

Abstract
Protein disulfide isomerase (PDI) catalyzes the formation of native disulfide pairings in secretory proteins. The ability of PDI to act as a disulfide isomerase makes it an essential enzyme in eukaryotes. PDI also fulfills other important roles. Recent studies have emphasized the importance of PDI as an oxidant in the endoplasmic reticulum. Intriguing questions remain regarding how PDI is able to catalyze both isomerization and oxidation in vivo. Studies of PDI and its homologs have led to the development of small-molecule folding catalysts that are able to accelerate disulfide isomerization in vitro and in vivo. PDI will continue to provide both an inspiration for the design of such artificial foldases and a benchmark with which to gauge the success of those designs. Here, we review current understanding of the chemistry and biology of PDI, its homologs, and small molecules that mimic its catalytic activity.
INTRODUCTION
Protein disulfide isomerase (PDI; EC 5.3.4.1) was first identified 40 years ago as an enzyme that catalyzes the activation of reduced, and thus inactive, ribonuclease A (29). Anfinsen and his coworkers went on to hypothesize that the primary role of PDI is to be a “general and nonspecific catalyst for disulfide interchange in proteins containing disulfide bonds” (28). From their experiments with reduced RNase A (rRNase A), they suggested that the formation of native disulfide bonds in proteins begins with the uncatalyzed air oxidation of dithiols, followed by the PDI-catalyzed isomerization of nonnative disulfide bonds. More recent studies have revealed that an ensemble of enzymes and chaperones are involved in the pathway of native disulfide formation (95, 101). Still, PDI remains at the center of this complex biological process.
PROPERTIES OF PROTEIN DISULFIDE ISOMERASE
PDI is a 57-kDa protein that resides in the endoplasmic reticulum (ER) of eukaryotic cells. There, PDI catalyzes the formation, reduction, and isomerization of disulfide bonds in newly synthesized proteins (25). Independent of its catalytic activity, PDI exhibits chaperone activity by inhibiting the aggregation of unfolded proteins (6, 71) and is a member of at least two multimeric enzyme complexes: prolyl 4-hydroxylase (45, 69) and microsomal triglyceride transfer protein (51, 89).
PDI has five distinct structural domains (a, a′, b, b′ and c; Figure 1a) as deduced from its primary and tertiary structure (15). The catalytic a and a′ domains are homologous, and each contains a Cys–Gly–His–Cys (CGHC) active-site sequence (Figure 1b). The b and b′ domains are also homologous to the a and a′ domains (15, 42). The role of the b′ domain is to bind substrate proteins (7, 44). The cationic c domain is not required for enzymatic activity (46) and ends with a C-terminal ER retention signal (KDEL in rat and human; HDEL in yeast) (60). The three-dimensional structure of intact PDI is not known, but structures of the individual a, a′, b, and b′ domains reveal that each has a fold characteristic of thioredoxin (Trx; Figure 1b) (41, 42).
Figure 1.

PDI is composed of four thioredoxin-like domains. a. The a and a′ domains each contain a CGHC active site. The b and b′ domains are likely involved in substrate binding. The c domain is cationic and ends with a C-terminal ER-retention sequence. The numbering system used here is based on the sequence of rat PDI (15). b. The thioredoxin fold of the oxidized a domain of PDI as revealed by NMR spectroscopy (41). The CGHC active site is at the N-terminus of an α helix.
PDI is a member of the Trx family of proteins, which is characterized by the ability to catalyze thiol–disulfide interchange reactions. Proteins of this family all share a common Cys–Xaa–Xaa–Cys (CXXC, where X refers to any amino acid) motif in their active site(s). The N-terminal cysteine residue in this motif has high reactivity at physiological pH, due in part to its location on the surface of the protein (Figure 1b) and its depressed pKa value (10). The family contains proteins with redox properties that range from strongly reducing (Trx, E°′ = –0.270 V (48)) to strongly oxidizing (DsbA, E°′ = –0.120 V (35, 36)). The proteins are therefore equipped to perform specialized roles in a variety of cellular compartments.
MECHANISM OF DISULFIDE ISOMERIZATION
A simple mechanism for disulfide isomerization requires only one reactive thiolate in the active site of PDI. In this scenario (Figure 2), the more reactive N-terminal cysteine residue of its CXXC motif (pKa = 6.7 (34)) provides the active-site thiolate. Nucleophilic attack of the thiolate on a nonnative disulfide bond results in the formation of a mixed-disulfide intermediate between enzyme and substrate (11, 12). Conformational changes in the substrate then allow a substrate thiolate to initiate disulfide rearrangements that lead, ultimately, to the formation of native disulfide bonds and the release of PDI. Based on this mechanism, a CXXS motif should be as efficient as a CXXC motif in catalysis of disulfide isomerization. Yet, experimental evidence indicates otherwise.
Figure 2.

Simple mechanism of disulfide isomerization (11). Isomerization begins with the nucleophilic attack of a thiolate provided by the catalyst (such as PDI) to form a mixed disulfide intermediate between catalyst and substrate. Additional intramolecular thiol–disulfide interchange reactions are performed by substrate thiolates. If these interchange reactions are slow, then a second thiol in the catalyst can provide an escape route, releasing trapped intermediates by reduction of the mixed disulfide bond (86). Native disulfide bonds can then be formed by reoxidation.
Eug1p, a homolog of PDI, is a nonessential lumenal protein in Saccharomyces cerevisiae that contains the active-site sequences CLHS and CIHS (80). The in vitro isomerase activity of wild-type Eug1p is low using both reduced procarboxypeptidase Y (proCPY) and scrambled proCPY (which contains non-native disulfide bonds) as substrates (63). In contrast, a variant of Eug1p in which the CXXS motifs are replaced with CXXC motifs is an efficient catalyst of the folding of scrambled proCPY (63). In addition, CXXC Eug1p catalyzes the folding of scrambled proCPY and reduced proCPY folding at an almost identical rate (63). These experiments support the earlier proposal that isomerization limits catalysis of oxidative folding by enzymes containing a CXXC motif (28), and indicate that the second cysteine residue enhances catalysis.
The second active-site cysteine residue is likely to be important in rescuing trapped intermediates that can accumulate during catalysis (Figure 2) (86). Complex protein substrates can be slow to rearrange and become trapped in nonnative configurations that prevent successful folding. The C-terminal CXXC cysteine residue provides a mechanism for escape from this obstacle to efficient folding. The presence of two cysteine residues permits PDI to reduce trapped nonnative disulfide bonds and then re-oxidize the substrate to form, ultimately, the native disulfide bonds. Recent results indeed indicate that the isomerization of scrambled RNase A (sRNase A, a complex substrate with 8 randomly oxidized cysteine residues) by PDI involves cycles of reduction and reoxidation (77).
REGULATING E°′ AND pKa
The ability of an oxidoreductase to be an efficient isomerase is governed by the reduction potential of its active-site disulfide bond (E°′) and the acid dissociation constant of its nucleophilic active-site thiol (Ka) (8). For efficient substrate turnover and regeneration of catalyst, an isomerase must strike a balance between the dithiol and disulfide forms. A E°′ value that is too low (that is, too negative) will destabilize the dithiol form and provide too little reactive thiolate to initiate attack of a nonnative disulfide bond. The CGPC active site of Trx, for example, has E°′ = –0.27 V (48) and is an effective reducing agent. An enzyme that contains an active site with a E°′ value that is too high will not be able to release trapped intermediates. The CPHC active site of DsbA has a E°′ = –0.12 V and is an effective oxidizing agent, but is inefficient at disulfide isomerization (Table 1) (35, 36, 99).
Table 1.
Properties of Oxidoreductases.
| Protein | Active-Site Sequence | Eo′ of CXXC (V) | pKa of CXXC | Primary role in the cell |
|---|---|---|---|---|
| Rat PDI | WCGHCK | –0.180a | 6.7b | oxidase/isomerase |
| E. coli Trx | WCGPCK | –0.270c | 7.5d | reductase |
| E. coli DsbA | FCPHCY | –0.120e | 3.3f | oxidase |
Determined from the equilibrium constant with GSH/GSSG and Trx (53).
Determined from the rate of inactivation by alkylation (34).
Determined from the equilibrium constant for the thioredoxin reductase-catalyzed reaction with NADPH/NADP+ (48).
Determined by 13C-NMR spectroscopy for the state in which Asp26 and Cys35 are protonated (10).
Determined by ultraviolet spectroscopy (36).
The thiol pKa, like the disulfide E°′, must be in balance. An active-site cysteine residue not only acts as a nucleophilic thiolate to initiate catalysis, but also acts as a leaving group to allow for thiol–disulfide interchange (27). Hence, an isomerase will be optimal if its thiol pKa equals the pH of the environment, which is near 7.0 in the ER (37). Although the pKa of a typical cysteine residue is 8.7 (79), a thiol pKa that is closer to 7.0 is more optimal for an oxidoreductase in the ER.
The N-terminal cysteine residue of the CXXC motif in thioredoxin-like enzymes consistently has a depressed pKa value (6.7 for PDI (34)). This pKa depression is consistent with the location of this residue at the N-terminus of an α-helix (Figure 1b) (21). The negative charge on the thiolate of the N-terminal cysteine residue is stabilized by interactions with the positive end of the α-helix dipole (47). Changes in the intervening –XX– residues can enhance or diminish this Coulombic interaction and thereby decrease or increase, respectively, the pKa of the N-terminal cysteine residue (Table 1) (8).
Variations in the –XX– residues of thioredoxin-like enzymes can also modulate the reduction potential of its active sites (Table 2). The variants can differ in thiol–disulfide interchange activity. Altering the residues in Trx can produce a more efficient oxidant (8, 32, 39, 48), while changing the residues in DsbA can make it a more potent reductant (32). The values of pKa and E°′ are related, as a more acidic thiol (lower pKa) produces a less stable disulfide bond (higher E°′) (9). For –XX– variants of DsbA, changes in the disulfide E°′ correlate well with changes in the pKa of the N-terminal cysteine residue of the active site (32). On the other hand, active-site variants of Trx with different intervening residues reveal that thiol pKa does not exclusively determine disulfide E°′ (Table 2) (8, 10). Three active-site variants of Trx (CGHC Trx, CVWC Trx, and CWGC Trx) are able to catalyze the isomerization of disulfide bonds in vivo (8). For these variants, the changes in E°′ do not correlate with changes in pKa (Table 2), which is consistent with the presence of a pKa-independent term in the relevant form of the Nernst equation (9, 10):
| (1) |
where E° is independent of pKa and refers to the standard reduction potential, αo refers to the fraction of fully protonated dithiol species, and and are the formal (i.e., total) concentrations for the reduced and oxidized molecules, respectively.
Table 2.
Properties of active-site variants of thioredoxin.
| Protein | pKa of CXXC | Eo′ of CXXC (V) | Relative doubling time of complemented pdi1Δ yeast |
|---|---|---|---|
| Yeast PDI | ND | ND | 1.0 |
| Rat PDI | 6.7a | –0.180b | 1.8 ± 0.2c |
| CGPC Trx | 7.5d | –0.270e | NC |
| CGPS Trx | ND | – | 4.3 ± 0.5f |
| CGHC Trx | ND | –0.235e | 4.4 ± 0.8f |
| CVWC Trx | 6.2d | –0.230g | 3.8 ± 0.4f |
| CWGC Trx | 6.1d | –0.200g | 2.2 ± 0.2f |
ND, not determined; NC, no complementation.
Determined from the rate of inactivation by alkylation (34).
Determined from the equilibrium constant with GSH/GSSG and Trx (53).
Data from ref (49).
Determined by 13C-NMR spectroscopy for the state in which Asp26 and Cys35 are protonated (10).
Determined from the equilibrium constant of the thioredoxin reductase-catalyzed reaction with NADPH/NADP+ (48).
Data from ref (8).
Determined from the equilibrium constant of the thioredoxin reductase-catalyzed reaction with NADPH/NADP+ (8).
The effective concentration of two thiols reports on their tendency to form an intramolecular disulfide bond. In thioredoxin-like proteins, the effective concentration of thiols in the active site is determined largely by the three-dimensional structure of the protein (3, 9). The insertion of a tryptophan residue in CVWC Trx and CWGC Trx could destabilize the active-site disulfide bond simply by providing steric hindrance to its formation. Alternatively, removal of the proline residue in the CVWC and CGHC variants could increase the reduction potential by increasing the conformational entropy of the polypeptide chain, and thereby stabilizing the reduced form relative to the oxidized form. From such changes, a set of homologous oxidoreductases has evolved to carry out a variety of redox functions in different cellular environments (Table 1).
DISULFIDE ISOMERIZATION—THE ESSENTIAL FUNCTION
PDI is required for the viability of S. cerevisiae (20, 50, 75, 81). Of all the cellular roles of PDI, its most important function is the isomerization of nonnative disulfide bonds (11, 49). Replacing PDI with a variant in which both active-site sequences are CGHS, instead of CGHC, restores viability to pdi1Δ S. cerevisiae (Table 3). Although this PDI variant has low dithiol oxidation activity and low disulfide reduction activity, it is proficient in its catalysis of disulfide isomerization (Table 3) (49, 85). A PDI variant that contains SGHC active-site sequences is unable to catalyze the formation, reduction, or isomerization of disulfide bonds, and does not complement a pdi1 deletion (Table 3).
Table 3.
Properties of active-site variants of protein disulfide isomerase.
| PDIa | Dithiol oxidation activityb | Disulfide reduction activityc | Disulfide isomerization activityd | Doubling time of complemented pdi1Δ S. cerevisiaee |
|---|---|---|---|---|
| CGHC | 100 | 100 | 100 | 1.8 ± 0.2 |
| CGHS | 3 | 6 | 93 | 2.3 ± 0.6 |
| SGHC | 0 | 3 | 4 | NC |
Data from ref (49).
NC, no complementation.
For each protein, the sequence indicated is present in both active sites of rat PDI.
Percentage of wild-type PDI activity for the activation of reduced RNase A.
Percentage of wild-type PDI activity for the reduction of the disulfide bonds in insulin.
Percentage of wild-type PDI activity for the activation of scrambled RNase A.
Relative to cells complemented with S. cerevisiae PDI.
Further support for the conclusion that isomerization is the essential function of PDI comes from in vivo studies with PDI homologs. Overproduction of Eug1p (with its CLHS and CIHS active sites) is able to rescue pdi1Δ S. cerevisiae, even though the isomerization activity of Eug1p is less than that of PDI (80). Trx is unable to restore viability to pdi1Δ yeast because of its low E°′ value (Table 2) (8). A Trx variant in which the active site is replaced with CGPS is, however, able to complement a pdi1 deletion (Table 2) (8). In Eug1p and CGPS Trx, catalysis of dithiol oxidation and disulfide reduction is not efficient because neither enzyme can form a disulfide bond in its active site. Nonetheless, both enzymes are able to catalyze disulfide isomerization and thereby endow a cell with the activity that is necessary for its survival.
DITHIOL OXIDATION
The process of native disulfide formation involves both the oxidation of dithiols to disulfides and the rearrangement of nonnative to native disulfide bonds. PDI is involved in both of these steps, but its contribution to each is not yet fully understood. In recent years, much focus has been on the oxidative role of PDI in the cell (24, 58, 83).
Ero1p (and its human homologs Ero1-Lα and Ero1-Lβ (4, 66)) is a membrane-associated ER protein that was discovered in a screen for proteins that confer resistance to dithiothreitol (DTT) when overproduced or cause sensitivity to DTT when altered (22, 70). Ero1p is an essential enzyme in yeast that oxidizes proteins in the ER (22, 70). Ero1p is specific in its choice of substrates, interacting with only a few proteins (23). Most importantly, Ero1p and PDI form a mixed disulfide in vivo (23). Ero1 in mammalian cells specifically oxidizes the C-terminal active site of PDI during the PDI-catalyzed retrotranslocation of cholera toxin (82). Mutational analysis of Ero1p has suggested that a CXXXXC motif is responsible for oxidizing PDI and other substrates, while a distinct CXXC motif reoxidizes its catalytic site (24). This finding, combined with evidence that PDI is involved in intermolecular disulfide bonds with its substrates (23, 59), suggests a pathway for the formation of disulfide bonds in newly synthesized proteins (Figure 3). In the lumen of the ER, FAD-bound Ero1p oxidizes PDI (83), which accumulates in its reduced form in the absence of Ero1p. Oxidized PDI then transfers oxidizing equivalents from Ero1p to reduced substrate proteins. Ero1p is then reoxidized by molecular oxygen (84). The oxidative power of Ero1p, and therefore the oxidative folding cycle, is regulated by levels of free FAD in the ER (84).
Figure 3.

The pathway of native disulfide formation in the lumen of the ER. FAD-bound Ero1p (83) (and presumably Ero1-Lα and Ero-Lβ in humans) specifically oxidizes PDI as well as ERp44 (human cells), Mpd2p (yeast), and perhaps other proteins (1, 23). Ero1p uses molecular oxygen to reoxidize itself for further folding cycles (84). Oxidized PDI catalyzes the formation of disulfide bonds in newly synthesized proteins; the thiolate form of reduced PDI catalyzes the isomerization of nonnative disulfide bonds (49). Proteins that do not achieve the native state are degraded rather than secreted. For simplicity, only one of the two active site of Ero1p and PDI is shown.
Contrary to previous assumptions, studies with Ero1p have revealed that the majority of PDI in the ER of S. cerevisiae exists in the oxidized state (23). To catalyze disulfide isomerization, PDI must be in its reduced form. The ER, which has a Esolution = –0.18 V (37), seems to be the optimum environment for PDI to carry out oxidative protein folding. With E°′ = –0.18 V, PDI at equilibrium in the ER would exist as an equimolar mixture of reduced and oxidized forms (53). The finding that most PDI in the ER of yeast is in the oxidized state suggests, however, that the majority of the enzyme is involved in disulfide formation rather than isomerization.
The situation appears to be quite different for human PDI. In three human cell lines (HeLa, COS, and U937) virtually all of the cellular PDI is reduced (58). The oxidized form of PDI only becomes detectable upon exposure of the cells to high levels of DTT. Perhaps, the redox system in human cells is more complex than is that in S. cerevisiae cells. For example, additional PDI and Ero homologs in human cells could allow PDI to be a specific catalyst of isomerization, as other enzymes take up the slack in oxidation.
BALANCING ISOMERIZATION AND OXIDATION
Although the majority of PDI in S. cerevisiae appears to be devoted to catalyzing the formation of disulfide bonds, isomerization of disulfide bonds limits the rate of native disulfide formation. This task is not accomplished by another enzyme in the absence of PDI. Yet, other proteins fulfill the oxidative role of PDI in pdi1Δ yeast, perhaps via the yeast unfolded protein response (UPR) pathway (8, 49, 62).
Only a small amount of isomerization activity is necessary for viability. The isolated a and a′ domains of rat PDI possess isomerization activity that is only 8.5% and 14%, respectively, that of wild-type PDI (13). These values agree well with the finding that having only a single reactive cysteine residue in the full-length protein results in a rate of sRNase A folding that is 10–12% lower than that of wild-type PDI (86). Even with this low activity, the isolated a and a′ domains are able to rescue pdi1Δ S. cerevisiae with growth rates that are comparable to those with wild-type PDI (98). When overproduced, Eug1p is able to suppress a PDI deficiency in yeast, even though the in vitro isomerase activity of this CXXS enzyme is poor (63, 80). Trx variants that complement a PDI deficiency are likely to be largely oxidized in the ER, and hence poor catalysts of isomerization (Table 2) (8, 10). Taken together, the available data indicate that a minimal amount of reactive thiolate is required to initiate isomerization reactions in misfolded proteins in the ER.
The physical properties of PDI are ideal for catalysis of disulfide formation and isomerization in the ER of eukaryotic cells (Figure 3). In contrast, bacteria have evolved two distinct systems to accomplish these tasks. In bacteria, the DsbA/DsbB system is responsible for the formation of disulfide bonds, while the DsbC/DsbD system accomplishes nonnative disulfide isomerization (16). DsbB is the oxidant for DsbA which can accept electrons from reduced substrates. If substrates form nonnative disulfide bonds, the periplasmic DsbC protein is responsible for their rearrangement. DsbC is maintained in a reduced state by the membrane bound protein, DsbD. The function of bacterial oxidoreductases is discussed further in the accompanying review by Ortenberg and Beckwith (64).
SMALL-MOLECULE MIMICS OF PDI
The primary determinants of isomerization efficiency are an E°′ near –0.18 V and a thiol pKa near 7.0. Using these criteria, it should be possible to design small-molecule mimics of Nature's enzymic isomerase. Such a small-molecule “foldase” would have a number of practical applications. Large-scale production of secretory proteins in E. coli is often complicated by aggregation or proteolysis associated with unfavorable conditions for disulfide formation in the bacterial cytosol (27, 55). Insoluble protein aggregates must be solublized and folded in vitro in an appropriate redox buffer. Glutathione is often used for this task, but is not an efficient catalyst of oxidative protein folding. PDI is not a practical catalyst for large-scale protein production due to the high cost of its production, its instability, and the necessity to separate it from a target protein. An effective small-molecule catalyst would be desirable for oxidative protein folding in vitro, as well as for addition directly to cell cultures to improve heterologous protein production (74).
Although only one cysteine residue is required for catalysis of disulfide isomerization, the presence of two cysteine residues in the active site enhances the overall effectiveness of PDI in the formation of native disulfide bonds (63, 86). Hence, it is reasonable to assume that a small-molecule dithiol would be a more active catalyst of protein folding than would an analogous monothiol. This assumption has been confirmed by experiments. The dithiol (±)-trans-1,2-bis(mercaptoacetamido)cyclohexane (BMC or Vectrase™-P) has thiol pKa1 = 8.3 and pKa2 = 9.9 and E°′ = –0.24 V (Table 4) (94). These values approach those of the CXXC motif in PDI, suggesting that this molecule may be efficient in catalyzing disulfide isomerization. Indeed, BMC increases both the rate of reactivation of sRNase A and the final yield of native RNase A over that attainable with glutathione (94). In addition, BMC (a dithiol) shows an increase in activity and recovery over that with N-methylmercaptoacetamide (NMA, a monothiol) (94). The presence of two sulfhydryl groups allows rescue from trapped mixed disulfides between catalyst and substrate, resulting in greater recovery of active RNase A and an increased rate of isomerization. An immobilized analog of BMC has similar attributes and provides another strategy for preparative protein folding in vitro (96).
Table 4.
Properties of small-molecule dithiol catalysts of disulfide isomerization.
| Molecule | Structure | First pKa | Eo′ (V) | Specific Activity (U/mol)a |
|---|---|---|---|---|
| BMC | 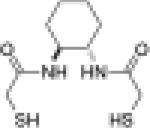 |
8.3b | –0.240b | 56c |
| CGC | 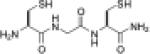 |
8.7d | –0.167d | 132c |
| CGC Trx | ND | ≥–0.200e | 3300c | |
| CGPC Trx | 7.5f | –0.270g | 0c |
ND, not determined.
One unit (U) of reduced catalyst activates 1 mmol of sRNase A per min in 0.1 M Tris–HCl buffer, pH 7.6, containing EDTA (1 mM).
pKa value was determined by ultraviolet spectroscopy; Eo′ value was determined by equilibration with β-mercaptoethanol/β-hydroxyethyldisulfide (94).
Data from ref (97).
pKa value was determined by ultraviolet spectroscopy; Eo′ value was determined by equilibration with β-mercaptoethanol/β-hydroxyethyldisulfide (97).
Determined from the equilibrium constant for the thioredoxin reductase-catalyzed reaction with NADPH/NADP+ (97).
Determined by 13C-NMR spectroscopy for the state in which Asp26 and Cys35 are protonated (10).
Determined from the equilibrium constant of the thioredoxin reductase-catalyzed reaction with NADPH/NADP+ (48).
BMC is also an effective disulfide isomerase in vivo. S. pombe acid phosphatase is a 30-kDa homodimer with eight disulfide bonds (74, 93). S. cerevisiae cells grown in the presence of BMC (0.1 g/L) secrete 3-fold more acid phosphatase, an increase equivalent to that achieved with 15-fold overproduction of PDI (94). Likewise, the presence of BMC (2 μg/L) in an E. coli culture medium increases the yield of proinsulin, which has three disulfide bonds, by 60% (92). These data indicate that a small-molecule mimic of PDI can enhance the yield of heterologous protein production in both eukaryotic and prokaryotic systems.
Aromatic thiols are also efficient small-molecule foldases. Aromatic thiols have low pKa values (pKa = 3–7) compared to their aliphatic counterparts (pKa = 7–11) and are more reactive for thiol–disulfide interchange reactions at physiological pH (90, 91). Of particular interest is 4-mercaptobenzeneacetate, which has a pKa = 6.6 close to that of active-site thiols in PDI (17). This reactive monothiol is able to catalyze the reactivation of sRNase A at a rate that is 5–6-fold higher than that of glutathione (31). Further rate enhancement could be attainable with an aromatic dithiol.
Other design strategies for small-molecule foldases are based more literally on the active site of PDI. Linear CXXC peptides that mimic the active sites of a variety of oxidoreductases exhibit reduction potentials that do not reflect the enzyme from which they were derived. The reduction potential of the disulfide bond in the W–CGPC–KHI peptide, for example, is 70 mV lower than that of native CGPC Trx (78). Varying the –XX– sequence of linear CXXC octapeptides results in minimal changes in its redox properties with E°′ values (calculated from equilibrium constants and a E°′ of –0.252 for glutathione (52)) in the range of –0.220 V to –0.200 V (78).
On the other hand, by restricting the conformational freedom of the active-site motif in a cyclic hexapeptide, CXXC-containing peptides can be more efficient oxidative protein folding catalysts (5). Three cyclic peptides, with sequences corresponding to glutaredoxin reductase (Grx), Trx, and PDI, show increasing activity and yield of native protein in a rRNase A assay. The enhancement in catalytic efficiency corresponds to a decrease in thiol pKa and an increase in E°′.
An alternative to changing the identity of intervening residues in a CXXC-containing peptide is to change the number of intervening residues in a C(X)nC-containing peptide. To obtain an effective isomerase, it is desirable to have a fairly unstable disulfide bond. CXC peptides, with only one intervening residue, form 11-membered disulfide-bonded rings and typically have E°′ values that are 30–40 mV lower than CXXC peptides (43, 102). A CGC peptide is a promising candidate for a small-molecule isomerase (97). Although the pKa of the reactive thiolate of CGC is higher than that of BMC, the reduction potential matches that of the PDI active site (Table 4). The CGC peptide is more efficient at folding sRNase A than is BMC. Even without imposing conformational constraints, a linear CGC is an effective catalyst of disulfide isomerization due to its favorable redox properties.
Small-molecule dithiols can act as isomerases, yet their activities lie far below that of PDI. Although the presence of a CXXC (or CXXS) motif is the only absolute requirement for isomerase activity, the protein scaffold provided by PDI increases the catalytic efficiency of the motif (87). Not only does the protein scaffold form a conformationally constrained active site that serves to regulate its redox properties, it also provides a means of noncovalent interaction. In addition to covalent rearrangements, catalysis of disulfide isomerization relies on noncovalent interactions between PDI and its substrates. The b′ domain of PDI provides a binding site for unfolded proteins (7, 44, 61). Mutagenesis studies of individual PDI domains have shown that when combined with the a′ domain, the b′ domain improves the rate of BPTI isomerization by 75% over that with the a′ domain alone (14). Further, full-length PDI is 7–12 fold more active in the folding of sRNase A than is the a or a′ domain alone (13).
Can an improved isomerase—one that combines the benefits of covalent and noncovalent interactions—be obtained by inserting a chemical catalyst into the context of a stable protein that has affinity for unfolded proteins? To answer this question, a CGC motif has been placed at the active site of Trx. Wild-type CGPC Trx has low activity in the catalysis of native disulfide formation due to the stability of its active-site disulfide bond (68). The disulfide bond destabilization imposed by deletion of a proline residue in the active site endows reduced CGC Trx with the ability to reactivate sRNase A in vitro (Table 4) (97).
The development of chemical catalysts that mimic the properties of PDI will be beneficial to both biotechnology and biomedicine. The production of eukaryotic proteins in E. coli often requires additional steps to obtain properly folded protein (55). Current techniques are neither time- nor cost-efficient. Small-molecule catalysts like BMC and CGC will provide better options for native disulfide formation during heterologous protein expression and purification from inclusion bodies. Defects in protein folding have been implicated in several diseases (18). For example, Alzheimer's disease coincides with a down-regulation of the ER stress response (38, 67), in which PDI is a key player (19). A small-molecule mimic of PDI could thus be a useful chemotherapeutic. Conversely, the catalytic activity of cell-surface PDI is necessary for the entry of HIV-1 virus into T lymphocytes (2, 26, 56). Hence, cell-surface PDI is a valid target for inhibitor development.
NEED FOR MORE MECHANISTIC UNDERSTANDING
What is the precise chemical mechanism by which PDI achieves disulfide formation and isomerization? What is the substrate specificity of PDI, and its molecular basis? How is PDI able to balance its role as a disulfide isomerase and dithiol oxidase? Two major obstacles hinder researchers seeking answers to these and related questions. First, the lack of a three-dimensional structure of intact PDI makes it difficult to obtain a clear picture of the active-site residues within the enzyme, as well as between the enzyme and its substrates. Second, substrates now available for studying disulfide isomerization are too complex to delineate the details of the chemical mechanism.
The structures of individual a and b domains of PDI are known (Figure 1b) (41, 42) and provide evidence for structural similarity between all four of the major domains. It is not yet understood either how the four domains interact with each other or how the two active sites communicate with each other. At saturating concentrations of substrate, the two active sites play different roles. The N-terminal active site is responsible for most of the catalytic activity, while the C-terminal active site contributes more to substrate binding (54). At substrate concentrations near the value of KM (where the rate is half-maximal) the two active sites are equally efficient catalysts and function independently (85). In contrast, communication between the two active sites plays an integral role in catalysis by E. coli DsbB (40) and S. cerevisiae Ero1p (24).
Structures of PDI homologs provide some intriguing proposals for the arrangement of individual PDI domains. CVWC Trx crystallizes in a dimer-like structure (76). This dimer is connected through a continuous β-sheet across both monomers and interlocked α-helices that cap the β-strands. Similarly, the structure of a disulfide oxidoreductase from Pyrococcus furiosus contains two CXXC-containing thioredoxin-like units that are joined by a continuous β-sheet (73). In both of these examples, adjacent thioredoxin-like units are arranged colinearly.
In PDI, the two CXXC motifs do not form disulfide bonds of equivalent stabilities. Circular dichroism (CD) studies of isolated catalytic domains suggest that the a′ domain contains a very unstable disulfide bond, whereas the disulfide bond of the a domain is relatively stable (13). Similarly, the structure of the P. furiosis enzyme reveals differences between the stabilities of the two active-site disulfide bonds (73). The more N-terminal disulfide bond is highly flexible due to conformational strain imposed by unfavorable dihedral angles. This flexibility results in a destabilized disulfide bond. The more C-terminal disulfide bond is more stable and has dihedral angles resembling that in other Trx homologs. The difference in stability between the two active-site disulfide bonds in PDI could reflect a difference in their physiological roles.
The structure of DsbC provides useful information on that of PDI (57). DsbC is the enzyme responsible for catalyzing disulfide isomerization in the bacterial periplasm (72, 100). DsbC is a dimeric protein, and, like the enzymes described above, its monomers are joined through a continuous β-sheet. Each monomer is composed of a catalytic N-terminal domain and a C-terminal dimerization domain. This arrangement is similar to the arrangement of domains in PDI in which the a and a′ domains are catalytic and the b and b′ domains serve another role. In the DsbC crystal structure (57), the N- and C-terminal domains are connected by a hinged α-helix that allows for flexibility of the catalytic domain. Between the two active sites is a broad uncharged cleft that can accommodate the nonspecific binding of misfolded substrate proteins. The flexibility provided by the hinged region allows for variation in the size of the cleft to allow binding of large or small substrate proteins. This structure provides a model for how the domains of PDI might interact to allow the nonspecific binding of substrates.
The structure and function of DsbC in a complex with the N-terminal domain of DsbD (DsbDα) provides insight on how a disulfide isomerase interacts with a protein substrate (30, 33). DsbDα binds in the cleft of DsbC, causing conformational changes in both the substrate and catalyst. Both active sites of DsbC contact DsbDα cysteine residues. The primary noncovalent interactions are between hydrophobic or uncharged polar groups.
Even with more structural information, a proper substrate for detailed mechanistic work is essential. Currently, the most common in vitro substrates for studying catalysis by PDI are RNase A and BPTI (11), neither of which is ideal. RNase A is a 124-residue protein with 8 cysteine residues that can be randomly oxidized under denaturing conditions to give sRNase A, a mixture of up to 105 (= 8C8 × 7 × 5 × 3) distinct fully oxidized species, only one of which is native. Moreover, RNase A can from 764 (= 8C8 × 7 × 5 × 3 + 8C6 × 5 × 3 + 8C4 × 3 + 8C2 + 8C0) different oxidized and reduced species, altogether. BPTI, a 58-residue protein, is a less complex substrate with only 6 cysteine residues. Still, BPTI can form 15 (= 6C6 × 5 × 3) distinct fully oxidized species.
Simple peptides have been used to study disulfide formation and reduction by PDI. A 28-residue peptide, which is based on the sequence of BPTI but with only one disulfide bond, is a substrate for catalysis of oxidation by PDI in the presence of glutathione (12). A fluorescently-labeled peptide of seven residues and one disulfide bond has been used to study the oxidation and reduction activities of PDI (88). Recently, a variant of green fluorescent protein (GFP) with a redox-sensitive disulfide bond has been developed as a tool to monitor disulfide bond formation in living cells (65).
Likewise, a simple substrate could be used to characterize disulfide isomerization by PDI in an in vitro assay. The ideal substrate would contain only two disulfide bonds and thus only three fully oxidized forms. To be a substrate for characterizing disulfide isomerization activity, the two disulfide bonds in the native form must be both stable under the conditions of a typical folding assay (Esolution = –0.18 V) and more stable (higher E°′) than those in the other two fully oxidized forms. A continuous folding assay could be used with such a substrate. For example, by modifiying the substrate to contain an appropriate donor and acceptor, fluorescence resonance energy transfer (FRET) could be used to monitor any conformational change that accompanies disulfide isomerization. Unlike the discontinuous assays currently available, a continuous folding assay would permit rapid, detailed analysis of the chemical and kinetic mechanism by which PDI catalyzes the isomerization of nonnative disulfide bonds.
CONCLUSIONS
Although disulfide bonds are responsible for stabilizing the native, active structure of many secretory proteins, the formation of the correct disulfide pairs is not always a simple task. As the number of cysteine residues in a protein increases, the chance of forming nonnative disulfide bonds also increases. PDI is an essential enzyme in eukaryotes that prevents the accumulation of nonnative and hence inactive species. By providing a reactive thiolate, PDI is able to catalyze disulfide isomerization, its essential function. With help from Ero1p, PDI is also able to play an important role in oxidizing newly synthesized proteins. An interesting topic for future research will be to determine how PDI is able to balance its roles as an isomerase and oxidase in the ER. Understanding the mechanism of disulfide isomerization will allow further development of small-molecule and enzymic isomerases that can be used to improve heterologous protein production and be adapted for biomedical purposes. Although the reduction potential and pKa are good indicators of thiolate reactivity, they do not fully explain the differences in effectiveness of catalysts of disulfide isomerization. A three-dimensional structure of the complete PDI protein would be invaluable in understanding how PDI recognizes, interacts, and converts substrates to native (active) proteins in the ER. Further mechanistic studies will also be aided by the development of simple two-disulfide substrates that could be used to test a variety of catalysts in a continuous disulfide isomerization assay.
ACKNOWLEDGMENT
Work on oxidative protein folding in the Raines laboratory was supported by grant BES04563 (NSF). E.A.K. was supported by WARF and Steenbock predoctoral fellowships, and Biotechnology Training Grant GM08349 (NIH). We are grateful to K. J. Woycechowsky for contributive discussions.
Abbreviations used
- BMC
(±)-trans-1,2-bis(mercaptoacetamido)cyclohexane
- BPTI
bovine pancreatic trypsin inhibitor
- DsbA
periplasmic protein thiol:disulfide oxidoreductase from Escherichia coli
- DsbDα
N-terminal domain of DsbD
- ER
endoplasmic reticulum
- FAD
flavin adenine dinucleotide
- Grx
glutaredoxin
- GSH
reduced glutathione (γ-Glu–Cys–Gly)
- GSSG
oxidized glutathione
- NADP+
nicotinamide adenine dinucleotide diphosphate, oxidized form
- NADPH
nicotinamide adenine dinucleotide diphosphate, reduced form
- NMA
N-methylmercaptoacetamide
- NMR
nuclear magnetic resonance
- PDI
protein disulfide isomerase
- proCPY
procarboxypeptidase Y
- RNase A
ribonuclease A
- rRNase A
reduced RNase A
- sRNase A
scrambled RNase A
- Trx
thioredoxin
REFERENCES
- 1.Anelli T, Alessio M, Mezghrani A, Simmen T, Talamo F, Bachi A, Sitia R. ERp44, a novel endoplasmic reticulum folding assistant of the thioredoxin family. EMBO J. 2002;21:835–844. doi: 10.1093/emboj/21.4.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barbouche R, Miquelis R, Jones IM, Fenouillet E. Protein-disulfide isomerase-mediated reduction of two disulfide bonds of HIV envelope glycoprotein 120 occurs post-CXCR4 binding and is required for fusion. J Biol Chem. 2003;278:3131–3136. doi: 10.1074/jbc.M205467200. [DOI] [PubMed] [Google Scholar]
- 3.Burns JA, Whitesides GM. Predicting the stability of cyclic disulfides by molecular modeling: “Effective concentrations” in thiol–disulfide interchange and the design of strongly reducing thiols. J Am Chem Soc. 1990;112:6296–6303. [Google Scholar]
- 4.Cabibbo A, Pagani M, Fabbri M, Rocchi M, Farmery M, Bulleid N, Sitia R. ERO1-L, a human protein that favors disulfide bond formation in the endoplasmic reticulum. J Biol Chem. 2000;275:4827–4833. doi: 10.1074/jbc.275.7.4827. [DOI] [PubMed] [Google Scholar]
- 5.Cabrele C, Fiori S, Pegorano S, Moroder L. Redox-active cyclic bis(cysteinyl) peptides as catalysts for in vitro oxidative protein folding. Chem Biol. 2002;9:731–740. doi: 10.1016/s1074-5521(02)00152-7. [DOI] [PubMed] [Google Scholar]
- 6.Cai H, Wang CC, Tsou CL. Chaperone-like activity of protein disulfide isomerase in the refolding of a protein with no disulfide bonds. J Biol Chem. 1994;2697:24550–24552. [PubMed] [Google Scholar]
- 7.Cheung PY, Churchich JE. Recognition of protein substrates by protein-disulfide isomerase. J Biol Chem. 1999;274:32757–32761. doi: 10.1074/jbc.274.46.32757. [DOI] [PubMed] [Google Scholar]
- 8.Chivers PT, Laboissière MCA, Raines RT. The CXXC motif: Imperatives for the formation of native disulfide bonds in the cell. EMBO J. 1996;16:2659–2667. [PMC free article] [PubMed] [Google Scholar]
- 9.Chivers PT, Prehoda KE, Raines RT. The CXXC motif: A rheostat in the active site. Biochemistry. 1997;36:4061–4066. doi: 10.1021/bi9628580. [DOI] [PubMed] [Google Scholar]
- 10.Chivers PT, Prehoda KE, Volkman B, Kim B-M, Markley JL, Raines RT. Microscopic pKa values of Escherichia coli thioredoxin. Biochemistry. 1997;36:14985–14991. doi: 10.1021/bi970071j. [DOI] [PubMed] [Google Scholar]
- 11.Chivers PT, Laboissière MCA, Raines RT. Protein disulfide isomerase: Cellular enzymology of the CXXC motif. In: Guzman NA, editor. Prolyl Hydroxylase, Protein Disulfide Isomerase, and Other Structurally-Related Proteins. Marcel Dekker; New York: 1998. pp. 487–505. [Google Scholar]
- 12.Darby NJ, Freedman RB, Creighton TE. Dissecting the mechanism of protein disulfide isomerase: Catalysis of disulfide bond formation in a model peptide. Biochemistry. 1994;33:7937–7947. doi: 10.1021/bi00191a022. [DOI] [PubMed] [Google Scholar]
- 13.Darby NJ, Creighton TE. Functional properties of the individual thioredoxin-like domains of protein disulfide isomerase. Biochemistry. 1995;34:11725–11735. doi: 10.1021/bi00037a009. [DOI] [PubMed] [Google Scholar]
- 14.Darby NJ, Penka E, Vincentelli R. The multi-domain structure of protein disulfide isomerase is essential for high catalytic efficiency. J Mol Biol. 1998;276:239–247. doi: 10.1006/jmbi.1997.1504. [DOI] [PubMed] [Google Scholar]
- 15.Darby NJ, van Straaten M, Penka E, Vincentelli R, Kemmink J. Identifying and characterizing a second structural domain of protein disulfide isomerase. FEBS Lett. 1999;448:167–172. doi: 10.1016/s0014-5793(99)00374-9. [DOI] [PubMed] [Google Scholar]
- 16.Debarbieux L, Beckwith J. Electron avenue: Pathways of disulfide bond formation and isomerization. Cell. 1999;99:117–119. doi: 10.1016/s0092-8674(00)81642-6. [DOI] [PubMed] [Google Scholar]
- 17.DeCollo TV, Lees WJ. Effects of aromatic thiols on thiol–disulfide interchange reactions that occur during protein folding. J Org Chem. 2001;66:4244–4249. doi: 10.1021/jo015600a. [DOI] [PubMed] [Google Scholar]
- 18.Dobson CM. Protein folding and its links with human disease. Biochem Soc Symp. 2001:1–26. [PubMed] [Google Scholar]
- 19.Dorner AJ, Wasley LC, Raney P, Haugejorden S, Green M, Kaufman RJ. The stress response in Chinese hamster ovary cells. Regulation of ERp72 and protein disulfide isomerase expression and secretion. J Biol Chem. 1990;265:22029–22034. [PubMed] [Google Scholar]
- 20.Farquhar R, Honey H, Murant SJ, Bossier P, Schultz L, Montgomery D, Ellis RW, Freedman RB, Tuite MF. Protein disulfide isomerase is essential for viability in Saccharomyces cerevisiae. Gene. 1991;108:81–89. doi: 10.1016/0378-1119(91)90490-3. [DOI] [PubMed] [Google Scholar]
- 21.Forman-Kay JD, Clore GM, Wingfield PT, Gronenborn AM. High-resolution three-dimensional structure of reduced recombinant human thioredoxin in solution. Biochemistry. 1991;30:2685–2698. doi: 10.1021/bi00224a017. [DOI] [PubMed] [Google Scholar]
- 22.Frand AR, Kaiser CA. The ERO1 gene of yeast is required for oxidation of protein dithiols in the endoplasmic reticulum. Mol Cell. 1998;1:161–170. doi: 10.1016/s1097-2765(00)80017-9. [DOI] [PubMed] [Google Scholar]
- 23.Frand AR, Kaiser CA. Ero1p oxidizes protein disulfide isomerase in a pathway for disulfide bond formation in the endoplasmic reticulum. Mol Cell. 1999;4:469–477. doi: 10.1016/s1097-2765(00)80198-7. [DOI] [PubMed] [Google Scholar]
- 24.Frand AR, Kaiser CA. Two pairs of conserved cysteines are required for the oxidative activity of Ero1p in protein disulfide bond formation in the endoplasmic reticulum. Mol Biol Cell. 2000;11:2833–2843. doi: 10.1091/mbc.11.9.2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Freedman RB, Hirst TR, Tuite MF. Protein disulphide isomerase: Building bridges in protein folding. Trends Biochem Sci. 1994;19:331–336. doi: 10.1016/0968-0004(94)90072-8. [DOI] [PubMed] [Google Scholar]
- 26.Gallina A, Hanley TM, Mandel R, Trahey M, Broder CC, Viglianti GA, Ryser HJ-P. Inhibitors of protein-disulfide isomerase prevent cleavage of disulfide bonds in receptor-bound glycoprotein 120 and prevent HIV-1 entry. J Biol Chem. 2002;277:50579–50588. doi: 10.1074/jbc.M204547200. [DOI] [PubMed] [Google Scholar]
- 27.Gilbert HF. Molecular and cellular aspects of thiol–disulfide exchange. Adv Enzymol. 1990;63:69–172. doi: 10.1002/9780470123096.ch2. [DOI] [PubMed] [Google Scholar]
- 28.Givol D, Goldberger RF, Anfinsen CB. Oxidation and disulfide interchange in the reactivation of reduced ribonuclease. J Biol Chem. 1964;239:PC3114–3116. [PubMed] [Google Scholar]
- 29.Goldberger RF, Epstein CJ, Anfinsen CB. Acceleration of reactivation of reduced bovine pancreatic ribonuclease by a microsomal system from rat liver. J Biol Chem. 1963;238:628–635. [PubMed] [Google Scholar]
- 30.Goldstone D, Haebel PW, Katzen F, Bader MW, Bardwell JC, Beckwith J, Metcalf P. DsbC activation by the N-terminal domain of DsbD. Proc Natl Acad Sci USA. 2001;98:9551–9556. doi: 10.1073/pnas.171315498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gough JD, Williams JRH, Donofrio AE, Lees WJ. Folding disulfide-containing proteins faster with an aromatic thiol. J Am Chem Soc. 2002;124:3885–3892. doi: 10.1021/ja016938p. [DOI] [PubMed] [Google Scholar]
- 32.Grauschopf U, Winther JR, Korber P, Zander T, Dallinger P, Bardwell JCA. Why is DsbA such an oxidizing disulfide catalyst? Cell. 1995;83:947–955. doi: 10.1016/0092-8674(95)90210-4. [DOI] [PubMed] [Google Scholar]
- 33.Haebel PW, Goldstone D, Katzen F, Beckwith J, Metcalf P. The disulfide bond isomerase DsbC is activated by an immunoglobulin-fold thiol oxidoreductase: Crystal structure of the DsbC–DsbDα complex. EMBO J. 2002;21:4774–4784. doi: 10.1093/emboj/cdf489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hawkins HC, Freedman RB. The reactivities and ionization properties of the active-site dithiol groups of mammalian protein disulphide-isomerase. Biochem J. 1991;275:335–339. doi: 10.1042/bj2750335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hennecke J, Sillen A, Huber-Wunderlich M, Engelborghs Y, Glockshuber R. Quenching of tryptophan fluorescence by the active-site disulfide bridge in the DsbA protein from Escherichia coli. Biochemistry. 1997;36:6391–6400. doi: 10.1021/bi963017w. [DOI] [PubMed] [Google Scholar]
- 36.Huber-Wunderlich M, Glockshuber R. A single dipeptide sequence modulates the redox properties of a whole enzyme family. Fold Des. 1998;3:161–171. doi: 10.1016/S1359-0278(98)00024-8. [DOI] [PubMed] [Google Scholar]
- 37.Hwang C, Sinskey AJ, Lodish HF. Oxidized redox state of glutathione in the endoplasmic reticulum. Science. 1992;257:1496–1502. doi: 10.1126/science.1523409. [DOI] [PubMed] [Google Scholar]
- 38.Imaizumi K, Miyoshi K, Katayama T, Yoneda T, Taniguchi M, Kudo T, Tohyama M. The unfolded protein response and Alzheimer's disease. Biochim Biophys Acta. 2001;1536:85–96. doi: 10.1016/s0925-4439(01)00049-7. [DOI] [PubMed] [Google Scholar]
- 39.Joelson T, Sjöberg BM, Eklund H. Modifications of the active center of T4 thioredoxin by site-directed mutagenesis. J Biol Chem. 1990;265:3183–3188. [PubMed] [Google Scholar]
- 40.Kadokura H, Beckwith J. Four cysteines of the membrane protein DsbB act in concert to oxidize its substrate DsbA. EMBO J. 2002;21:2354–2363. doi: 10.1093/emboj/21.10.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kemmink J, Darby NJ, Dijkstra K, Nilges M, Creighton TE. Structure determination of the N-terminal thioredoxin-like domain of protein disulfide isomerase using multidimensional heteronuclear 13C/15N NMR spectroscopy. Biochemistry. 1996;35:7684–7691. doi: 10.1021/bi960335m. [DOI] [PubMed] [Google Scholar]
- 42.Kemmink J, Darby NJ, Dijkstra K, Nilges M, Creighton TE. The folding catalyst protein disulfide isomerase is constructed of active and inactive thioredoxin modules. Curr Biol. 1997;7:239–245. doi: 10.1016/s0960-9822(06)00119-9. [DOI] [PubMed] [Google Scholar]
- 43.Kishore R, Balaram P. Stabilization of γ-turn conformations in peptides by disulfide bridging. Biopolymers. 1985;24:2041–2043. doi: 10.1002/bip.360241104. [DOI] [PubMed] [Google Scholar]
- 44.Klappa P, Ruddock LW, Darby NJ, Freedman RB. The b’ domain provides the principal peptide-binding site of protein disulfide isomerase but all domains contribute to binding misfolded proteins. EMBO J. 1998;17:927–935. doi: 10.1093/emboj/17.4.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koivu J, Myllylä R, Helaakoski T, Pihlajaniemi T, Tasanen K, Kivirikko KI. A single polypeptide acts both as the β subunit of prolyl 4-hydroxylase and as a protein disulfide isomerase. J Biol Chem. 1987;262:6447–6449. [PubMed] [Google Scholar]
- 46.Koivunen P, Pirneskoski A, Karvonen P, Ljung J, Helaakoski T, Notbohm H, Kivirikko KI. The acidic C-terminal domain of protein disulfide isomerase is not critical for the enzyme subunit function or for the chaperone or disulfide isomerase activities of the polypeptide. EMBO J. 1999;18:65–74. doi: 10.1093/emboj/18.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kortemme T, Creighton TE. Ionisation of cysteine residues at the termini of model α-helical peptides. Relevance to unusual thiol pKa values in proteins of the thioredoxin family. J Mol Biol. 1995;253:799–812. doi: 10.1006/jmbi.1995.0592. [DOI] [PubMed] [Google Scholar]
- 48.Krause G, Lundström J, Barea JL, Pueyo de la Cuesta C, Holmgren A. Mimicking the active site of protein disulfide-isomerase by substitution of proline 34 in Escherichia coli thioredoxin. J Biol Chem. 1991;266:9494–9500. [PubMed] [Google Scholar]
- 49.Laboissière MCA, Sturley SL, Raines RT. The essential function of protein-disulfide isomerase is to unscramble non-native disulfide bonds. J Biol Chem. 1995;270:28006–28009. doi: 10.1074/jbc.270.47.28006. [DOI] [PubMed] [Google Scholar]
- 50.LaMantia M, Miura T, Tachikawa H, Kaplan HW, Lennarz WJ, Mizunaga T. Glycosylation site binding protein and protein disulfide isomerase are identical and essential for cell viability in yeast. Proc Natl Acad Sci USA. 1991;88:4453–4457. doi: 10.1073/pnas.88.10.4453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lamberg A, Jauhiainen M, Metso J, Ehnholm C, Shoulders C, Scott J, Pihlajaniemi T, Kivirikko KI. The role of protein disulphide isomerase in the microsomal triacylglycerol transfer protein does not reside in its isomerase activity. Biochem J. 1996;315:533–536. doi: 10.1042/bj3150533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lees WJ, Whitesides GM. Equilibrium constants for thiol–disulfide interchange reactions: A coherent, corrected set. J Org Chem. 1993;58:642–647. [Google Scholar]
- 53.Lundström J, Holmgren A. Determination of the reduction-oxidation potential of the thioredoxin-like domains of protein disulfide-isomerase from the equilibrium with glutathione and thioredoxin. Biochemistry. 1993;32:6649–6655. doi: 10.1021/bi00077a018. [DOI] [PubMed] [Google Scholar]
- 54.Lyles MM, Gilbert HF. Mutations in the thioredoxin sites of protein disulfide isomerase reveal functional nonequivalence of the N- and C-terminal domains. J Biol Chem. 1994;269:30946–30952. [PubMed] [Google Scholar]
- 55.Marston FAO. The purification of eukaryotic proteins synthesized in Escherichia coli. Biochem J. 1986;240:1–12. doi: 10.1042/bj2400001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Matthias LJ, Yam PTW, Jiang X-M, Vandegraaff N, Li P, Poumbourios P, Donoghue N, Hogg PJ. Disulfide exchange in domain 2 of CD4 is required for entry of HIV-1. Nature Immunol. 2002;3:727–710. doi: 10.1038/ni815. [DOI] [PubMed] [Google Scholar]
- 57.McCarthy AA, Haebel PW, Törrönen A, Rybin V, Baker EN, Metcalf P. Crystal structure of the protein disulfide bond isomerase DsbC from Escherichia coli. Nat Struct Biol. 2000;7:196–199. doi: 10.1038/73295. [DOI] [PubMed] [Google Scholar]
- 58.Mezghrani A, Fassio A, Benham A, Simmen T, Braakman I, Sitia R. Manipulation of oxidative protein folding and PDI redox state in mammalian cells. EMBO J. 2001;20:6288–6296. doi: 10.1093/emboj/20.22.6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Molinari M, Helenius A. Glycoproteins form mixed disulfides with oxidoreductases during folding in living cells. Nature. 1999;402:90–93. doi: 10.1038/47062. [DOI] [PubMed] [Google Scholar]
- 60.Munro S, Pelham HRB. A C-terminal signal prevents secretion of luminal ER proteins. Cell. 1987;48:899–907. doi: 10.1016/0092-8674(87)90086-9. [DOI] [PubMed] [Google Scholar]
- 61.Noiva R, Freedman RB, Lennarz WJ. Peptide binding to protein disulfide isomerase occurs at a site distinct from the active sites. J Biol Chem. 1993;268:19210–19217. [PubMed] [Google Scholar]
- 62.Nørgaard P, Westphal V, Tachibana C, Alsoe L, Holst B, Winther JR. Functional differences in yeast protein disulfide isomerases. J Cell Biol. 2001;152:553–562. doi: 10.1083/jcb.152.3.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nørgaard P, Winther JR. Mutation of yeast Eug1p CXXS active sites to CXXC results in a dramatic increase in protein disulphide isomerase activity. Biochem J. 2001;358:269–274. doi: 10.1042/0264-6021:3580269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ortenberg R, Beckwith J. Antioxid Redox Signal. 2003;5 doi: 10.1089/152308603768295140. In Press. [DOI] [PubMed] [Google Scholar]
- 65.Østergaard H, Henriksen A, Hansen FG, Winther JR. Shedding light on disulfide bond formation: Engineering a redox switch in green fluorescent protein. EMBO J. 2001;20:5853–5862. doi: 10.1093/emboj/20.21.5853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pagani M, Fabbri M, Benedetti C, Fassio A, Pilati S, Bulleid NJ, Cabbibo A, Sitia R. Endoplasmic reticulum oxidoreductin 1-Lβ (ERO1-Lβ), a human gene iduced in the course of the unfolded protein response. J Biol Chem. 2000;275:23685–23692. doi: 10.1074/jbc.M003061200. [DOI] [PubMed] [Google Scholar]
- 67.Paschen W, Frandsen A. Endoplasmic reticulum dysfunction—a common denominator for cell injury in acute and degenerative diseases of the brain? J Neurochem. 2001;79:719–725. doi: 10.1046/j.1471-4159.2001.00623.x. [DOI] [PubMed] [Google Scholar]
- 68.Pigiet VP, Schuster BJ. Thioredoxin-catalyzed refolding of disulfide-containing proteins. Proc Natl Acad Sci USA. 1986;83:7643–7647. doi: 10.1073/pnas.83.20.7643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pihlajaniemi T, Helaakoski T, Tasanen K, Myllylä R, Huhtala M-L, Koivu J, Kivirikko KI. Molecular cloning of the β-subunit of human prolyl 4-hydroxylase. This subunit and protein disulphide isomerase are products of the same gene. EMBO J. 1987;6:643–649. doi: 10.1002/j.1460-2075.1987.tb04803.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pollard MG, Travers KJ, Weissman JS. Ero1p: A novel and ubiquitous protein with an essential role in oxidative protein folding in the endoplasmic reticulum. Mol Cell. 1998;1:171–182. doi: 10.1016/s1097-2765(00)80018-0. [DOI] [PubMed] [Google Scholar]
- 71.Puig A, Gilbert HF. Protein disulfide isomerase exhibits chaperone and anti-chaperone activity in the oxidative refolding of lysozyme. J Biol Chem. 1994;269:7764–7771. [PubMed] [Google Scholar]
- 72.Reitch A, Belin D, Martin N, Beckwith J. An in vivo pathway for disulfide bond isomerization in Escherichia coli. Proc Natl Acad Sci USA. 1996;93:13048–13053. doi: 10.1073/pnas.93.23.13048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ren B, Tibbelin G, Pascale Dd, Rossi M, Bartolucci S, Ladenstein R. A protein disulfide oxidoreductase from the archaeon Pyrococcus furiosus contains two thioredoxin fold units. Nature Struct Biol. 1998;5:602–611. doi: 10.1038/862. [DOI] [PubMed] [Google Scholar]
- 74.Robinson AS, Hines V, Wittrup KD. Protein disulfide isomerase overexpression increases secretion of foreign proteins in Saccharomyces cerevisiae. BioTechnology. 1994;12:381–384. doi: 10.1038/nbt0494-381. [DOI] [PubMed] [Google Scholar]
- 75.Scherens B, Dubois E, Messenguy F. Determination of the sequence of the yeast YCL313 gene localized on chromosome III. Homology with the protein disulfide isomerase (PDI gene product) of other organisms. Yeast. 1991;7:185–193. doi: 10.1002/yea.320070212. [DOI] [PubMed] [Google Scholar]
- 76.Schultz LW, Chivers PT, Raines RT. The CXXC motif: Crystal structure of an active-site variant of Escherichia coli thioredoxin. Acta Cryst. 1999;D55:1533–1538. doi: 10.1107/s0907444999008756. [DOI] [PubMed] [Google Scholar]
- 77.Schwaller M, Wilkinson B, Gilbert HF. Reduction–reoxidation cycles contribute to catalysis of disulfide isomerization by protein-disulfide isomerase. J Biol Chem. 2003;278:7154–7159. doi: 10.1074/jbc.M211036200. [DOI] [PubMed] [Google Scholar]
- 78.Siedler F, Rudolph-Böhner S, Doi M, Musiol H-J, Moroder L. Redox potentials of active-site bis(cysteinyl) fragments of thiol-protein oxidoreductases. Biochemistry. 1993;32:7488–7495. doi: 10.1021/bi00080a021. [DOI] [PubMed] [Google Scholar]
- 79.Szajewski RP, Whitesides GM. Rate constants and equilibrium constants for thiol–disulfide interchange reactions involving oxidized glutathione. J Am Chem Soc. 1980;102:2011–2026. [Google Scholar]
- 80.Tachibana C, Stevens TH. The yeast EUG1 gene encodes an endoplasmic reticulum protein that is functionally related to protein disulfide isomerase. Mol Cell Biol. 1992;12:4601–4611. doi: 10.1128/mcb.12.10.4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tachikawa H, Miura T, Katakura Y, Mizunaga T. Molecular structure of a yeast gene, PDI1, encoding protein disulfide isomerase that is essential for cell growth. J Biochem. 1991;110:306–313. doi: 10.1093/oxfordjournals.jbchem.a123576. [DOI] [PubMed] [Google Scholar]
- 82.Tsai B, Rapoport TA. Unfolded cholera toxin is transferred to the ER membrane and released from protein disulfide isomerase upon oxidation by Ero1. J Cell Biol. 2002;159:207–216. doi: 10.1083/jcb.200207120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tu BP, Ho-Schleyer SC, Travers KJ, Weissman JS. Biochemical basis of oxidative protein folding in the endoplasmic reticulum. Science. 2000;290:1571–1574. doi: 10.1126/science.290.5496.1571. [DOI] [PubMed] [Google Scholar]
- 84.Tu BP, Weissman JS. The FAD- and O2-dependent reaction cycle of Ero1-mediated oxidative protein folding in the endoplasmic reticulum. Mol Cell. 2002;10:983–994. doi: 10.1016/s1097-2765(02)00696-2. [DOI] [PubMed] [Google Scholar]
- 85.Walker KW, Lyles MM, Gilbert HF. Catalysis of oxidative protein folding by mutants of protein disulfide isomerase with a single active-site cysteine. Biochemistry. 1996;35:1972–1980. doi: 10.1021/bi952157n. [DOI] [PubMed] [Google Scholar]
- 86.Walker KW, Gilbert HF. Scanning and escape during protein-disulfide isomerase-assisted protein folding. J Biol Chem. 1997;272:8845–8848. doi: 10.1074/jbc.272.14.8845. [DOI] [PubMed] [Google Scholar]
- 87.Weissman JS, Kim PS. Efficient catalysis of disulphide bond rearrangements by protein disulphide isomerase. Nature. 1993;365:185–188. doi: 10.1038/365185a0. [DOI] [PubMed] [Google Scholar]
- 88.Westphal V, Spetzler JC, Meldal M, Christensen U, Winther JR. Kinetic analysis of the mechanism and specificity of protein-disulfide isomerase using fluorescence-quenched peptides. J Biol Chem. 1998;273:24992–24999. doi: 10.1074/jbc.273.39.24992. [DOI] [PubMed] [Google Scholar]
- 89.Wetterau JR, Combs KA, Spinner SN, Joiner BJ. Protein disulfide isomerase is a component of the microsomal triglyceride transfer protein complex. J Biol Chem. 1990;265:9800–9807. [PubMed] [Google Scholar]
- 90.Whitesides GM, Lilburn JE, Szajewski RP. Rates of thiol–disulfide interchange reactions between mono- and dithiols and Ellman's reagent. J Org Chem. 1977;42:332–338. [Google Scholar]
- 91.Wilson JM, Bayer RJ, Hupe DJ. Structure–reactivity correlations for the thiol–disulfide interchange reaction. J Am Chem Soc. 1977;99:7922–7926. [Google Scholar]
- 92.Winter J, Lilie H, Rudolph R. Recombinant expression and in vitro folding of proinsulin are stimulated by the synthetic dithiol Vectrase-P. FEMS Microbiol Lett. 2002;213:225–230. doi: 10.1111/j.1574-6968.2002.tb11310.x. [DOI] [PubMed] [Google Scholar]
- 93.Wittrup KD. Disulfide bond formation and eukaryotic secretory productivity. Curr Opin Biotechnol. 1995;6:203–208. doi: 10.1016/0958-1669(95)80033-6. [DOI] [PubMed] [Google Scholar]
- 94.Woycechowsky KJ, Wittrup KD, Raines RT. A small-molecule catalyst of protein folding in vitro and in vivo. Chem Biol. 1999;6:871–879. doi: 10.1016/s1074-5521(00)80006-x. [DOI] [PubMed] [Google Scholar]
- 95.Woycechowsky KJ, Raines RT. Native disulfide bond formation in proteins. Curr Opin Chem Biol. 2000;4:533–539. doi: 10.1016/s1367-5931(00)00128-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Woycechowsky KJ, Hook BA, Raines RT. Catalysis of protein folding by an immobilized small-molecule dithiol. Biotechnol Prog. 2003;19 doi: 10.1021/bp0257123. In Press. [DOI] [PubMed] [Google Scholar]
- 97.Woycechowsky KJ, Raines RT. The CXC motif: A functional mimic of protein disulfide isomerase. Biochemistry. 2003;42 doi: 10.1021/bi026993q. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Xiao R, Solovyov A, Gilbert HF, Holmgren A, Lundström-Ljung J. Combinations of protein-disulfide isomerase domains show that there is little correlation between isomerase activity and wild-type growth. J Biol Chem. 2001;276:27975–27980. doi: 10.1074/jbc.M104203200. [DOI] [PubMed] [Google Scholar]
- 99.Zapun A, Bardwell JC, Creighton TE. The reactive and destabilizing disulfide bond of DsbA, a protein required for protein disulfide bond formation in vivo. Biochemistry. 1993;32:5083–5092. doi: 10.1021/bi00070a016. [DOI] [PubMed] [Google Scholar]
- 100.Zapun A, Missiakas D, Raina S, Creighton TE. Structural and functional characterization of DsbC, a protein involved in disulfide bond formation in Escherichia coli. Biochemistry. 1995;34:5075–5089. doi: 10.1021/bi00015a019. [DOI] [PubMed] [Google Scholar]
- 101.Zapun A, Jakob CA, Thomas DY, Bergeron JJ. Protein folding in a specialized compartment: The endoplasmic reticulum. Structure Fold Des. 1999;7:R173–182. doi: 10.1016/s0969-2126(99)80112-9. [DOI] [PubMed] [Google Scholar]
- 102.Zhang R, Snyder GH. Dependence of formation of small disulfide loops in two-cysteine peptides on the number and types of intervening amino acids. J Biol Chem. 1989;264:18472–18479. [PubMed] [Google Scholar]