syn-conformation (B.). The opposing strand is omitted for clarity.
Interstrand cross-link formation upon irradiation of iodinated nucleotide analogues
Recently, γ- and UV-irradiation of 5-bromo-2'-deoxyuridine was shown to produce interstrand cross-links in non-base paired regions of duplex DNA.16,17,50 ICL formation from irradiation of halogenated nucleotides (or analogues) in paired regions would be even more significant because these regions are more plentiful in DNA. γ-Radiolysis of 5'-32P-15a-d under anaerobic conditions produced interstrand cross-links without a significant dependence on the identity of the opposing nucleotide, although the ICL yield was slightly higher when IT was opposed by a pyrimidine than a purine (Figure 9, Table 1). Addition of the hydroxyl radical scavenger, t-BuOH (10 mM) did not affect the cross-link yield (Figure 9). However, ICL formation was reduced to <1% in the presence of O2, which is known to scavenge solvated electrons (data not shown).51 These observations suggest that cross-linking is induced via solvated electron capture and subsequent aryl radical formation via fragmentation of the radical anion.
Figure 9.

The effect of t-BuOH on interstrand cross-link (ICL) yield in 137Cs irradiated 5'-32P-15a-d as a function of nucleotide (Y) opposite IT.
Table 1.
Interstrand cross-link yields following 137Cs irradiation (700 Gy).a
| 5'-d(CGA GTA CTG C A A X AA CGT GTA CAG C) | ||||
|---|---|---|---|---|
| 3'-d(GCT CAT GAC GT15T14 Y13TT GCA CAT GTC G) | ||||
| 15-18 (a-d) | ||||
| ICL Yield (%) | ||||
| X | Y = A | Y = G | Y = C | Y = T |
| IT | 4.8 ± 0.8 | 4.7 ± 0.4 | 5.2 ± 0.2 | 5.6 ± 0.1 |
| I2 | 1.6 ± 0.5 | 1.3 ± 0.3 | 2.1 ± 0.1 | 2.2 ± 0.2 |
| I3 | 6.4 ± 0.7 | 7.7 ± 0.7 | 9.7 ± 0.7 | 10.4 ± 0.6 |
| I4 | 9.3 ± 1.0 | 8.4 ± 0.3 | 15.7 ± 0.4 | 10.6 ± 0.6 |
Yields are the average ± std. dev. of 3 samples.
137Cs irradiation of the monoiodide nucleotide analogues also produced interstrand cross-links (Table 1). The ortho-iodoaryl precursor (I2) was the least effective at producing cross-links and the para-iodoaryl nucleotide analogue (I4) produced the highest ICL yields. The cross-link yields reached almost 16% when I4 (18a-d) was irradiated. Adding t-BuOH generally had little effect on cross-link yield, although inexplicably when I4 was opposed by dA, the ICL yield almost doubled.42 The high yields of ICLs produced from I4 were also observed when 18a was photolyzed (Table 2). However, a control duplex (19) containing a T:A base pair in place of the nucleotide analogue yielded <2 % ICL. Cross-link yields were higher when the nucleotide analogue was opposed by pyrimidines than when purines were present at these positions. This trend is the same as when the duplex was exposed to γ-radiolysis (Table 1). UV-induced cross-link yields were modestly reduced when photolyses were carried out under aerobic conditions. One possible explanation is that O2 partially quenches the excited state. Alternatively, assuming that cross-links result from aryl radical addition into a π-bond of the opposing nucleotide, O2 (kO2 ~4 × 105 s−1 when [O2] = 0.2 mM) trapping may compete with this reaction. If so, the anticipated rate constant for O2 trapping would indicate that cross-linking by the aryl radical is considerably faster than that estimated for cross-linking by the 5-(2'-deoxyuridinyl)methyl radical (20), whose rate constant for ICL formation (~103 s−1) is limited by rotation about the nucleotide's glycosidic bond.52,53 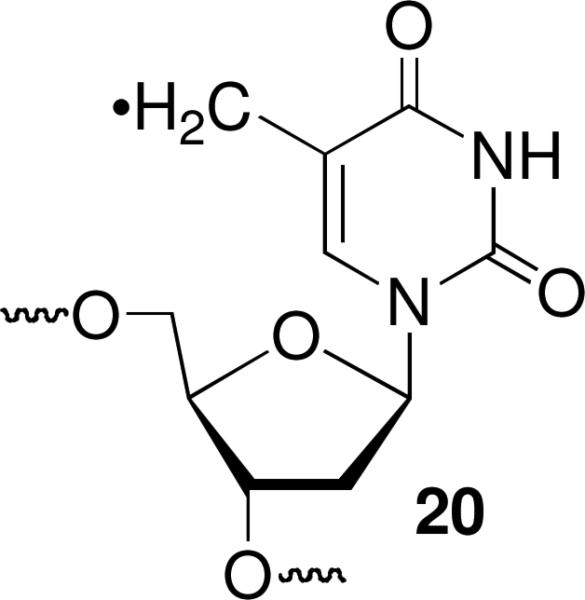
Table 2.
Interstrand cross-link yields following UV-irradiation of DNA containing I4.a
| 5'-d(CGA GTA CTG C A A X AA CGT GTA CAG C) | |||
|---|---|---|---|
| 3'-d(GCT CAT GAC GT15T14 Y13TT GCA CAT GTC G) | |||
| 18a-d, 19 | |||
| ICL Yield (%) | |||
| Duplex | X : Y | Aerobic | Anaerobic |
| 18a | I4 : A | 7.0 ± 0.8 | 10.0 ± 2.2 |
| 18b | I4 : G | 8.4 ± 1.0 | 9.4 ± 0.3 |
| 18c | I4 : C | 18.5 ± 2.4 | 21.1 ± 0.7 |
| 18d | I4 : T | 14.1 ± 1.8 | 19.2 ± 1.7 |
| 19 | T : A | 1.5 ± 0.3 | 1.6 ± 0.2 |
Yields are the average ± std. dev. of 3 samples.
Although cross-linking by the nucleotide analogues may be considerably faster than by 20, the latter is more selective. Cross-links derived from 20 exclusively involve the opposing nucleotide.54 Hydroxyl radical cleavage experiments were carried out on a subset of the cross-linked products from the para- and meta-aryl radicals, which gave the highest yields of ICLs in order to determine which nucleotide(s) on the opposing strand was cross-linked.42,55 Cross-links produced from photolysis of I4 in 18a under anaerobic conditions occurred exclusively with the opposing dA. When I4 was paired opposite dC (18c), the opposing nucleotide was still the major position cross-linked. However, a small amount of cross-linking with T14 was also detected. Similar behavior was observed when I3 was opposite dT. The majority of cross-links were formed with the opposing thymidine (T13) in 17d, along with small amounts with T14. Cross-linking in photolyzed duplex containing I3 opposite dG (17b) was least specific. Although ICLs were produced with the opposing purine, considerable cross-linking was observed with T14 and T15. The less selective cross-linking observed when I3 was opposite dG may be a consequence of greater disruption of base pairing in the vicinity of the nucleotide analogue when it is paired with the larger purine. Assuming that cross-linking occurs via σ-radical addition to a π-bond, dG may also be inherently less reactive with the incipient aryl radical because its major tautomer does not contain a π-bond proximal to the aryl radical.
Summary
The studies described above demonstrate a new application of aryl iodide nucleotide analogues. UV-irradiation (300 nm) of duplexes containing these molecules produces a mixture of direct strand breaks and alkali-labile lesions. The strand damage produced is consistent with generation of highly reactive aryl radicals. However, unlike the 5-halopyrimidines we do not know if the aryl radicals are formed via direct excitation and/or photoinduced electron transfer from a neighboring nucleotide. Additional studies are necessary to more fully characterize the strand products of these reactions, as well as the mechanisms of their formation.
The formation of interstrand cross-links upon irradiation of the iodinated nucleotide analogues in base paired regions of DNA is distinctive. The 5-(2'-deoxyuridinyl)methyl radical (20) produces ICLs.52-54 However, a synthetic molecule that generates this reactive species when exposed to γ-radiolysis has not been reported.56 5-Bromo-2'-deoxyuridine (BrdU) produces ICLs, but only in non-base paired regions of DNA. We speculate that Watson-Crick base pairing involving BrdU imposes a barrier to rotation about the glycosidic bond into the syn-conformation required for cross-linking that prevents this reaction from competing with others of the σ-radical. The non-hydrogen bonding nucleotide analogues investigated here probably face a lower barrier to glycosidic bond rotation due to the absence of hydrogen bonds with the opposing nucleotide. Furthermore, their destabilization of the duplex may facilitate the conformational freedom that facilitates cross-link formation. Cross-link formation in γ-irradiated samples containing hydroxyl radical quencher indicates that the aryl iodides do not react with this species. We hypothesize that the aryl iodides produce cross-links via reaction with solvated electrons. This is consistent with ICL quenching by O2, which traps this species. This suggests that these molecules, especially I3 and I4 could be useful mechanistic tools for probing excess electron transfer in DNA. One could imagine using cross-link formation in a PAGE experiment as a simple read out for excess electron transfer. Furthermore, their ability to produce cross-links under anaerobic conditions suggests that the iodinated nucleotide analogues could be useful as radiosensitizing agents. Their ultimate utility as radiosensitizing agents in cells requires that they be incorporated into cellular DNA. Although it is unlikely that the I3 or I4 would be incorporated in DNA by existing polymerases, one could hope that in the future engineered polymerases may accept such substrates.57 Alternatively, one could utilize advances made in the design of nucleotides analogues that are accepted as substrates by DNA polymerases as inspiration for designing new halogenated radiosensitizing agents that produce interstrand cross-links.58-63
Experimental
General Methods
Oligonucleotides were synthesized via standard automated DNA synthesis on an Applied Biosystems model 394 instrument. The coupling time for the phosphoramidites of modified nucleotides IT, I2, I3, and I4 was 5 min. For synthesis of oligonucleotides containing IT, fast-deprotecting phosphoramidites were used and 1 M TBHP in toluene was used as the oxidation reagent. Oligonucleotides were deprotected using 1:1 methylamine (40% in water) – concentrated NH4OH at 65 °C for 75 min (oligonucleotides containing I2, I3 and I4), or concentrated NH4OH at 25 °C for 9 h (oligonucleotides containing IT). Oligonucleotides were purified by 20 % denaturing polyacrylamide gel electrophoresis (PAGE). All oligonucleotides containing modified nucleotides were characterized by ESI-MS. Radiolabeled oligonucleotides were hybridized with 1.5 eq. of complementary oligonucleotides in 10 mM potassium phosphate (pH 7.2) and 100 mM NaCl at 90 °C for 5 min and cooled to room temperature. The concentration of duplexes was 50 nM in all experiments involving radiolabeled samples. All anaerobic reactions were carried out in sealed Pyrex tubes, which were degassed and sealed using freeze-pump-thaw (three cycles, 3 min each) degassing techniques. Experiments involving radiolabeled oligonucleotides were analyzed following PAGE using a Storm 840 phosphorimager.
Preparation of 2
A pressure tube was charged with CuI (57 mg, 0.3 mmol), NaI (1.8 g, 12 mmol), and sealed with a rubber septum. It was evacuated and backfilled with argon (repeat three times). A solution of 139 (163 mg, 0.59 mmol) and trans-N, N′-dimethyl-1, 2-cyclohexanediamine (86 mg, 0.6 mmol) in 1-pentanol (1.6 mL) was added to the pressure tube via syringe. The reaction mixture was sealed with a Teflon stopper and stirred at 130 °C for 40 h. The resulting suspension was cooled to 25 °C, at which time saturated NaHCO3 (20 mL) was added and the resulting mixture was extracted with diethyl ether. The combined organic phases were washed with saturated NaHCO3, water, and brine. After drying over anhydrous Na2SO4, the solution was concentrated to give the crude product which was purified by flash chromatography (30 : 1 EtOAc/MeOH) to yield an oil. 1H NMR showed it is a mixture (1.1:1) of the desired product and 1′, 2′-dideoxy-β-1′-phenyl-D-ribofuranose. The mixture was further purified by HPLC to give 2 (45 mg, 24% yield). HPLC conditions: Waters Delta Pak C18 column (300 × 7.8 mm); solution A: water; solution B: acetonitrile; increase of B from 2% to 50% in 15 min at a flow rate of 4 mL/min. 1H NMR (acetone-d6) δ 7.83 (d, J = 8.0 Hz, 1H), 7.70 (d, J = 7.8 Hz, 1H), 7.39 (dd, J = 7.8, 7.8 Hz, 1H), 7.03 (dd, J = 7.6, 7.6 Hz, 1H), 5.26 (ddd, J = 9.9, 5.6, 5.6 Hz, 1H), 4.44-4.38 (m, 1H), 4.32 (br s, 1H), 4.04-3.86 (m, 2H), 3.80-3.66 (m, 2H), 2.51 (ddd, J = 12.8, 5.6, 2.0 Hz, 1H), 1.70 (ddd, J = 12.8, 9.9, 5.8 Hz, 1H); 13C NMR (acetone-d6) δ 145.3, 138.9, 129.1, 128.4, 127.2, 96.1, 88.2, 83.1, 73.0, 62.9, 42.8; IR (film): 3384, 2916, 1650, 1615, 1559, 1512, 1459, 1179, 1047, 754 cm−1; HRMS-FAB (m/z) [M-H]+ calcd. for C11H12IO3, 318.9837; found, 318.9830.
Preparation of 3
C-Nucleoside 2 (42 mg, 0.13 mmol) was coevaporated with pyridine three times and then dissolved in pyridine (1.2 mL). To the solution was added 4,4-dimethoxytrityl chloride (67 mg, 0.2 mmol). The mixture was stirred at 25 °C for 16 h and concentrated. The residue was loaded onto a silica gel (oven-dried) column and eluted (2:1 hexanes/EtOAc) to give 3 as a colorless foam (75 mg, 91%). 1H NMR (acetone-d6) δ 7.85 (d, J = 8.0 Hz, 1H), 7.71 (d, J = 8.0 Hz, 1H), 7.60-7.52 (m, 2H), 7.47-6.62 (m, 13H), 5.31 (ddd, J = 9.6, 5.9, 5.9 Hz, 1H), 4.46-4.33 (m, 2H), 4.18-4.10 (m, 1H), 3.79 (s, 6H), 3.41-3.35 (m, 2H), 2.55 (ddd, J = 12.8, 5.8, 2.2 Hz, 1H), 1.78 (ddd, J = 15.5, 9.6, 5.9 Hz, 1H); 13C NMR (acetone-d6) δ 158.7, 145.4, 145.3, 139.1, 136.1, 130.2, 129.2, 128.5, 128.2, 127.7, 126.7, 113.0, 96.1, 86.7, 85.9, 83.2, 73.1, 64.2, 54.6, 42.9; IR (film): 3446, 2933, 2836, 1608, 1509, 1463, 1300, 1251, 1177, 1034, 754 cm−1; HRMS-FAB (m/z) [M-H]+ calcd. for C32H30IO5, 621.1137; found, 621.1174.
Preparation of 4
To a solution of 3 (74 mg, 0.118 mmol) in CH2Cl2 (1.2 mL), was added diisopropylethylamine (31 mg, 0.24 mmol) and 2-cyanoethyl-N,N′-diisopropylchlorophosphoramidite (45 mg, 0.19 mmol). The reaction mixture was stirred at 25 °C for 4 h and concentrated. The residue was loaded onto a silica gel (oven-dried) column and eluted (3:1 hexanes/EtOAc) to afford 4 as a colorless foam (62 mg, 64%). 1H NMR (acetone-d6) δ 7.85 (d, J = 8.0 Hz), 7.76-7.68 (m), 7.61-7.54 (m), 7.49-7.21 (m), 7.10-7.03 (m), 6.97-6.87 (m), 5.34-5.26 (m), 4.67-4.55 (m), 4.32-4.23 (m), 3.99-3.61 (m), 3.49-3.34 (m), 2.83-2.62 (m), 1.92-1.77 (m), 1.33-0.85(m); 31P NMR (acetone-d6) δ 148.0, 147.9; IR (film): 2966, 2836, 2252, 1607, 1508, 1462, 1178, 1034, 895, 754 cm−1; HRMS-FAB (m/z) [M+Na]+ calcd. for C41H48N2O6NaPI, 845.2187; found, 845.2203.
Preparation of 7
n-BuLi (1.6 M in hexanes, 2.1 mL, 3.36 mmol) was added via syringe to a solution of the 1,4-diiodobezene (1.07 g, 3.25 mmol) in anhydrous THF (3.0 mL) cooled at –78 °C under argon. After 30 min, a solution of 3′,5′-O-((1,1,3,3-tetraisopropyl)disiloxanediyl)-2′-deoxy-D-ribono-1,4-lactone40 (1.2 g, 3.12 mmol) in anhydrous THF (5.0 mL) was added to the solution via cannula at –78 °C. After 1 h, the reaction mixture was quenched at –78°C with saturated aqueous NH4Cl (3 mL). The mixture was extracted with diethyl ether. The combined ether phases were washed with saturated aqueous NH4Cl, water, and brine. The organic phase was dried over anhydrous Na2SO4 and concentrated to give an oil that was used without further purification.
A solution of the crude oil in CH2Cl2 (5.1 mL) under argon and at –78 °C was treated with Et3SiH (0.72 g, 6.5 mmol) and BF3•Et2O (0.91 g, 6.5 mmol). The reaction mixture was stirred at –78 °C for 3 h, and quenched at –78 °C by the addition of saturated aqueous NaHCO3 (3 mL). The mixture was extracted with diethyl ether. The combined organic phases were washed with saturated aqueous NaHCO3, water, and brine. This solution was dried over anhydrous Na2SO4, concentrated and purified by flash chromatography (3 : 2 to 1 : 1 hexanes/toluene) to yield the desired β isomer of 7 (163 mg, 0.29 mmol, 9%) as an oil (~6 : 1 β/α mixture in the crude product; 1.6% isolated yield of α–7; the total yield is 13%). β-7: 1H NMR (CDCl3) δ 7.67 (d, J = 8.2 Hz, 2H), 7.11 (d, J = 8.4 Hz, 2H), 5.06 (dd, J = 14.4, 7.2 Hz, 1H), 4.52 (ddd, J = 7.7, 4.7, 4.7 Hz, 1H), 4.18-4.10 (m, 1H), 3.94-3.86 (m, 2H), 3.39 (ddd, J = 12.4, 7.0, 4.7 Hz, 1H), 2.03 (ddd, J = 15.4, 7.7, 7.7 Hz, 1H), 1.28-0.76 (m, 28H); 13C NMR (CDCl3) δ 142.0, 137.4, 127.8, 92.8, 86.4, 78.4, 72.9, 63.5, 43.0, 17.6, 17.5, 17.39, 17.3, 17.11, 17.08, 16.99, 13.5, 13.4, 13.3, 12.6; IR (film): 2994, 2867, 1464, 1386, 1116, 1089, 1037, 885 cm−1; HRMS-FAB (m/z) [M-H]+ calcd. for C23H39IO4Si2, 561.1353; found, 561.1355.
α–7
1H NMR (CDCl3) δ 7.68 (d, J = 8.3 Hz, 2H), 7.14 (d, J = 8.3 Hz, 2H), 4.98 (ddd, J = 9.6, 6.4, 6.4 Hz, 1H), 4.63-4.56 (m, 1H), 4.09-4.02 (m, 1H), 3.97-3.88 (m, 2H), 2.66 (ddd, J = 12.8, 6.4, 6.4 Hz, 1H), 2.09 (ddd, J = 9.6, 9.6, 9.6 Hz, 1H), 1.24-0.88 (m, 28H); 13C NMR (CDCl3) δ 142.6, 137.4, 127.9, 92.7, 83.7 77.8, 73.5, 63.1, 42.9, 17.5, 17.44, 17.43, 17.4, 17.3, 17.2, 17.0, 13.5, 13.3, 12.9, 12.6; IR (film): 2944, 2867, 1462, 1388, 1247, 1141, 1034, 1005, 782 cm−1; HRMS-FAB (m/z) [M-H]+ calcd. for C23H39IO4Si2, 561.1353; found, 561.1336.
Preparation of 8
To a solution of 7 (163 mg, 0.29 mmol) in THF (5 mL) was added Bu4N+F− hydrate (280 mg, 0.89 mmol). The resulting mixture was stirred at 25 °C for 18 h. It was concentrated and purified by flash column chromatography (15 : 1 CH2Cl2-MeOH) to give 8 as a colorless oil (79 mg, 85%). 1H NMR (MeOH-d4) δ 7.68 (d, J = 8.2 Hz, 2H), 7.20 (d, J = 8.4 Hz, 2H), 5.09 (ddd, J = 10.8, 5.4, 5.4 Hz, 1H), 4.35-4.29 (m, 1H), 3.99-3.93 (m, 1H), 3.68 (d, J = 3.8 Hz, 2H), 2.24-2.15 (m, 1H), 1.97-1.84 (m, 1H); 13C NMR (MeOH-d4) δ 141.9, 137.1, 127.8, 92.0, 87.9, 79.5, 73.0, 62.6, 43.6; IR (film): 3333, 2923, 2891, 1682, 1559, 1458, 1066, 997, 815 cm−1; HRMS-FAB (m/z) [M+NH4]+ calcd. for C11H17NO3I, 338.0248; found, 338.0248.
Preparation of 9
C-Nucleoside 8 (79 mg, 0.246 mmol) was coevaporated with pyridine three times and dissolved in pyridine (2.0 mL). To the solution was added 4,4-dimethoxytrityl chloride (114 mg, 0.34 mmol). The mixture was stirred at 25 °C for 20 h and concentrated. The residue was loaded onto a silica gel (oven-dried) column and eluted (2:1 hexanes/EtOAc) to give 9 as a colorless foam (102 mg, 67%). 1H NMR (acetone-d6) δ 7.72 (d, J = 8.1 Hz, 2H), 7.52 (d, J = 7.5 Hz, 2H), 7.42-7.19 (m, 9H), 6.91-6.87 (m, 4H), 5.14 (ddd, J = 9.6, 4.8, 4.8 Hz, 1H), 4.39 (s, 1H), 4.34-4.26 (m, 1H), 4.13-4.05 (m, 1H), 3.80 (s, 6H), 3.28-3.24 (m, 2H), 2.31-2.23 (m, 1H), 1.98-1.88 (m, 1H); 13C NMR (acetone-d6) δ 158.7, 145.4, 143.1, 137.2, 136.1, 130.1, 128.2, 127.7, 126.6, 113.0, 91.8, 86.9, 85.9, 792, 73.4, 64.6, 54.5, 44.2; IR (film): 3425,2967, 1607, 1508, 1459, 1300, 1250, 1177, 1080, 1034, 1004, 827 cm−1; HRMS-FAB (m/z) [M+Na]+ calcd. for C32H31IO5Na, 645.1108; found, 645.1099.
Preparation of 10
To a solution of 9 (102 mg, 0.16 mmol) in CH2Cl2 (1.6 mL), was added diisopropylethylamine (42 mg, 0.33 mmol) and 2-cyanoethyl-N,N′-diisopropylchlorophosphoramidite (59 mg, 0.25 mmol). The reaction mixture was stirred at 25 °C for 4 h and concentrated. The residue was loaded onto a silica gel (oven-dried) column and eluted (3:1 hexanes/EtOAc) to afford 10 as a colorless foam (73 mg, 54%). 1H NMR (CDCl3) δ 7.76-7.62 (m), 7.56-7.45 (m), 7.44-7.15 (m), 6.92-6.76 (m), 5.22-5.08 (m), 4.61-4.49 (m), 4.32-4.19 (m), 3.91-3.54 (m), 3.43-3.20 (m), 2.68-2.55 (m), 2.54-2.28 (m), 2.10-1.95 (m), 1.32-0.88 (m); 31P NMR (CDCl3) δ 148.1, 148.0; IR (film): 2967, 2932, 2250, 1608, 1509, 1462, 1365, 1251, 1178, 1034, 1003, 828 cm−1; HRMS-FAB (m/z) [M+Na]+ calcd. for C41H48N2O6NaPI, 845.2187; found, 845.2181.
Photoreactions
Photoreactions of the duplexes were carried out in Pyrex tubes in a Rayonet photoreactor fitted with 16 lamps having a maximum output at 300 nm. All photoreactions were carried out for 30 min in 10 mM potassium phosphate (pH 7.2) and 100 mM NaCl. After reaction, each sample (20 μL) was aliquoted into three 0.6-mL eppendorf tubes, A (6 μL), B (8 μL) and C (6 μL). A was mixed with formamide loading buffer. B was incubated with 0.5 M NaOH (2 μL) at 37 °C for 20 min, then treated with 1 M AcOH (1 μL) and mixed with formamide loading buffer. C was treated with 2 M piperidine (6 μL) at 90 °C for 20 min, dried, coevaporated with H2O (3 × 10 μL) and resuspended in formamide loading buffer. Finally, they were subjected to 20% PAGE analysis.
Thiol effect on strand damage
The appropriate radiolabeled duplex (11-14) was prepared in 10 mM potassium phosphate (pH 7.2) and 100 mM NaCl in the presence of varying concentrations of BME (0, 1, and 100 mM). Anaerobic samples were degassed and sealed. Photoreactions were carried out at 300 nm for 30 min. After reaction, each sample (12 μL) was distributed in two 0.6 mL eppendorf tubes A (6 μL) and B (6 μL). A was mixed with formamide loading buffer. B was dried, treated with 1 M piperidine at 90 °C for 20 min, dried again, coevaporated with H2O (3 × 10 μL) and resuspended in formamide loading buffer, and subjected to 20% PAGE analysis.
γ-Radiolysis
γ-Radiolysis of the duplexes were carried out in Pyrex tubes in a J. L. Shepherd Mark I 137Cs irradiator that has an output of 25 Gray/min. After reaction (700 Gy), samples were lyophilized, resuspended in formamide loading buffer, and subjected to 20% PAGE analysis.
Fe(II)-EDTA digestion of cross-linked DNA
Fe(II)-EDTA cleavage reactions of ICLs were carried out in 50 μM (NH4)2Fe(SO4)2, 100 μM EDTA, 1 mM sodium ascorbate, 5.0 mM H2O2, 100 mM NaCl and 10 mM potassium phosphate (pH 7.2), for 1 min at 25 °C (total volume of 20 μL each). The reactions were quenched with 100 mM thiourea (10 μL). Samples were lyophilized, resuspended in formamide loading buffer and subjected to 20% PAGE analysis.
Supplementary Material
Acknowledgement
We are grateful for support of this research from the National Institute of General Medical Sciences (GM-054996).
Footnotes
Supporting Information Available. Strand damage data for 3'-32P-labeled duplexes. Hydroxyl radical digestion analysis of cross-linked products. Sample autoradiogram of UV-irradiation of 5'- and 3'-32P-11 showing cleavage pattern and comigration with Maxam-Gilbert sequencing reactions. Spectral data for previously unreported compounds, UV absorption spectra of aryl iodide nucleosides, and ESI-MS for oligonucleotides containing nucleotide analogues. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Ito T, Rokita SE. J. Am. Chem. Soc. 2004;126:15552–15559. doi: 10.1021/ja045637n. [DOI] [PubMed] [Google Scholar]
- 2.Ito T, Rokita SE. Angew. Chem. Int. Ed. 2004;43:1839–1842. doi: 10.1002/anie.200353038. [DOI] [PubMed] [Google Scholar]
- 3.Xu Y, Tashiro R, Sugiyama H. Nat. Protoc. 2007;2:78–87. doi: 10.1038/nprot.2006.467. [DOI] [PubMed] [Google Scholar]
- 4.Xu Y, Sugiyama H. Angew. Chem. Int. Ed. 2006;45:1354–1362. doi: 10.1002/anie.200501962. [DOI] [PubMed] [Google Scholar]
- 5.Tashiro R, Nakamura K, Sugiyama H. Tetrahedron Lett. 2008;49:428–431. [Google Scholar]
- 6.Watanabe T, Bando T, Xu Y, Tashiro R, Sugiyama H. J. Am. Chem. Soc. 2005;127:44–45. doi: 10.1021/ja0454743. [DOI] [PubMed] [Google Scholar]
- 7.Xu Y, Sugiyama H. J. Am. Chem. Soc. 2004;126:6274–6279. doi: 10.1021/ja031942h. [DOI] [PubMed] [Google Scholar]
- 8.Kawai K, Saito I, Sugiyama H. J. Am. Chem. Soc. 1999;121:1391–1392. [Google Scholar]
- 9.Sugiyama H, Saito I. J. Am. Chem. Soc. 1990;112:6720. [Google Scholar]
- 10.Chen T, Cook GP, Koppisch AT, Greenberg MM. J. Am. Chem. Soc. 2000;122:3861–3866. [Google Scholar]
- 11.Cook GP, Chen T, Koppisch AT, Greenberg MM. Chem. Biol. 1999;6:451–459. doi: 10.1016/s1074-5521(99)80063-5. [DOI] [PubMed] [Google Scholar]
- 12.Cook GP, Greenberg MM. J. Am. Chem. Soc. 1996;118:10025–10030. [Google Scholar]
- 13.Li Y, Owusu A, Lehnert S. Int. J. Radiat. Oncol. Biol. Phys. 2004;58:519–527. doi: 10.1016/j.ijrobp.2003.09.054. [DOI] [PubMed] [Google Scholar]
- 14.Berry SE, Davis TW, Schupp JE, Hwang HS, de Wind N, Kinsella TJ. Cancer Res. 2000;60:5773–5780. [PubMed] [Google Scholar]
- 15.Jones GDD, Ward JF, Limoli CL, Moyer DJ, Aguilera JA. Int. J. Radiat. Biol. 1995;67:647–653. doi: 10.1080/09553009514550761. [DOI] [PubMed] [Google Scholar]
- 16.Dextraze M-E, Cecchini S, Bergeron F. o., Girouard S, Turcotte K, Wagner JR, Hunting DJ. Biochemistry. 2009;48:2005–2011. doi: 10.1021/bi801684t. [DOI] [PubMed] [Google Scholar]
- 17.Dextraze M-E, Wagner JR, Hunting DJ. Biochemistry. 2007;46:9089–9097. doi: 10.1021/bi062114e. [DOI] [PubMed] [Google Scholar]
- 18.Cecchini S, Girouard S, Huels MA, Sanche L, Hunting DJ. Biochemistry. 2005;44:1932–1940. doi: 10.1021/bi048105s. [DOI] [PubMed] [Google Scholar]
- 19.Noll DM, Mason TM, Miller PS. Chem. Rev. 2006;106:277–301. doi: 10.1021/cr040478b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schärer OD. Chem. Bio. Chem. 2005;6:27–32. doi: 10.1002/cbic.200400287. [DOI] [PubMed] [Google Scholar]
- 21.Dronkert ML, Kanaar R. Mutat. Res. 2001;486:217–247. doi: 10.1016/s0921-8777(01)00092-1. [DOI] [PubMed] [Google Scholar]
- 22.Sczepanski J, Jacobs AC, Van Houten B, Greenberg MM. Biochemistry. 2009:7565–7567. doi: 10.1021/bi901006b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garden SJ, Avila DV, Beckwith ALJ, Bowry VW, Ingold KU, Lusztyk J. J. Org. Chem. 1996;61:805–809. doi: 10.1021/jo951245a. [DOI] [PubMed] [Google Scholar]
- 24.Braslau R, Anderson MO. Tetrahedron Lett. 1998;39:4227–4230. [Google Scholar]
- 25.Chami Z, Gareil M, Pinson J, Saveant JM, Thiebault A. J. Org. Chem. 1991;56:586–95. [Google Scholar]
- 26.Citterio A, Minisci F, Vismara E. J. Org. Chem. 1982;47:81–8. [Google Scholar]
- 27.M'Halla F, Pinson J, Savéant JM. J. Am. Chem. Soc. 1980;102:4120–4127. [Google Scholar]
- 28.Lee HR, Helquist SS, Kool ET, Johnson KA. J. Biol. Chem. 2008;283:14411–14416. doi: 10.1074/jbc.M705006200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee HR, Helquist SA, Kool ET, Johnson KA. J. Biol. Chem. 2008;283:14402–14410. doi: 10.1074/jbc.M705007200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Delaney JC, Henderson PT, Helquist SA, Morales JC, Essigmann JM, Kool ET. Proc. Natl. Acad. Sci. USA. 2003;100:4469–4473. doi: 10.1073/pnas.0837277100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kool ET. Ann. Rev. Biochemistry. 2002;71:191–219. doi: 10.1146/annurev.biochem.71.110601.135453. [DOI] [PubMed] [Google Scholar]
- 32.Pallan PS, Egli M. J. Am. Chem. Soc. 2009;131:12548–12549. doi: 10.1021/ja905739j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jarchow-Choy SK, Sjuvarsson E, Sintim HO, Eriksson S, Kool ET. J. Am. Chem. Soc. 2009;131:5488–5494. doi: 10.1021/ja808244t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Silverman AP, Garforth SJ, Prasad VR, Kool ET. Biochemistry. 2008;47:4800–4807. doi: 10.1021/bi702427y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mizukami S, Kim TW, Helquist SA, Kool ET. Biochemistry. 2006;45:2772–2778. doi: 10.1021/bi051961z. [DOI] [PubMed] [Google Scholar]
- 36.Kim TW, Delaney JC, Essigmann JM, Kool ET. Proc. Natl. Acad. Sci. USA. 2005;102:15803–15808. doi: 10.1073/pnas.0505113102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tawarada R, Seio K, Sekine M. J. Org. Chem. 2008;73:383–390. doi: 10.1021/jo701634t. [DOI] [PubMed] [Google Scholar]
- 38.Kim TW, Kool ET. Org. Lett. 2004;6:3949–3952. doi: 10.1021/ol048487u. [DOI] [PubMed] [Google Scholar]
- 39.Hwang GT, Romesberg FE. Nucl. Acids Res. 2006;34:2037–2045. doi: 10.1093/nar/gkl049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wichai U, Woski SA. Org. Lett. 1999;1:1173–1175. doi: 10.1021/ol990813w. [DOI] [PubMed] [Google Scholar]
- 41.Reddy MP, Hanna NB, Faroqui F. Tetrahedron Lett. 1994;35:4311–4314. [Google Scholar]
- 42. Information., S. S.
- 43.Hong IS, Carter KN, Sato K, Greenberg MM. J. Am. Chem. Soc. 2007;129:4089–4098. doi: 10.1021/ja0692276. [DOI] [PubMed] [Google Scholar]
- 44.Kim TW, Kool ET. J. Org. Chem. 2005;70:2048–2053. doi: 10.1021/jo048061t. [DOI] [PubMed] [Google Scholar]
- 45.Crean C, Uvaydov Y, Geacintov NE, Shafirovich V. Nucleic Acids Res. 2008;36:742–755. doi: 10.1093/nar/gkm1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carter KN, Greenberg MM. J. Am. Chem. Soc. 2003;125:13376–13378. doi: 10.1021/ja036629u. [DOI] [PubMed] [Google Scholar]
- 47.Greenberg MM. Org. Biomol. Chem. 2007;5:18–30. doi: 10.1039/b612729k. [DOI] [PubMed] [Google Scholar]
- 48.Newcomb M. Tetrahedron. 1993;49:1151–1176. [Google Scholar]
- 49.Giese B, Dussy A, Meggers E, Petretta M, Schwitter U. J. Am. Chem. Soc. 1997;119:11130–11131. [Google Scholar]
- 50.Cecchini S, Masson C, La Madeleine C, Huels MA, Sanche L, Wagner JR, Hunting DJ. Biochemistry. 2005;44:16957–16966. doi: 10.1021/bi050799x. [DOI] [PubMed] [Google Scholar]
- 51.von Sonntag C. Free-Radical-Induced DNA Damage and Its Repair. Springer-Verlag; Berlin: 2006. [Google Scholar]
- 52.Hong IS, Ding H, Greenberg MM. J. Am. Chem. Soc. 2006;128:485–491. doi: 10.1021/ja0563657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ding H, Majumdar A, Tolman JR, Greenberg MM. J. Am. Chem. Soc. 2008;130:17981–17987. doi: 10.1021/ja807845n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hong IS, Greenberg MM. J. Am. Chem. Soc. 2005;127:3692–3693. doi: 10.1021/ja042434q. [DOI] [PubMed] [Google Scholar]
- 55.Millard JT, Weidner MF, Kirchner JJ, Ribeiro S, Hopkins PB. Nucleic Acids Res. 1991;19:1885–1892. doi: 10.1093/nar/19.8.1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hong IS, Ding H, Greenberg MM. J. Am. Chem Soc. 2006;128:2230–2231. doi: 10.1021/ja058290c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Loakes D, Gallego J, Pinheiro VB, Kool ET, Holliger P. J. Am. Chem. Soc. 2009;131:14827–14837. doi: 10.1021/ja9039696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hirao I, Kimoto M, Mitsui T, Fujiwara T, Kawai R, Sato A, Harada Y, Yokoyama S. Nature Methods. 2006;3:729–735. doi: 10.1038/nmeth915. [DOI] [PubMed] [Google Scholar]
- 59.Silverman AP, Jiang Q, Goodman MF, Kool ET. Biochemistry. 2007;46:13874–13881. doi: 10.1021/bi700851z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Leconte AM, Matsuda S, Romesberg FE. J. Am. Chem Soc. 2006;128:6780–6781. doi: 10.1021/ja060853c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Young Jun S, Floyd ER. ChemBioChem. 2009;10:2394–2400. [Google Scholar]
- 62.Hwang GT, Romesberg FE. J. Am. Chem. Soc. 2008;130:14872–14882. doi: 10.1021/ja803833h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Seo YJ, Hwang GT, Ordoukhanian P, Romesberg FE. J. Am. Chem. Soc. 2009;131:3246–3252. doi: 10.1021/ja807853m. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.