

Abstract
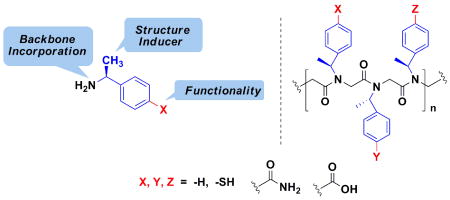
Peptoids, oligo-N-substituted glycines, can fold into well-defined helical secondary structures. The design and synthesis of new peptoid building blocks that are capable of both (a) inducing a helical secondary structure, and (b) decorating the helices with chemical functionalities is reported. Peptoid heptamers containing carboxamide, carboxylic acid or thiol functionalities were synthesized, and the resulting peptoids were shown to form stable helices. A thiol-containing peptoid readily formed the homo-disulfide, providing a convenient route to prepare peptoid helix homodimers.
One of the hallmarks of protein structure is precise specificity of heteropolymer sequence. A set of chemically diverse amino acid building blocks comprise the sequence and encode basic information; then, the sequence undergoes folding to obtain a distinct structure and a function.1 For decades, scientists have attempted to mimic the exquisite natural system and to generate protein-like structures and functions from non-natural heteropolymers.2 To that end, a prerequisite would be to have equivalents of amino acids that have a secondary structure-inducing element as well as a variety of chemical functional groups.
Peptoids are a class of biomimetic polymers based on oligo-N-substituted glycine backbones, designed to mimic peptides and proteins. 3 Peptoids can be efficiently synthesized with precisely defined sequences and chain lengths (up to ∼50 monomers)4 employing solid-phase submonomer synthesis protocol (Figure 2(a)).5 Peptoids can form well-defined three-dimensional folds in solution: (1) peptoid oligomers with α-chiral sidechains were shown to adopt helical structures;6 (2) a threaded loop structure was formed by intramolecular hydrogen bonds in peptoid nonamers;7 and (3) head-to-tail macrocyclizations provided conformationally restricted cyclic peptoids.8
Figure 2.

Synthesis of functionalized peptoid heptamers.
Understanding the factors that influence peptoid conformation has been a central theme of peptoid research. Previously, we elucidated the dependence of oligomer sequence,9 chain length,10 and solvent composition7,11 on the formation of the helical or threaded loop conformations. More recently, Blackwell and co-workers demonstrated the ability to control peptoid secondary structure using electron-deficient submonomers such as (S)-1-(pentafluorophenyl)ethylamine 12 or (S)-1-(2-nitro-phenyl)ethylamine.13 In particular, they provided strong evidence that an electronic n→π* interaction at the monomer level was essential to the backbone cis/trans isomerism 14 and, potentially, to the global peptoid conformation. The role of monomer units on peptoid folding was also investigated by Kirshenbaum et al. They introduced novel submonomers such as (1) (S)-N-(1-carboxy-2-phenylethyl)glycine 15 that provided pH dependent conformational change in peptoid secondary structure and (2) N-aryl glycines16 that could control the backbone cis/trans isomerism.
Utilizing the relationship between peptoid monomer sequence and adopted secondary structure, functional peptoid foldamers have been developed including antimicrobial peptoids, 17 pulmonary surfactant protein mimics, 18 asymmetric catalysts, 19 and zinc binding peptoids.20 These studies demonstrate the importance of (1) access to chemically diverse monomer units and (2) precise control of secondary structures to expand applications of peptoid helices.
To form secondary and tertiary structures, natural proteins utilize non-covalent interactions (i.e. hydrophobic, electrostatic, and hydrogen bonds) and covalent bonds (i.e. disulfides). Inspired by the natural system, our goal is to build higher-order structures by modulating monomer functionality and sequence.2, 21 In this study, we present the synthesis of several novel peptoid submonomers that are capable of displaying functional groups as well as inducing a helical conformation. A sophisticated decoration of the peptoid helix can promote interactions between helices and can therefore be used to create novel peptoid-based ordered constructs.
As shown in Figure 1, three new peptoid submonomers were designed.22 These new submonomers are derivatives of (S)-1-phenylethylamine, or Nspe, which can be readily incorporated into a peptoid and induces a stable helical fold.6a Functionalized peptoids containing the new submonomers can form non-covalent bonds (i.e., hydrogen bonds) as well as covalent bonds (i.e., disulfide bonds, metal-ligand interactions). In addition, they can modulate the hydrophobicity and water solubility11,15 of the peptoid helix.
Figure 1.

Peptoid submonomer design.
Thiol submonomer 6 was synthesized as shown in Scheme 1. For the asymmetric synthesis of the α-branched amine, we employed Ellman's N-tert-butanesulfinyl imine as a key intermediate.23 A condensation of commercially available (+)-tert-butanesulfinamide (1) and 4-(methylthio)benzaldehyde with Ti(OEt)4 as a Lewis acid catalyst24 provided sulfinimine 2 in a 94% yield. S-Methyl thioether protecting group was well-tolerated in the Grignard reaction conditions, and the addition of methylmagnesium bromide to sulfinimine 2 proceeded efficiently. After recrystallization, sulfinamide 3 was isolated in a 95% yield, and the crystal was confirmed as a single diastereomer by 1H NMR.
Scheme 1.

Synthesis of 6 (Nspe-thiol).
Typical deprotection of an S-methyl group requires two step reactions: first, oxidation of the thioether to a sulfoxide; second, Pummerer rearrangement of the sulfoxide and subsequent methanolysis to a thiol. 25 Initially, we focused on carrying out the S-methyl deprotection on solid-phase. Oxidation to sulfoxide proceeded smoothly, but we found the following Pummerer rearrangement gave inconsistent results. Therefore, we decided to switch the S-methyl to an acid-labile S-trityl protecting group. First, tert-butanesulfinyl group was removed by HCl in 1,4-dioxane, and trifluoroacetamide protecting group was introduced to provide 4. S-Methyl group was then removed by oxidation,26 Pummerer rearrangement,27 and methanolysis in a good yield to provide an aromatic thiol. Without isolating the aromatic thiol, we proceeded the thiol protection reaction using trityl chloride and pyridine to provide 5; a basic hydrolysis of 5 removed trifluoroacetamide protecting group, and free amine 6 was obtained in a quantititive yield. Notably the synthesis of 6 did not require a single chromatographic purification; all the intermediates could be easily recrystallized, and a gram scale synthesis of 6 was efficiently accomplished.
Scheme 2 depicts the synthesis of Nspe-acid (11) and Nspe-amide (12). The two submonomers can be accessed from common intermediate 9. Aromatic nitrile 8 was prepared employing a similar strategy as the one in Scheme 1. Unlike thioether 3, aromatic nitrile 8 was an oily residue and not recrystallizable. The diastereomeric impurity was identified by 1H NMR (dr = 86:14). Although the diastereomers were separable by chromatography, we went on to the next hydrolysis without purification of 8. Several conditions were tried for the hydrolysis of aromatic nitrile 8 to acid 9. The best result was obtained when the alkaline hydrolysis was performed in refluxing water/ethylene glycol mixture.28 Pleasantly, acid 9 could be recrystallized, yielding the desired diastereomeric isomer in a 74% yield. Amino acid 10 was obtained after deprotection of tert-butanesulfinyl group. Subsequent Cbz protection, tert-butyl ester formation, and Cbz deprotection 29 provided 11. Carboxamide 12 was prepared by an amidation of 9 followed by the deprotection of tert-butanesulfinyl group. We found the synthetic route to be highly reproducible. We prepared key intermediate 9 in a multi-gram scale, and 11 and 12 were synthesized in a gram scale.
Scheme 2.

Synthesis of 11 (Nspe-acid) and 12 (Nspe-amide).
Submonomers 6 (Nspe-thiol), 11 (Nspe-acid), and 12 (Nspe-amide) were then employed to synthesize peptoid oligomers (14-20, Figure 2). Non-functionalized peptoid heptamer 13 was synthesized as a control. Peptoid 14 contains one amide in the middle; 15 has two amides on the same face of the helix (three residues per turn), while 16 has two amides placed on different faces. 17 has four amides, two of which are facing the same side of the helix. 18 has amides on all three faces. Interestingly, peptoid 18 was soluble in water (∼2 mg/mL). Peptoid 19 is an acid equivalent of peptoid 15. Peptoid 20 contains one thiol in the middle, and N-terminus was acetylated.
Isolated thiol peptoid 20 readily formed peptoid disulfide by air oxidation in methanol over a prolonged time (i.e. 2-3 days). The homodimeric peptoid helix 21 (see Supporting Information S20 - S21) was purified by preparative HPLC and characterized by MALDI-TOF mass spectrometry ([M + Na]+ = 2458.4). A number of natural helices function as sulfhydryl-dependent homodimers. 30 Hence, our method to synthesize homodimeric peptoid helices will be a useful tool to study protein mimicry.
The maintenance of helical folds was demonstrated by circular dichroism (CD) spectroscopy for peptoid heptamers 13 – 16 and 19 – 20, and peptoid homodimer 21 (Figure 3). 31 The amide functionalized peptoids exhibited decreased spectral intensity compared to non-functionalized peptoid 13 (Figure 3 (A)).6a CD spectra of 15 and 19 were almost identical indicating that the influences of amides and acids on the helical integrity were similar. Thiol peptoid 20 and peptoid homodimer 21 also exhibited identical CD spectra, which suggests the dimer formation did not affect the secondary structure of each helix. N-Termini of 20 and 21 are acetylated, and direct comparison with other peptoid heptamers was not attempted.
Figure 3.

CD spectra of peptoids in acetonitrile (13, 14, 15, 16, and 19: 50 μM; 20 and 21: 46 μM) were recorded as per-residue molar ellipticity (deg cm2/dmol). Data were aquired at room temperature. (A) Comparison of amide functionalized peptoids 14 – 16 and control peptoid 13. (B) Comparison of amide peptoid 15 and acid peptoid 19. Also the effect of peptoid homodimerization via disulfide (20 and 21).
In summary, we described the efficient synthesis of new peptoid submonomers that can incorporate into peptoids, induce helical conformations, and present a variety of functional groups on the helix. Our synthetic strategy can be applied to the synthesis of various functionalized aromatic helix-inducing peptoid submonomers. The functionalized peptoids showed helical conformations, and we demonstrated the ability to create homodimeric peptoid helices by using thiol submonomer 6. The new submonomers should provide versatility to peptoid helices, and the functionalized peptoid helices should find numerous applications in the field of biomedicine and nanoscience.
Supplementary Material
Acknowledgments
Work at the Molecular Foundry was supported by the Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. We are also grateful to the National Institute of Health (R01 AI072666 to A. E. B.) for the support. J.S. thanks Dr. Byoung-Chul Lee (Lawrence Berkeley National Laboratory) and Dr. Modi Wetzler (Stanford University) for valuable discussions.
Footnotes
Supporting Information Available: Detailed procedures for the synthesis, characterization, and purification of all new compounds and peptoids. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Branden C, Tooze J. Introduction to Protein Structure. 2nd. Garland Publishing Inc.; New York, NY: 1999. [Google Scholar]
- 2.Barron AE, Zuckermann RN. Curr Opin Chem Biol. 1999;3:681–687. doi: 10.1016/s1367-5931(99)00026-5. [DOI] [PubMed] [Google Scholar]
- 3.(a) Simons RJ, Kania RS, Zuckermann RN, Huebner VD, Jewell DA, Banville S, Ng S, Wang L, Rosenberg S, Marlowe CK, Spellmeyer DC, Tan R, Frankel AD, Santi DV, Cohen FE, Bartlett PA. Proc Natl Acad Sci USA. 1992;89:9367–9371. doi: 10.1073/pnas.89.20.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Fowler SA, Blackwell HE. Org Biomol Chem. 2009;7:1508–1524. doi: 10.1039/b817980h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murphy JE, Uno T, Hamer JD, Cohen FE, Dwarki V, Zuckermann RN. Proc Natl Acad Sci USA. 1998;95:1517–1522. doi: 10.1073/pnas.95.4.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zuckermann RN, Kerr JM, Kent SBH, Moos WH. J Am Chem Soc. 1992;114:10646–10647. [Google Scholar]
- 6.(a) Kirshenbaum K, Barron AE, Goldsmith RA, Armand P, Bradley EK, Truong KTV, Dill KA, Cohen FE, Zuckermann RN. Proc Natl Acad Sci USA. 1998;95:4303–4308. doi: 10.1073/pnas.95.8.4303. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Armand P, Kirshenbaum K, Goldsmith RA, Farr-Jones S, Barron AE, Truong KTV, Dill KA, Mierke DF, Cohen FE, Zuckermann RN, Bradley EK. Proc Natl Acad Sci USA. 1998;95:4309–4314. doi: 10.1073/pnas.95.8.4309. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wu CW, Kirshenbaum K, Sanborn TJ, Patch JA, Huang K, Dill KA, Zuckermann RN, Barron AE. J Am Chem Soc. 2003;125:13525–13530. doi: 10.1021/ja037540r. [DOI] [PubMed] [Google Scholar]
- 7.Hunag K, Wu CW, Sanborn TJ, Patch JA, Kirshenbaum K, Zuckermann RN, Barron AE, Radhakrishnan I. J Am Chem Soc. 2006;128:1733–1738. doi: 10.1021/ja0574318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shin SBY, Yoo B, Todaro LJ, Kirshenbaum K. J Am Chem Soc. 2007;129:3218–3225. doi: 10.1021/ja066960o. [DOI] [PubMed] [Google Scholar]
- 9.Wu CW, Sanborn TJ, Kai H, Zuckermann RN, Barron AE. J Am Chem Soc. 2001;123:6778–6784. doi: 10.1021/ja003154n. [DOI] [PubMed] [Google Scholar]
- 10.Wu CW, Sanborn TJ, Zuckermann RN, Barron AE. J Am Chem Soc. 2001;123:2958–2963. doi: 10.1021/ja003153v. [DOI] [PubMed] [Google Scholar]
- 11.Sanborn TJ, Wu CW, Zuckermann RN, Barron AE. Biopolymers. 2002;63:12–20. doi: 10.1002/bip.1058. [DOI] [PubMed] [Google Scholar]
- 12.Gorske BC, Blackwell HE. J Am Chem Soc. 2006;128:14378–14387. doi: 10.1021/ja065248o. [DOI] [PubMed] [Google Scholar]
- 13.Fowler SA, Luechapanichkul R, Blackwell HE. J Org Chem. 2009;74:1440–1449. doi: 10.1021/jo8023363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gorske BC, Bastian BL, Gaske GD, Blackwell HE. J Am Chem Soc. 2007;129:8928–8929. doi: 10.1021/ja071310l. [DOI] [PubMed] [Google Scholar]
- 15.Shin SBY, Kirshenbaum K. Org Lett. 2007;9:5003–5006. doi: 10.1021/ol702207n. [DOI] [PubMed] [Google Scholar]
- 16.Shah NH, Butterfoss GL, Nguyen K, Yoo B, Bonneau R, Rabenstein DL, Kirshenbaum K. J Am Chem Soc. 2008;130:16622–16632. doi: 10.1021/ja804580n. [DOI] [PubMed] [Google Scholar]
- 17.(a) Patch JA, Barron AE. J Am Chem Soc. 2003;125:12092–12093. doi: 10.1021/ja037320d. [DOI] [PubMed] [Google Scholar]; (b) Chongsiriwatana NP, Patch JA, Czyzewski AM, Dohm MT, Ivankin A, Gidalevitz D, Zuckermann RN, Barron AE. Proc Natl Acad Sci USA. 2008;105:2794–2799. doi: 10.1073/pnas.0708254105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seurynck-Servoss SL, Dohm MT, Barron AE. Biochemistry. 2006;45:11809–11818. doi: 10.1021/bi060617e. [DOI] [PubMed] [Google Scholar]
- 19.Maayan G, Ward MD, Kirshenbaum K. Proc Natl Acad Sci USA. 2009;106:13679–13684. doi: 10.1073/pnas.0903187106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee BC, Chu TK, Dill KA, Zuckermann RN. J Am Chem Soc. 2008;130:8847–8855. doi: 10.1021/ja802125x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gellman SH. Acc Chem Res. 1998;31:173–180. [Google Scholar]
- 22.A guanidine functionalized submonomer was also designed and synthesized, but sidechain cleavage was observed after incorporation into peptoid. See Supporting Information S13 - S16 for detailed discussion.
- 23.(a) Liu G, Cogan DA, Ellman JA. J Am Chem Soc. 1997;119:9913–9914. [Google Scholar]; (b) Liu G, Cogan DA, Owens TD, Tang TP, Ellman JA. J Org Chem. 1999;64:1278–1284. [Google Scholar]; (c) Ellman JA, Owens TD, Tang TP. Acc Chem Res. 2002;35:984–995. doi: 10.1021/ar020066u. [DOI] [PubMed] [Google Scholar]; (d) Higashibayashi S, Tohmiya H, Mori T, Hashimoto K, Nakata M. Synlett. 2004;3:457–460. [Google Scholar]
- 24.Initially, we used anhydrous CuSO4 as a catalyst; however, the reaction proceeded sluggishly. After 5 days at room temperature, only 64% isolated yield was obtained. Alternatively, Cs2CO3 is known to be an effective catalyst and water scavenger (ref. 23(d)). Cs2CO3 is especially amenable for large scale reactions due to the ease of reaction workup and catalyst removal.
- 25.Young RN, Gauthier JY, Coombs W. Tetrahedron Lett. 1984;25:1753–1756. [Google Scholar]
- 26.(a) Vincent SP, Burkart MD, Tsai C, Zhang Z, Wong C. J Org Chem. 1999;64:5264–5279. doi: 10.1021/jo990686h. [DOI] [PubMed] [Google Scholar]; (b) Jing Y, Huang X. Tetrahedron Lett. 2004;45:4615–4618. [Google Scholar]
- 27.(a) Sugihara S, Tanikaga R, Kaji A. Synthesis. 1978:881. [Google Scholar]; (b) Gryko DT, Clausen C, Lindsey JS. J Org Chem. 1999;64:8635–8647. [Google Scholar]
- 28.(a) Mederski WWKR, Dorsch D, Bokel H, Beier N, Lues I, Schelling P. J Med Chem. 1994;37:1632–1645. doi: 10.1021/jm00037a014. [DOI] [PubMed] [Google Scholar]; (b) Ismail MAH, Barker S, Alou El Ella DA, Abouzid KAM, Toubar RA, Todd MH. J Med Chem. 2006;49:1526–1535. doi: 10.1021/jm050232e. [DOI] [PubMed] [Google Scholar]
- 29.(a) Filira F, Biondi L, Gobbo M, Rocchi R. Tetrahedron Lett. 1991;32:7463–7464. [Google Scholar]; (b) Mazaleyrat J, Xie J, Wakselman M. Tetrahedron Lett. 1992;33:4301–4302. [Google Scholar]
- 30.(a) Johansson J, Curstedt T, Joernvall H. Biochemistry. 1991;30:6917–6921. doi: 10.1021/bi00242a015. [DOI] [PubMed] [Google Scholar]; (b) Benezra R. Cell. 1994;79:1057–1067. doi: 10.1016/0092-8674(94)90036-1. [DOI] [PubMed] [Google Scholar]
- 31.The multiple sidechain carboxamides complicated CD analysis of peptoid 17 and 18.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.