

Abstract
Holoprosencephaly (or HPE) has captivated the imagination of Man for millennia because its most extreme manifestation, the single-eyed cyclopic newborn infant, brings to mind the fantastical creature Cyclops from Greek mythology. Attempting to understand this common malformation of the forebrain in modern medical terms requires a systematic synthesis of genetic, cytogenetic and environmental information typical for studies of a complex disorder. However, even with the advances in our understanding of HPE in recent years, there are significant obstacles remaining to fully understand its heterogeneity and extensive variability in phenotype. General lessons learned from HPE will likely be applicable to other malformation syndromes. Here we outline the common, and rare, genetic and environmental influences on this conserved developmental program of forebrain development and illustrate the similarities and differences between these malformations in humans and those of animal models.
Keywords: holoprosencephaly, HPE, disease genes, multifactorial inheritance
Introduction
Holoprosencephaly (HPE) represents a virtually continuous clinical spectrum of disorders ranging from simple microform features, such as closely spaced eyes or a single central maxillary incisor, to the extreme of a single cyclopic eye and superiorly placed proboscis. It is the most common malformation of the brain and face in humans [Muenke and Beachy, 2001; Cohen 2006; Dubourg et al., 2007; El-Jaick et al., 2007a]. While there has been considerable progress in our understanding of HPE over the past decade on both a genetic and mechanistic level, there has also been a growing appreciation for its etiologic heterogeneity and molecular complexity [Krauss 2007; Monuki 2008]. Furthermore, while similar defective genes can lead to HPE in humans as those that cause cyclopia in animals, there are fundamental differences between the universally observed heterozygous mutations in human subjects and the typically homozygous null model systems. Here we will describe these differences of gene number and context that may suggest a working model to account for some of these disparities.
The Mapping of HPE Genetic Loci in Human Chromosomes
Most investigators consider HPE to result from the genetic loss or mutational dysfunction of any one of at least 13 different autosomal dominant loci that serve as core susceptibility factors for humans (Table I). The initial clue to presence and location of these loci was derived from the systematic collection of HPE patients with consistent cytogenetic alterations that resulted in the loss, or gain, of critical chromosomal regions. Almost half of new HPE cases result from genetic inbalances generated by cytogenetic rearrangements occurring either de novo or following inheritance of a translocated chromosome with resulting aneupoidy [see Bendavid et al., 2005a, 2005b; Tyshchenko et al., 2008]. These original studies firmly established the concept that only a single genetic insult was sufficient to trigger HPE pathologies. The minimal critical regions defined by these relatively uncommon cytogenetic rearrangements were subsequently demonstrated to contain a principal risk factor gene for new mutations at each key locus: SHH at 7q36, SIX3 at 2p21, ZIC2 at 13q3.2 and TGIF at 18p11.3. To date, these four genes are the established targets of novel mutation leading to HPE susceptibility in hundreds of families ascertained world-wide [Dubourg et al., 2004, 2007]. When examined carefully, mutations in these, or related genes, have been shown to result in proteins with diminished biological function [(SHH) Schell-Apacik et al., 2003; Traiffort et al., 2004; Maity et al., 2005; Goetz et al., 2006; Singh et al., 2009; reviewed in Roessler et al., 2009c; (ZIC2) Brown et al., 2005; reviewed in Roessler et al., 2009a; (SIX3) Domené et al., 2008; Geng et al., 2008; (TGIF) Knepper et al., 2006; El-Jaick et al., 2007; (GLI2) Roessler et al., 2003, 2005; (NODAL pathway) Roessler et al., 2008, 2009d; (DISP) Roessler et al., 2009b]. Despite the proven utility of mutation screening of these genes, one must note that nearly 75% of cases of HPE with normal chromosomes do not have identified mutations. Hence, many additional factors related to HPE pathogenesis are uncharacterized, and likely include both environmental agents and additional genetic factors.
Table I. Genes (loci) contributing to HPE.
| Human gene | (Human locus) | Chromosome | Molecular function |
|---|---|---|---|
| ---- | HPE1 | 21q22.3 | (unknown) |
| SIX3 | HPE2 | 2p21 | Forebrain and eye development |
| SHH | HPE3 | 7q36 | Ventral CNS patterning |
| TGIF | HPE4 | 18p11.3 | Transcriptional repressor including retinoids |
| ZIC2 | HPE5 | 13q32 | Axis formation and dorsal brain development |
| ---- | HPE6 | 2q37.1-q37.3 | (unknown) |
| PTCH1 | HPE7 | 9q22.3 | Receptor for hedgehog ligands |
| ---- | HPE8 | 14q13 | (unknown) |
| GLI2 | HPE9 | 2q14 | Transcription factor mediating hedghog signaling |
| ---- | HPE10 | ---- | (unknown) |
| DISP1 | ---- | 1q42 | Release of hedgehog ligands |
| NODAL | ---- | 10q | TGFβ-like ligand involved in midline and laterality establishment |
| FOXH1 | ---- | 8q24.3 | Transcription factor for NODAL signaling |
Note that not all chromosomal loci implicated in HPE have genes that are considered firmly established as contributory. Furthermore, additional loci will undoubtedly be characterized by comparative hybridization strategies or other methods.
Despite the fact that the “glass is only one quarter full” at this stage, we can begin to draw important conclusions that should pertain to any new HPE gene(s) in the future. This extensive genetic heterogeneity suggests that there is a large set of genes that when structurally altered, or lost, can lead to HPE spectrum disorders. Thus, the population incidence of HPE should be the sum of many individual risk target loci. As described in Table 1, there are at least 10 named HPE loci, including four with the responsible gene yet to be identified. Mutations in at least 9 genes have been described to occur among HPE probands and these heterozygous mutations are usually the only molecularly significant variation detected by routine molecular diagnostics. The number of HPE loci is likely to increase as new genes are evaluated. In general, it is a new mutation, or gene loss/gain, that creates the risk for any given HPE family. Importantly, extrapolation from one family to another is problematic since different mutation(s) are at the core of each case. Finally, the typical variable expressivity of the same mutation among affected family members invokes additional co-morbid factors that can contribute to the ultimate phenotype [Roessler et al., 1996, 1997; Ming and Muenke, 2002].
The Role of Model Organisms in Candidate Gene Selection
A second important source of knowledge and insight about the genetics of HPE derives from the characterization of key genes through their manipulation in model organisms, such as the mouse, chick, frog, or zebrafish. Cyclopic phenotypes are relatively easy to produce by disturbances in the highly conserved process of gastrulation [Muenke and Beachy, 2001; Krauss 2007; Schachter and Krauss 2008]. At the same time, it should be remembered that the tools used to manipulate genes in animal systems are typically not intended to accurately model human disease; rather, these manipulations are designed to demonstrate the effects of complete loss, or over-expression, of these factors. Neither extreme would be expected to be typical of a human HPE patient. However, since these developmental programs of brain development are, in evolutionary terms, quite ancient, it is possible and profitable to extrapolate from animals to humans for defects in related gene function. This is generally helpful both for human candidate gene selection, as well as for functional analysis, of related genes by their introduction in a zeno-transplant experiment.
The Division of the Eye Field is an Active Process
The establishment of the axial midline in vertebrates creates a series of important signaling centers topologically oriented to reinforce conserved fundamental aspects of telecephalic patterning [see Fig.1 A and A′, adapted from: Rubenstein et al., 1998; Beddington and Robertson, 1999; Roessler and Muenke, 2001; Wilson and Houart, 2004]. One of the key signaling centers crucial for the pathogenesis of HPE is the most anterior extent of the midline mesoderm, called the prechordal plate (PCP). Several signals emanate from the PCP and trigger a secondary patterning center in the ventral forebrain, including Sonic hedgehog, the molecule most closely related to HPE (see below) and one of the major sources of ventralizing signals during forebrain development. As described in Fig. 1A to A′, the future forebrain acquires its regional specialization under the influence of several patterning centers. At the most rostral position in the forebrain, the anterior neural ridge (ANR) secretes mitogenic factors, such as Fibrobalst growth factor 8 (Fgf8), as well as Wnt inhibitors, such as Tlc [Houart et al., 2002], that prevent caudalizing Wnt signals from the posterior neuraxis from influencing the development of the telencephalon (see Fig. 1F to F′). As we will see shortly, one of the key HPE gene products, SIX3, has both anti-Bmp and anti-Wnt biological activity and creates a zone in the eye field and forebrain where these signals are neutralized. Although this process normally occurs in three dimensions in vivo, it can be studied by using Wnt and TGFβ inhibitors in stem cell culture [Watanabe et al., 2005]. A second major signaling center is the midbrain-hindbrain boundary that secretes a number of signals such as fibroblast growth factors and Wnts (Fig. 1A). Wnt ligands are important factors that are actively neutralized in the anterior neural plate but have essential functions in the hindbrain development. Retinoic acid is yet another posteriorizing factor that is produced in the trunk paraxial mesoderm and is crucial for the patterning of the hindbrain, but is actively neutralized by a cytochrome p450 enzyme (Cyp26) in the anterior neural plate that helps to define the MHB territories [Kudoh et al., 2002; White et al., 2007]. These in vivo and in vitro systems demonstrate both the intrinsic tendency for neural induction, given the appropriate circumstances, and the requirement for a delicate balancing of numerous key influences: hedgehogs, fibroblast growth factors (Fgf), bone morphogenic proteins (Bmp), retinoic acid, and canonical and non-canonical Wnt signals. Therefore, while basic mechanisms of forebrain patterning are intelligible, they are complex and require the simultaneous integration of a large number of factors.
Figure 1.
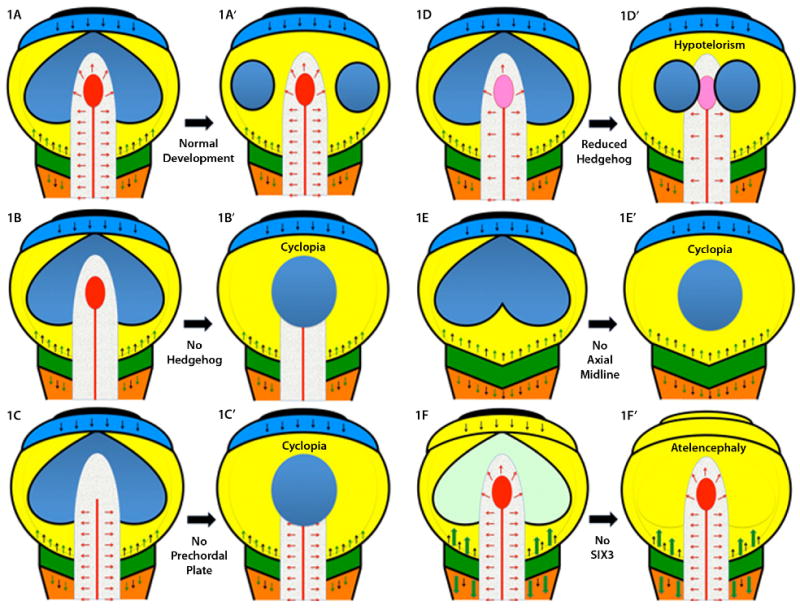
A: A schematic representation of a typical flat neural plate prior to the neurulation stage, viewed from above, is shown with key signaling centers indicated by various colors. Anterior is at the top and the hindbrain (orange) and spinal chord at the bottom. Each pair of figures illustrates the effects of the loss of key genes/structures in a simplified developmental progression. The most rostral center, the anterior neural ridge (ANR), is in black and secretes FGF8 (black arrows, also produced at the midbrain-hindbrain boundary [MHB], which is dark green) and the anti-WNT factor TLC (not shown) [Houart et al., 2002]. The ANR promotes the growth and expansion of the telencephalon (shown as an undivided light blue strip of tissue). Note that the eye field, shown in dark blue, is also contiguous across the midline just caudal to the telencephalon within the mesencephalon/midbrain territory (yellow). The MHB produces several important caudalizing factors, including WNTs (green arrows), whose actions are contained by rostral inhibitors (DKK1, TLC, SIX3, etc). The paraxial mesoderm (caudal to the MHB) is a source of retinoic acid that patterns the hindbrain, but is eliminated anterior to the MHB by the enzyme Cyp26. The axial midline (notochord as red line, and prechordal plate, PCP as red ellipse) is a source of hedgehog ligands (red arrows) as well as anti-BMP factors such as chordin, follistatin and noggin (not shown, see Klingensmith chapter). Definitive ectoderm is a source of BMP activity (thin black line surrounding each diagram) and assumes its exterior location, with neural tissue interior, following neurulation [England et al., 2006]. A′: If the PCP correctly migrates beneath the telencephalon, then secondary signals, including a secondary domain of hedgehog activity in the ventral telencephalon, accompanies the division of the eye field into two discrete eyes. B: If hedgehog signals (red arrows) are eliminated, genetically or pharmacologically, the eye field fails to divide resulting in cyclopia, see B′. Hedgehog activity is also required in the developing face [Cordero et al., 2004] linking additional signaling centers to the function of the PCP. C: Surgical ablation of the PCP also results in cyclopia [Shih and Fraser, 1995], see C′. D and D′: Attenuated hedgehog signaling is compatible with some eye field separation, although the eyes are closely spaced (hypotelorism). E and E′: If the axial midline fails to form, or degenerates, the resulting animals are uniformly cyclopic [Israeli et al., 1999; Karkera et al., 2002]. F: The importance of WNT inhibition is illustrated by model organisms lacking Six3 where the telencephalon is truncated under the un-restrained activity (large green arrows) of caudalization signals. To the extent that anterior eye field forms under these circumstances (faint green) these anterior structures soon disappear, see F′. Interestingly, one WNT inhibitor, DKK1, is not a common mutational target in humans [Roessler et al., 2000]. Excessive exposure to retinoids can also result in atelencephaly, or HPE [Sulik et al., 1995], possibly through a similar mechanism of caudalization.
The eye field begins as a single structure that spans the midline [Adelmann 1936; Li et al., 1997]. Under the influence of signals from the PCP, the vertebrate eye field splits into discrete left and right eyes [compare Fig. 1A to 1A′; Marlow et al., 1998; Varga et al., 1999]. In a recent study in zebrafish, this process has been directly measured under time-lapse photography [England et al., 2006]. Thus, the division of the eye field, and by implication the forebrain, is an active process involving directed cellular movements and the critical orientation of the midline PCP signaling center beneath the telencephalon. If these developmental steps are not completed correctly, for any of several reasons, the default result is cyclopia. For example, animals where the PCP is surgically removed (Fig. 1C to C′) or never forms (Fig. 1E to E′) consistently develop cyclopia [Shih and Fraser, 1995; Feldman et al., 1998; reviewed in Shen and Schier, 2000; Schier 2003]. Experimental evidence that hedgehog signals are both necessary and sufficient for the completion of the eye field separation (Fig. 1B to B′) comes from treatment of zebrafish with ethanol (a well known HPE teratogen) [Blader and Strähle, 1998; see also Aoto et al., 2008], or anti-hedgehog morphilinos (chemicals that result in gene-specific suppression of protein translation) [Nasevicious and Ekker, 2000] or chemical inhibitors of hedgehog signaling itself [Cordero et al., 2004].
Nodal Signaling and the Midline
Nodal was initially described as a gene essential for the establishment of the organizer, or node in higher vertebrates that was lethal in the homozygous null state [reviewed in Beddington and Robertson 1999; Schier 2003; Shen 2007]. As we have described previously, typical mice heterozygous for mutations in Nodal are normal. However, both the development of the axis in vertebrates and the establishment of organ laterality depend on Nodal signals. As these become progressively decreased below 50%, a range of phenotypes can result [Lowe et al., 2001; Vincent et al., 2003; reviewed in Roessler et al., 2001, 2008, 2009d]. The most consistent consequence of impaired Nodal signaling is disturbance in laterality. Only when the co-factors Gdfs also compromised is the axial midline affected [Andersson et al., 2006]. However, a compromised axial midline will inevitably lead to secondary changes in the PCP and factors secreted by this structure, such as Sonic hedgehog (Fig. 1E to E′).
Sonic Hedgehog Signaling and HPE
It is now widely accepted that holoprosencephaly is an example of a multi-factorial trait requiring the synergy between novel mutations in key genes, the interaction of these mutations with endogenous host variants, and the likely additional effect of environmental insults. A difference between human HPE and its closest mouse model was apparent from the first example. When mice are deleted for both copies of the Sonic hedgehog (Shh) gene, the animals display uniformly severe HPE-like features, growth retardation, limb anomolies, extreme cyclopia and defective axial patterning throughout the entire neuraxis. However, murine Shh +/- heterozygotes are phenotypically normal [Chiang et al., 1996]. Although this degree of clinical severity, evident in the homozygous null mouse embryos, can be seen in humans, it is not typical for these cases to survive to term. On the other hand, heterozygous variations in the SHH gene are the most commonly detected mutations in a live-born collection of HPE probands [Roessler et al., 1996, 2003, 2009c]. Furthermore, instances of two mutations in the human SHH gene in the same individual HPE patient have not been reported. With the passage of time, this gene dosage discrepancy has never been fully explicated. In one scenario, this paradox would be explained by invoking multiple different genetic alterations. However, these mutations, in our current view, would likely occur in several independent genes (in humans) instead of two identical mutations in the same gene (in mice).
Subsequent studies of model systems confirmed that dysfunction of hedgehog signaling was a common mechanism for the production of HPE-like phenotypes [reviewed in Roessler et al., 2003; Ingham 2008]. Three additional genes in the human SHH signaling pathway have been described as mutational targets in HPE patients, including the SHH receptor PTCH1 [Ming et al., 2001; Ribiero et al., 2006], the ligand transporter DISP1 [Ma et al., 2002; Roessler et al., 2009b] and the transcription factor GLI2 [Roessler et al., 2003, 2005; Rahimov et al., 2007]. Again, we detect salient differences between the mouse models and the human phenotypes of HPE probands with heterozygous mutations. These phenotypic differences suggest that the consequences of diminished hedgehog signaling are similar between mice and humans, but that number and types of genetic alterations that accomplish them are different.
A recurrent theme emerging from the comparison of mouse models of HPE with human pathologies is the notion that homozygous null animals serve as proof and illustration of the more severe phenotypic extremes but do not reliably reconstruct the genetic architecture of human HPE cases [Hayhurst and McConnell, 2003]. For example, mice lacking the key transducer of hedgehog signals, Smo, arrest early in embryonic development due to the elimination of all hedgehog signals [e.g. see 1B, fails to proceed to 1B′; see Zhang et al., 2003]. Similarly, mice homozygous null for Disp arrest at a nearly identical stage [Ma et al., 2002]; however, in both cases the murine heterozygotes were phenotypically normal. These differences between murine and human phenotypic pathologies from a given gene variant are common to almost all murine HPE models and suggest: 1) humans with two lesions in the same gene are likely to be uncommon, and, 2) if present, it would be expected to reflect only the severe end of the spectrum [reviewed in Krauss 2007]. The extreme variability that is characteristic of HPE is difficult to explain if both alleles of a key gene (genetically recessive) must be significantly impaired in most cases. Furthermore, heterozygous carriers should be prevalent in a control population; yet, this is contrary to the experience of molecular diagnostic centers. In contrast, gene-gene interactions between mutations and functionally linked factors and/or gene-environmental interactions would not be precluded in a model of novel heterozygous mutations interacting with other factors.
Genetic Modifiers Emerge as Key Modulators of Phenotype
As additional murine models of cyclopia have become available, the importance of strain-specific modifiers has emerged as the most likely explanation for discrepant HPE phenotypes. For example, the cell surface protein Cdo is a member of a family of hedgehog receptors that modulates signaling the target field [Cole et al., 2003]. In the original mouse strain examined, the Cdo -/- animals displayed a single central maxillary incisor, typical for microform HPE. Interestingly, when these animals are bred into another strain of mice, they begin to display increasingly severe phenotypes including cyclopia [Zhang et al., 2006]. Increasingly, murine models of similar craniofacial anomalies have exploited the potential interactions within hedgehog signaling pathways in compound mutants to more closely mimic human disease [Seppala et al., 2007; Tenzen et al., 2006; Allen et al., 2007]. This observation of strain-specific modifiers has been shown to be important in all of the HPE genes examined (reviewed in Krauss 2007; Schachter and Krauss, 2008; see below).
Both Early and Late Functions for ZIC2 in HPE
Mutation or deletion of the human ZIC2 gene is the second most common detectable alteration in HPE subjects [Brown et al., 1995, 1998, 2001, 2002, 2005; reviewed in Roessler et al., 2009a]. Although the precise functions of this transcription factor are still poorly understood, it encodes a classical Gli-type zinc finger DNA binding motif that recognizes exactly the same targets as the transcription factors mediating hedgehog signals [Redemann et al., 1988; Kinzler et al., 1990; Pavletich and Pabo, 1993]. The notion that Zic factors augment or co-regulate hedgehog targets was initially attractive but at variance with its observed dorsal expression pattern [Nagai et al., 1997; Brown et al., 2003; Elms et al., 2003, 2004; Aruga 2004; Aruga et al., 2006]. The initial murine model for Zic2 was a hypomorphic allele that was associated with a neurulation delay, monoventricle and spina bifida [Nagai et al., 2000].
A recent study has now demonstrated an early role for Zic2 in the axial midline that precedes the expression of Shh yet produces an extensive range of HPE phenotypes, from the mild to severe extent of the spectrum [Warr et al., 2008]. This variability in expressivity of the Zic2 -/- embryos is evidence of stochastic factors that can be important, particularly with early acting genes. Again, there is a link to Shh in that the degree of HPE features could be correlated with the extent of forebrain expression of the hedgehog protein. Thus, most of the abnormalities of HPE can be traced to the impact on forebrain expression of hedgehog signals.
Multiple Roles for Six3 in HPE
The murine Six3 gene is one of the earliest markers of the anterior forebrain and midline, structures known to be important in HPE pathogenesis [Oliver et al., 1995]. However, complete absence of the Six3 gene leads to anterior truncations of the forebrain, not classical HPE (see Fig. 1F to F′). In contrast, heterozygous mutations in human SIX3 are the third most commonly detected sequence variations among HPE patients [Wallis et al., 1999]. Genetic and biochemical studies of Six3 have demonstrated several essential roles at different times during development of the forebrain and eyes [Laguitin et al., 2003; Lavado et al., 2007] including anti-BMP [Gestri et al., 2005] and as a Groucho-dependent repressor of Wnt signals [Zhu et al., 2002]. The Six3 gene also encodes one of several transcription factors present in the eye field where it functions to inhibit caudalizing Wnt signals. An additional property of Six3 is its interaction with the cell cycle regulator, gemenin, where it can promote the continuing proliferation of retinal progenitors [del Bene et al., 2004]. Given these multiple roles, it was initially difficult to understand how Six3 fit into the HPE scheme.
A recent study has now clarified the role of SIX3 by confirming our independent results that mutations seen in HPE patients are of diminished function [Domené et al., 2008]; these investigators went on further to demonstrate that the introduction of a heterozygous human-type mutation into the mouse can lead to HPE-like phenotypes [Geng et al., 2008; see also Jeong et al., 2008]. A crucial observation is that the artificially mutated gene is sensitive to the genetic background of the mouse strain utilized and also exacerbated by the introduction of a dose reduction in the Shh +/- gene. These studies demonstrate that Six3 has an additional property of regulating Shh in the ventral forebrain. A long distance enhancer of the SHH gene had been postulated during the mapping of the HPE4 locus based on a cluster of translocations detected at a considerable distance from the coding region of the SHH gene [Roessler et al., 1997]. We now know that this SHH forebrain enhancer binds the SIX3 protein, and its expression in the ventral forebrain is compromised with diminished function of a mutant version of the protein. Thus, one key consequence of defective SIX3 function is to impair the expression of SHH in the ventral forebrain thus linking the two genes into the same developmental program.
18p-, TGIF and Retinoids
Deletions involving human chromosome 18p are among the most common chromosomal changes detected in HPE subjects [Overhauser et al., 1995]. By definition, most of these deletions encompass more than the TGIF gene where functionally abnormal mutations are detected [Gripp et al., 2000; El-Jaick et al., 2007b]. Three of the 13 mutations detected in HPE cases have been shown to be de novo. However, the penetrance of 18p deletions as a cause of HPE is as low as 10%, suggesting either that this is a weak HPE locus, or that additional co-morbid factors may be required. TGIF is a transcriptional co-repressor of TGFβ signaling and also inhibits the actions of retinoids [Wotton et al., 1999; Bartholin et al., 2006]. Despite intensive investigations in mouse models, the role for TGIF in HPE has remained obscure. Three different mouse models have failed to identify HPE-like phenotypes in Tgif null animals [Shen and Walshe, 2005; Bartholin et al., 2006; Jin et al., 2006]. A fourth mouse model has noted that a postulated dominant acting intragenic deletion of the murine Tgif locus can lead to forebrain defects and that the penetrance of these malformations are dependent on strain effects [Kuang et al., 2006].
Although it remains uncertain if this is the actual mechanism for HPE pathologies, mice lacking Tgif are modestly sensitized to external exposure to retinoids [Bartholin et al., 2006]. These agents are known teratogens causing anterior truncations and HPE-like malformations in mice [Sulik et al., 1995]. A possible mechanism for TGIF in human HPE could involve the presence of increased retinoid acid in the anterior forebrain (Fig. 1F to F′), which may then result in exceeding the enzymatic ability to degrade retinoic acid in this compartment [Gongal et al., 2008].
The Integration of Multiple Signaling Systems is Essential
It is now widely accepted that to attempt to explain the entire HPE spectrum of disease by focusing solely on an individual gene would be to grossly oversimplify what is clearly an elegant network of interacting genes and signaling centers [reviewed in Monuki 2007; Fernandez and Hébert, 2008]. The central importance of the midline signals, and Sonic hedgehog in particular, has continued to be emphasized in recent studies [Hayhurst et al., 2008], but clearly is not the sole potential cause of HPE phenotypes. For example, Bmp signaling in the dorsal regions of the telencephalon is crucial for the development of the hippocampus and cortical hem [Cheng et al., 2006; Fernandez et al., 2007; Hébert and Fishell, 2008]. Although the middle interhemispheric variant (MIHV) of HPE most closely resembles these types of defects, it is not yet known if defective BMP signals account for this type of malformation in humans. Furthermore, MIHV is not exclusively associated with ZIC2 mutations as is commonly presented [Fernandez and Hébert, 2008; Maurus and Harris, 2009]. Interactions among midline telencephalic centers have been emphasized repeatedly in recent HPE models [Storm et al., 2008]. Most of the factors described in Fig. 1 (Fgfs, Bmps, retinoids, hedgehogs, Wnts) have been shown to have cross-regulatory actions. Similarly, a recent study in zebrafish suggests that zic factors, in this system, can connect a wide range of signaling functions including Nodals, hedgehogs and retinoic acid [Maurus and Harris, 2009]. While this may be unique to zebrafish, the general principal may prove to extend to other organisms. We should be prepared for many new mechanistic surprises in the future, since only a fraction of HPE cases have even a single risk factor determined.
Summary
It is becoming increasingly likely that the integration of multiple defects will be required for the understanding of individual cases of HPE. While it is yet to be convincingly demonstrated that a digenic model of HPE is generally appropriate [Ming and Muenke, 2002], it would be naïve to attribute the variable expressivity of similar mutations in a HPE gene to anything other than co-morbid genetic or environmental modifiers. Since the proximate cause of HPE is typically due to a novel mutation, or gene gain/loss, these modifiers must already be present in the germline of the parents. A digenic model requiring two de novo mutations is unlikely to explain more than a handful of HPE cases, due the rarity of these events individually or collectively [Krykov et al., 2007]. Recent studies on ZIC2 mutations are notable for their high penetrance, frequent novelty of the mutations, and consistent phenotype that could not be readily explained by a digenic mechanism of divergent factors [Solomon, this volume]. However, a model of co-variations in genes that functionally interact with these novel mutations can help to explain the variability between mutation carriers within families. While functionally abnormal polymorphisms, such as the ones identified in the NODAL gene [Roessler et al., 2009d], have not been fully evaluated for their potential roles as modifiers, these are excellent candidates for context-dependent variations that can modify the effects of mutations in other genes. Just as the identical mutation(s) in mice can have dramatically different consequences in different mouse strains, the identical type of mutation in humans can also manifest itself differently depending on its genetic context. Finally, although the types of genetic interactions observed in animal models will often also be proven true for humans, this is almost certainly not absolute. Furthermore, there is no reason to believe that the individual variations/susceptibilities are identical across species. It is more likely that each vertebrate animal, including humans, has its own unique set of variations established within its population and these help to explain why a novel mutation can have such a wide range of consequences.
Acknowledgments
We would like to thank the clinicians from around the world for their continuing support of research investigations into the genetic basis of HPE and its clinical manifestations. This work is dedicated to the families who are confronting HPE as a diagnosis and the demands of a special needs child, and for their frequently amazing resilience and strength. This work was supported by the DIR of the NHGRI, NIH.
Biographies
Bio Sketches
Dr. Roessler is a faculty member in the Medical Genetics Branch National Human Genome Research Institute, National Institutes of Health, Bethesda, MD, USA. His research interests include malformations of the forebrain associated with holoprosencephaly (HPE) as well as disturbances in organ sidedness, or laterality.
Dr. Muenke is the Branch Chief of the same Medical Genetics Branch and his research interests include holoprosencephaly, craniofacial malformation syndromes and attention-deficit-hyperactivity disorder (ADHD).
References
- Adelmann HB. The problem of cyclopia. Quart Rev Biol. 1936;11:116–182. 284–364. [Google Scholar]
- Allen BL, Tenzen T, McMahon AP. The hedgehog-binding proteins Gas1 and Cdo cooperate to positively regulate Shh signaling during mouse development. Genes Dev. 2007;21:1244–1255. doi: 10.1101/gad.1543607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aruga J. The role of Zic genes in neural development. Mol Cell Neurosci. 2004;26:205–221. doi: 10.1016/j.mcn.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Aruga J, Kamiya A, Takahashi H, Fujimi TJ, Shimizu Y, Ohkawa K, Yazawa S, Umesono Y, Noguchi H, Shimizu T, Saitou N, Mikoshiba K, Sakaki Y, Agata K, Tyoda A. A wide-range phylogenetic analysis of Zic proteins: implications for correlations between protein structure conservation and body plan complexity. Genomics. 2006;87:783–792. doi: 10.1016/j.ygeno.2006.02.011. [DOI] [PubMed] [Google Scholar]
- Aoto K, Shikata Y, Higashiyama D, Shiota K, Motoyama J. Fetal ethanol exposure activates protein kinase A and impairs Shh expression in prechordal mesendoderm cells in the pathogenesis of holoprosencephaly. Birth Defects Res A. 2008;82:224–231. doi: 10.1002/bdra.20447. [DOI] [PubMed] [Google Scholar]
- Andersson O, Reissmann E, Jornvall H, Ibanez CF. Synergistic interaction between Gdf1 and Nodal during anterior axis development. Dev Biol. 2006;293:370–381. doi: 10.1016/j.ydbio.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Bartholin L, Powers SE, Melhuish TA, Lasse S, Weinstein M, Wotton D. TGIF inhibits retinoid signaling. Mol Cell Biol. 2006;26:990–1001. doi: 10.1128/MCB.26.3.990-1001.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beddington RSP, Robertson EJ. Axis development and early asymmetry in mammals. Cell. 1999;96:195–209. doi: 10.1016/s0092-8674(00)80560-7. [DOI] [PubMed] [Google Scholar]
- Bendavid C, Haddad BR, Griffin A, Huizing M, Dubourg C, Gicquel I, Cavalli LR, Pasquier L, Long R, Ouspenskaia M, Odent S, Lacbawan F, David V, Muenke M. Multicolor FISH and quantitative PCR can detect submicroscopic deletions in holoprosencephaly patients with a normal karyotype. J Med Genet. 2005a;43:496–500. doi: 10.1136/jmg.2005.037176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendavid C, Dubourg C, Gicquel I, Pasquier L, Saugler-Veber P, Durou MR, Jaillard S, Frebourg T, Haddad BR, Henry C, Odent S, David V. Molecular evaluation of foetuses with holoprosencephaly shows high incidence of microdeletions in the HPE genes. Hum Genet. 2005b;119:1–8. doi: 10.1007/s00439-005-0097-6. [DOI] [PubMed] [Google Scholar]
- Blader P, Strähle U. Ethanol impairs migration of the prechodal plate in the zebrafish embryo. Dev Biol. 1998;201:185–201. doi: 10.1006/dbio.1998.8995. [DOI] [PubMed] [Google Scholar]
- Brown S, Russo J, Chitayat D, Warburton D. The 13q- syndrome: the molecular definition of a critical deletion region in band 13q32. Amer J Hum Genet. 1995;57:859–866. [PMC free article] [PubMed] [Google Scholar]
- Brown SA, Warburton D, Brown LY, Yu Cy, Roeder ER, Stengel-Rutkowski S, Hennekam RCM, Muenke M. Holoprosencephaly due to mutations in ZIC2, a homologue of Drosophila odd-paired. Nat Genet. 1998;20:180–183. doi: 10.1038/2484. [DOI] [PubMed] [Google Scholar]
- Brown LY, Kottman AH, Brown S. Immunolocalization of Zic2 expression in the mouse forebrain. Gene Expr Patterns. 2003;3:361–367. doi: 10.1016/s1567-133x(03)00043-7. [DOI] [PubMed] [Google Scholar]
- Brown L, Paraso M, Arkell R, Brown S. In vitro analysis of partial loss-of-function ZIC2 mutations in holoprosencephaly: alanine tract expansion modulates DNA binding and transactivation. Hum Mol Genet. 2005;14:411–420. doi: 10.1093/hmg/ddi037. [DOI] [PubMed] [Google Scholar]
- Cheng X, Hsu CM, Currle DS, Hu JS, Barkovich AJ, Monuki ES. Central roles of the roof plate in telencephalic development and holoprosencephaly. J Neurosci. 2006;26:7640–7649. doi: 10.1523/JNEUROSCI.0714-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C, Littingtung Y, Lee E, Young KE, Corden JL, Westphal H, Beachy PA. Cyclopia and defective axial patterning in mice lacking sonic hedgehog gene function. Nature. 1996;383:407–413. doi: 10.1038/383407a0. [DOI] [PubMed] [Google Scholar]
- Cohen MM., Jr Holoprosencephaly: clinical, anatomic, and molecular dimensions. Birth Defects Res Part A Clin Mol Teratol. 2006;76:658–673. doi: 10.1002/bdra.20295. [DOI] [PubMed] [Google Scholar]
- Cole F, Krauss RS. Microform holoprosencephaly in mice that lack the Ig superfamily member Cdon. Cur Biol. 2003;13:411–415. doi: 10.1016/s0960-9822(03)00088-5. [DOI] [PubMed] [Google Scholar]
- Cordero D, Marcucio R, Hu D, Gaffield W, Tapadia M, Helms JA. Temporal perturbations in sonic hedgehog signaling elicit the spectrum of holoprosencephaly phenotypes. J Clin Invest. 2004;114:485–494. doi: 10.1172/JCI19596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Bene F, Tessmar-Raible K, Wittbrodt J. Direct interaction of geminin and Six3 in eye development. Nature. 2004;427:745–749. doi: 10.1038/nature02292. [DOI] [PubMed] [Google Scholar]
- Domené S, Roessler E, El-Jaick KB, Snir M, Brown JL, Vélez JI, Bale S, Lacbawan F, Muenke M, Feldman B. Mutations in the human SIX3 gene in holoprosencephaly are loss of function. Hum Mol Genet. 2008;17:3919–3928. doi: 10.1093/hmg/ddn294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubourg C, Bendavid C, Pasquier L, Henry C, Odent S, David V. Holoprosencephaly. Orphant J Rare Dis. 2007;2:8. doi: 10.1186/1750-1172-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubourg C, Lazaro L, Pasquier L, Bendavid C, Blayau M, Le Duff F, Durou MR, Odent S, David V. Molecular screening of SHH, ZIC2, SIX3 and TGIF genes in patients with features of holoprosencephaly spectrum: mutation review and genotype-phenotype correlations. Hum Mut. 2004;24:43–51. doi: 10.1002/humu.20056. [DOI] [PubMed] [Google Scholar]
- El-Jaick KB, Fonseca RF, Moreira MA, Ribeiro MG, Bolognese AM, Dias SO, Pereira ET, Castilla EE, Orioli IM. Single median maxillary central incisor: new data and mutation review. Birth Defects Res (PartA) 2007a;79:573–580. doi: 10.1002/bdra.20380. [DOI] [PubMed] [Google Scholar]
- El-Jaick KB, Powers SE, Bartholin L, Myers KR, Hahn J, Orioli IM, Ouspenskaia M, Lacbawan F, Roessler E, Wotton D, Muenke M. Functional analysis of mutations in TGIF associated with holoprosencephaly. Mol Genet Metab. 2007b;90:97–111. doi: 10.1016/j.ymgme.2006.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elms P, Siggers P, Napper D, Greenfield A, Arkell R. Zic2 is required for neural crest formation and hindbrain patterning during mouse development. Dev Biol. 2003;264:391–406. doi: 10.1016/j.ydbio.2003.09.005. [DOI] [PubMed] [Google Scholar]
- Elms P, Scurry A, Davies J, Willoughy C, Hacker T, Bogani D, Arkell R. Overlapping and distinct expression domains of Zic2 and Zic3 during mouse gastrulation. Gene Expr Patterns. 2004;4:505–511. doi: 10.1016/j.modgep.2004.03.003. [DOI] [PubMed] [Google Scholar]
- England SJ, Blanchard GB, Mahadevan L, Adams RJ. A dynamic fate map of the forebrain shows how vertebrate eyes form and explains two causes of cyclopia. Development. 2006;133:4613–4617. doi: 10.1242/dev.02678. [DOI] [PubMed] [Google Scholar]
- Feldman B, Gates MA, Egan ES, Dougan ST, Rennebeck G, Sirotkin HI, Schier AF, Talbot WS. Zebrafish organizer development and germ-layer formation require nodal-related signals. Nature. 1998;395:181–185. doi: 10.1038/26013. [DOI] [PubMed] [Google Scholar]
- Fernandes M, Gutin G, Alcorn H, McConnell SK, Hébert JM. Mutations in the BMP pathway in mice support the existence of two molecular classes of holprosencephaly. Development. 2007;134:3789–3794. doi: 10.1242/dev.004325. [DOI] [PubMed] [Google Scholar]
- Fernandes M, Hébert JM. The ups and downs of holoprosencephaly: dorsal versus ventral patterning forces. Clin Genet. 2008;73:413–423. doi: 10.1111/j.1399-0004.2008.00994.x. [DOI] [PubMed] [Google Scholar]
- Geng X, Speirs C, Laguitin O, Solnica-Krezel L, Jeong Y, Epstein D, Oliver G. Haploinsufficiency of Six3 fails to activate Sonic hedgehog expression in the ventral forebrain and causes holoprosencephaly. Dev Cell. 2008;15:236–247. doi: 10.1016/j.devcel.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gestri G, Carl M, Appolloni I, Wilson SW, Barsacchi G, Andreazzoli M. Six3 functions in anterior neural plate specification by promoting cell proliferation and inhibiting Bmp4 expression. Development. 2005;132:2401–2413. doi: 10.1242/dev.01814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz JA, Singh S, Suber LM, Kull FJ, Robbins DJ. A highly conserved amino-terminal region of Sonic hedgehog is required for the formation of its freely diffusible multimeric form. J Biol Chem. 2006;281:4087–4093. doi: 10.1074/jbc.M511427200. [DOI] [PubMed] [Google Scholar]
- Gongal PA, Waskiewicz AJ. Zebrafish model of holoprosencephaly demonstrates a key role for TGIF in regulating retinoic acid metabolism. Hum Mol Genet. 2008;17:525–538. doi: 10.1093/hmg/ddm328. [DOI] [PubMed] [Google Scholar]
- Gripp KW, Wotton D, Edwards MC, Roessler E, Ades L, Meinecke P, Richeri-Costa A, Zackai EH, Massague J, Muenke M, Elledge SJ. Mutations in TGIF cause holoprosencephaly and link NODAL signaling to human neural axis determination. Nat Genet. 2000;25:205–208. doi: 10.1038/76074. [DOI] [PubMed] [Google Scholar]
- Hayhurst M, McConnell SK. Mouse models of holoprosencephaly. Curr Opin Neurol. 2003;16:135–141. doi: 10.1097/01.wco.0000063761.15877.40. [DOI] [PubMed] [Google Scholar]
- Hayhurst M, Gore BB, Tessier-Lavigne M, McConnell SK. Ongoing sonic hedgehog signaling is required for dorsal midline formation in the developing forebrain. J Neurosci. 2008;26:83–100. doi: 10.1002/dneu.20576. [DOI] [PubMed] [Google Scholar]
- Hébert JM, Fishell G. The genetics of early telencephalon patterning: some assembly required. Nat Rev neurosci. 2008;9:678–685. doi: 10.1038/nrn2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houart C, Caneparo L, Heisenberg CP, Barth KA, Take-Uchi M, Wilson SW. Establishment of the telencephalon during gastrulation by local antagonisms of Wnt signaling. Neuron. 2002;35:255–265. doi: 10.1016/s0896-6273(02)00751-1. [DOI] [PubMed] [Google Scholar]
- Ingham PW. Hedgehog signaling. Cur Biol. 2008;18:R238–R241. doi: 10.1016/j.cub.2008.01.050. [DOI] [PubMed] [Google Scholar]
- Izraeli S, Lowe LA, Bertness VL, Good DJ, Kirsch IR, Kuehn MR. The SIL gene is required for mouse embryonic axial development and left-right specification. Nature. 1999;399:691–694. doi: 10.1038/21429. [DOI] [PubMed] [Google Scholar]
- Jeong Y, Leskow FC, El-Jaick K, Roessler E, Muenke M, Yocum A, Dubourg C, Li X, Geng X, Oliver G, Epstein DJ. Regulation of a remote Shh forebrain enhancer by the Six3 protein. Nat Genet. 2008;40:1348–1353. doi: 10.1038/ng.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin JZ, Gu S, McKinney P, Ding J. Expression and functional analysis of Tgif during mouse midline development. Dev Dyn. 2006;235:547–533. doi: 10.1002/dvdy.20642. [DOI] [PubMed] [Google Scholar]
- Kamnasaran D, Chen CP, Devriendt K, Mehta L, Cox DW. Defining a holoprosencephaly locus on human chromosome 14q13 and characterization of potential candidate genes. Genomics. 2005;85:608–621. doi: 10.1016/j.ygeno.2005.01.010. [DOI] [PubMed] [Google Scholar]
- Karkera JD, Izraeli S, Roessler E, Dutra A, Kirsch IR, Muenke M. The genomic structure, chromosomal localization, and analysis of SIL as a candidate gene for holoprosencephaly. Cytogenet Genome Res. 2002;97:62–67. doi: 10.1159/000064057. [DOI] [PubMed] [Google Scholar]
- Kinzler KW, Vogelstein B. The GLI gene encodes a nuclear protein which binds specific sequences in the human genome. Mol Cell Biol. 1990;10:634–642. doi: 10.1128/mcb.10.2.634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knepper JL, James AC, Ming JE. TGIF, a gene associated with human brain defects, regulates neuronal development. Dev Dyn. 2006;235:1482–1490. doi: 10.1002/dvdy.20725. [DOI] [PubMed] [Google Scholar]
- Krauss RS. Holoprosencephaly: new models, new insights. Expert Rev Mol Med. 2007;9:1–17. doi: 10.1017/S1462399407000440. [DOI] [PubMed] [Google Scholar]
- Krykov GV, Pennacchio LA, Sunyaev SR. Most rare missense alleles are deleterious in humans: implications for complex disease and association studies. Am J Hum Genet. 2007;80:727–739. doi: 10.1086/513473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang C, Xiao Y, Yang L, Chen Q, Wang Z, Conway SJ, Chen Y. Intragenic deletion of TGIF causes defects in brain development. Hum Mol Genet. 2006;15:3508–3519. doi: 10.1093/hmg/ddl427. [DOI] [PubMed] [Google Scholar]
- Kudoh T, Wilson SW, Dawid IB. Distinct roles for Fgf, Wnt and retinoic acid in posteriorizing the neural ectoderm. Development. 2002;129:4335–4348. doi: 10.1242/dev.129.18.4335. [DOI] [PubMed] [Google Scholar]
- Lagutin OV, Zhu CC, Kobayashi D, Topczewski J, Shimamura K, Puelles L, Russell HR, Mckinnon PJ, Solnica-Krezel L, Oliver G. Six3 repression of Wnt signaling in the anterior neuroectoderm is essential for vertebrate forebrain development. Genes Dev. 2003;17:368–379. doi: 10.1101/gad.1059403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavado A, Lagutin OV, Oliver G. Six3 inactivation causes progressive caudalization and aberrant patterning of the mammalian diencephalon. Development. 2007;135:441–450. doi: 10.1242/dev.010082. [DOI] [PubMed] [Google Scholar]
- Lehman NL, Zaleski DH, Sanger WG, Adickes ED. Holoprosencephaly associated with an apparent isolated 2q37.1-2q37.3 deletion. Am J Med Genet. 2001;100:179–181. doi: 10.1002/ajmg.1245. [DOI] [PubMed] [Google Scholar]
- Li Hs, Tierney C, Wen L, Wu JY, Rao Y. A single morphogenetic field gives rise to two retina primordial under the influence of the prechordal plate. Development. 1997;124:603–615. doi: 10.1242/dev.124.3.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe LA, Yamada S, Kuehn MA. Genetic dissection of nodal function in patterning the mouse embryo. Development. 2001;128:1831–1843. doi: 10.1242/dev.128.10.1831. [DOI] [PubMed] [Google Scholar]
- Ma Y, Erkner A, Gong R, Yao S, Taipale J, Basler K, Beachy PA. Hedgehog-mediated patterning of the mammalian embryo requires transporter-like function of Dispatched. Cell. 2002;111:63–75. doi: 10.1016/s0092-8674(02)00977-7. [DOI] [PubMed] [Google Scholar]
- Maity T, Fuse N, Beachy PA. Molecular mechanisms of Sonic hedgehog mutant effects in holoprosencephaly. Proc Natl Acad Sci, USA. 2005;102:17026–17031. doi: 10.1073/pnas.0507848102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marlow F, Zwartkruis F, Malicki J, Neuhaus SC, Abbas L, Weaver M, Driever W, Solnica-Krezel L. Functional interactions of genes mediating convergent extensión, knypek and trilobite, during the partitioning of the eye primordium in zebrafish. Dev Biol. 1998;203:382–399. doi: 10.1006/dbio.1998.9032. [DOI] [PubMed] [Google Scholar]
- Maurus D, Harris WA. Zic-associated holoprosencephaly: zebrafish Zic1 controls midline formation and forebrain patterning by regulating Nodal, Hedgehog, and retinoic acid signaling. Genes Dev. 2009;23:1461–1473. doi: 10.1101/gad.517009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming JE, Kaupas ME, Roessler E, Brunner HG, Golabi M, Tekin M, Stratton RF, Sujansky E, Bale SJ, Muenke M. Mutations in PATCHED-1, the receptor for SONIC HEDGEHOG, are associated with holoprosencephly. Hum Genet. 2002;110:297–301. doi: 10.1007/s00439-002-0695-5. [DOI] [PubMed] [Google Scholar]
- Ming JE, Muenke M. Multiple hits during early embryonic development: digenic disease and holoprosencephaly. Am J Hum Genet. 2002;71:1017–1032. doi: 10.1086/344412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizugishi K, Aruga J, Nakata K, Mikoshiba K. Molecular properties of Zic proteins as transcriptional regulators and their relationship to GLI proteins. J Biol Chem. 2001;276:2180–2188. doi: 10.1074/jbc.M004430200. [DOI] [PubMed] [Google Scholar]
- Monuki ES. The morphogen signaling network in forebrain development and holoprosencephaly. J Neuropath Exp Neurosci. 2007;66:566–575. doi: 10.1097/nen.0b013e3180986e1b. [DOI] [PubMed] [Google Scholar]
- Muenke M, Beachy PA. Holoprosencephaly. In: Scriver CR, et al., editors. The Metabolic & Molecular Bases of Inherited Disease. McGraw-Hill; New York: 2001. pp. 6203–6230. [Google Scholar]
- Nagai T, Aruga J, Takada S, Gunther T, Sorle R, Schughart K, Mikoshiba K. The expression of the mouse Zic1, Zic2, and Zic3 gene suggests and essential role for Zic genes in body pattern formation. Dev Biol. 1997;182:299–313. doi: 10.1006/dbio.1996.8449. [DOI] [PubMed] [Google Scholar]
- Nagai T, Aruga J, Minowa O, Sugimoto T, Ohno Y, Noda T, Mikoshiba K. Zic2 regulates the kinetics of neurulation. Proc Natl Acad Sci, USA. 2000;97:1618–1623. doi: 10.1073/pnas.97.4.1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasevicious A, Ekker SC. Effective targeted gene “knockdown” in zebrafish. Nat Genet. 2000;26:216–220. doi: 10.1038/79951. [DOI] [PubMed] [Google Scholar]
- Oliver G, Mailhos A, Wehr R, Copeland NG, Jenkins NA, Gruss P. Six3, a murine homlogue of the sine oculis gene, demarcates the most anterior border of the developing neural plate and is expressed during eye development. Development. 1995;121:4045–4055. doi: 10.1242/dev.121.12.4045. [DOI] [PubMed] [Google Scholar]
- Overhauser J, Mitchell HF, Zackai EH, Rojas K, Muenke M. Physical mapping of the holoprosencephaly critical region in 18p11.3. Am J Hum Genet. 1995;57:1080–1085. [PMC free article] [PubMed] [Google Scholar]
- Pavletich NP, Pabo CO. Crystal structure of a five-finger GLI-DNA complex: new perspectives on zinc fingers. Science. 1993;261:1701–1707. doi: 10.1126/science.8378770. [DOI] [PubMed] [Google Scholar]
- Rahimov F, Ribeiro LA, de Miranda E, Richieri-Costa A, Murray JC. GLI2 mutations in four Brazilian patients: how wide is the phenotypic spectrum? Am. J Med Genet. 2006;140A:2571–2576. doi: 10.1002/ajmg.a.31370. [DOI] [PubMed] [Google Scholar]
- Redemann N, Gaul U, Jäckle H. Disruption of a putative Cys-zinc interaction eliminates the biological activity of the Krüppel finger protein. Nature. 1988;332:90–92. doi: 10.1038/332090a0. [DOI] [PubMed] [Google Scholar]
- Ribeiro LA, Murray JC, Richieri-Costa A. PTCH mutations in four Brazilian patients with holoprosencephaly and in one with holoprosencephaly-like features and normal MRI. Am J Med Genet. 2006;140A:2584–2586. doi: 10.1002/ajmg.a.31369. [DOI] [PubMed] [Google Scholar]
- Rohr KB, Barth KA, Varga ZM, Wilson SW. The nodal pathway acts upstream of hedgehog signaling to specify ventral telencephalic identity. Neuron. 2001;29:341–351. doi: 10.1016/s0896-6273(01)00210-0. [DOI] [PubMed] [Google Scholar]
- Roessler E, Belloni E, Gaudenz K, Jay P, Berta P, Scherer SW, Tsui LC, Muenke M. Mutations in the human Sonic Hedgehog gene cause holoprosencephaly. Nat Genet. 1996;14:357–360. doi: 10.1038/ng1196-357. [DOI] [PubMed] [Google Scholar]
- Roessler E, Ward DE, Gaudenz K, Belloni E, Scherer SW, Donnai D, Siegel-Bartelt J, Tsui LC, Muenke M. Cytogenetic rearrangements involving the loss of the Sonic Hedgehog gene at 7q36 cause holoprosencephaly. Hum Genet. 1997;100:172–181. doi: 10.1007/s004390050486. [DOI] [PubMed] [Google Scholar]
- Roessler E, Du Y, Glinka A, Dutra A, Niehrs C, Muenke M. The gene structure, chromosomal location, and analysis of the human DKK1 head inducer gene as a candidate for holoprosencephaly. Cytogenet Cell Genet. 2000;89:220–224. doi: 10.1159/000015618. [DOI] [PubMed] [Google Scholar]
- Roessler E, Muenke M. Midline and laterality defects: left and right meet in the middle. BioEssays. 2001;23:888–900. doi: 10.1002/bies.1130. [DOI] [PubMed] [Google Scholar]
- Roessler E, Du YZ, Mullor JL, Casas E, Allen WP, Gillessen-Kaesbach G, Roeder ER, Ming JE, Ruiz i Altaba A, Muenke M. Loss-of-function mutations in the human GLI2 gene are associated with pituitary anomalies and holoprosencephaly-like features. Proc natl Acad Aci, USA. 2003;100:13424–13429. doi: 10.1073/pnas.2235734100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessler E, Muenke M. How a hedgehog might see holoprosencephaly. Hum Mol Genet. 2003;12 Spec No 1:R15–25. doi: 10.1093/hmg/ddg058. [DOI] [PubMed] [Google Scholar]
- Roessler E, Ermilov AN, Grange DK, Wang A, Grachtchouk M, Dluglosz AA, Muenke M. A previously unidentified amino terminal domain regulates transcriptional activity of wild-type and disease-associated human GLI2. Hum Mol Genet. 2005;14:2181–2188. doi: 10.1093/hmg/ddi222. [DOI] [PubMed] [Google Scholar]
- Roessler E, Ouspenskaia MV, Karkera JD, Vélez JI, Kantipong A, Lacbawan F, Bowers P, Belmont JW, Towbin JA, Goldmuntz E, Feldman B, Muenke M. Reduced NODAL signaling strength via mutation of several pathway members including FOXH1 is linked to human heart defects and holoprosencephaly. Am J Hum Genet. 2008;83:18–29. doi: 10.1016/j.ajhg.2008.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessler E, Lacbawan F, Dubourg C, Paulussen A, Herbergs J, Hehr U, Bendavid C, Zhou N, Ouspenskaia M, Bale S, Odent S, David V, Muenke M. The full spectrum of holoprosencephaly-associated mutations within the ZIC2 gene in humans predict loss-of-function as the predominant disease mechanism. Hum Mutat. 2009a;30:E541–544. doi: 10.1002/humu.20982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessler E, Ma Y, Ouspenskaia MV, Lacbawan F, Bendavid C, Dubourg C, Beachy PA, Muenke M. Truncating loss-of function mutations of DISP1 contribute to holoprosencephly-like microform features in humans. Hum Genet. 2009b;125:393–400. doi: 10.1007/s00439-009-0628-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessler E, El-Jaick KB, Dubourg C, Vélez JI, Solomon BD, Pineda-álvarez DE, Lacbawan F, Zhou N, Ouspenskaia M, Paulussen A, Smeets HJ, Hehr U, Bendavid C, Bale S, Odent S, David V, Muenke M. The mutational spectrum of holoprosencephaly-associated changes within the SHH gene in humans predicts loss-of-function through either key structural alterations of the ligand or its altered synthesis. Hum Mut. 2009c doi: 10.1002/humu.21090. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessler E, Pei W, Ouspenskaia MV, Karkera JD, Vélez JI, Banerjee-Basu S, Gibney G, Lupo PJ, Mitchell LE, Towbin JA, Bowers P, Belmont JW, Goldmuntz E, Baxevanis AD, Feldman B, Muenke M. Cumulative ligand activity of NODAL mutations and modifiers are linked to human heart defects and holoprosencephly. Mol Genet Metabol. 2009d doi: 10.1016/j.ymgme.2009.05.005. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein JL, Shimamura K, Martinez S, Puelles L. Regionalization of the prosencephalic neural plate. Ann Rev Neurosci. 1998;21:445–477. doi: 10.1146/annurev.neuro.21.1.445. [DOI] [PubMed] [Google Scholar]
- Schachter KA, Krauss RS. Murine models of holoprosencephaly. Curr Topics Dev Biol. 2008;84:140–170. doi: 10.1016/S0070-2153(08)00603-0. [DOI] [PubMed] [Google Scholar]
- Schell-Apacik C, Rivero M, Knepper JL, Roessler E, Muenke M, Ming JE. SONIC HEDGEHOG mutations causing human holoporsencephaly impair neural patterning activity. Hum Genet. 2003;113:170–177. doi: 10.1007/s00439-003-0950-4. [DOI] [PubMed] [Google Scholar]
- Schier AF. Nodal signaling in vertebrate development. Ann Rev Cell Dev Biol. 2003;19:589–621. doi: 10.1146/annurev.cellbio.19.041603.094522. [DOI] [PubMed] [Google Scholar]
- Seppala M, Depew MJ, Martinelli DC, Fan CM, Sharpe PT, Cobourne MT. Gas1 is a modifier for holoprosencephaly and genetically interacts with sonic hedgehog. J Clin Invest. 2007;117:1575–1584. doi: 10.1172/JCI32032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Walshe CA. Targeted disruption of Tgif, the mouse ortholog of the human holoprosencephaly gene, does not result in holoprosencephaly in mice. Mol Cell Biol. 2005;25:3639–3647. doi: 10.1128/MCB.25.9.3639-3647.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen MM, Schier AF. The EGF-CFC family in vertebrate development. Trends Genet. 2000;16:303–309. doi: 10.1016/s0168-9525(00)02006-0. [DOI] [PubMed] [Google Scholar]
- Shen MM. Nodal signaling: developmental roles and regulation. Development. 2007;134:1023–1034. doi: 10.1242/dev.000166. [DOI] [PubMed] [Google Scholar]
- Shih J, Fraser SE. Characterizing the zebrafish organizer: microsurgical analysis at the early shield stage. Development. 1996;122:131–1322. doi: 10.1242/dev.122.4.1313. [DOI] [PubMed] [Google Scholar]
- Singh S, Tokhunts R, Baubet V, Goetz JA, Huang ZJ, Schilling NS, Black KE, MacKenzie TA, Dahmane N, Robbins DJ. Sonic hedgehog mutations identified in holoprosencephaly patients can act in a dominant negative manner. Hum Genet. 2009;125:95–103. doi: 10.1007/s00439-008-0599-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm EE, Garel S, Borello U, Hebert JM, Martinez S, McConnell SK, Martin GR, Rubenstein JLR. Dose-dependent functions of Fgf8 in regulating telencephalic patterning centers. Development. 2006;133:1831–1844. doi: 10.1242/dev.02324. [DOI] [PubMed] [Google Scholar]
- Sulik KK, Dehart DB, Rogers JM, Chernoff N. Teratogenicity of low doses of all-trans retinoic acid in presomite mouse embryos. Teratology. 1995;51:398–403. doi: 10.1002/tera.1420510605. [DOI] [PubMed] [Google Scholar]
- Tenzen T, Allen BL, Cole F, Kang JS, Krauss RS, McMahon AP. The cell surface membrane protein Cdo and Boc are components and targets of the hedgehog signaling pathway and feedback network in mice. Dev Cell. 10:647–656. doi: 10.1016/j.devcel.2006.04.004. [DOI] [PubMed] [Google Scholar]
- Traiffort E, Dubourg C, Faure H, Rognan D, Odent S, Durou MR, David V, Ruat M. Functional characterization of Sonic Hedgehog mutations associated with holoprosencephaly. J Biol Chem. 2004;279:42889–42897. doi: 10.1074/jbc.M405161200. [DOI] [PubMed] [Google Scholar]
- Tyschenko N, Lurie I, Schinzel A. Chromosomal map of human brain malformations. Hum Genet. 2008;124:73–80. doi: 10.1007/s00439-008-0528-2. [DOI] [PubMed] [Google Scholar]
- Varga ZM, Wegner J, Westerfield M. Anterior movement of ventral diencephalic precursors separates the primordial eye field in the neural plate and requires Cyclops. Development. 1999;126:5533–5546. doi: 10.1242/dev.126.24.5533. [DOI] [PubMed] [Google Scholar]
- Vincent SD, Ray Dun N, Hayashi S, Norris DP, Robertson EJ. Cell fate decisions within the mouse organizer are governed by graded Nodal signals. Genes Dev. 2003;17:1646–1652. doi: 10.1101/gad.1100503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallis DE, Roessler E, Hehr U, Nanni L, Wiltshire T, Richieri-Costa A, Gillessen-Kaesbach G, Zackai EH, Rommens J, Muenke M. Missense mutations in the homeodomain of the human SIX3 gene cause holoprosencephaly. Nat Genet. 1999;22:196–198. doi: 10.1038/9718. [DOI] [PubMed] [Google Scholar]
- Watanabe K, Kamiya D, Nishiyama A, Katayama T, Nozaki S, Kawasaki H, Watanabe Y, Mizuseki K, Sasai Y. Directed differentiation of telecephalic precursors from embryonic stem cell. Nature Neurosci. 2005;8:288–296. doi: 10.1038/nn1402. [DOI] [PubMed] [Google Scholar]
- Warr N, Powles-Glover N, Chappell A, Robson J, Norris D, Arkell R. Zic2-associated holoprosencephaly is caused by a transient defect in the organizer region during gastrulation. Hum Mol Genet. 2008;17:2986–2996. doi: 10.1093/hmg/ddn197. [DOI] [PubMed] [Google Scholar]
- Wilson SW, Houart C. Early steps in the development of the forebrain. Dev Cell. 2004;6:167–181. doi: 10.1016/s1534-5807(04)00027-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RJ, Nie Q, Lander AD, Schilling TF. Complex regulation of cyp26a1 created a robust retinoic acid gradient in the zebrafish embryo. PLOS. 2007;5:2522–2533. doi: 10.1371/journal.pbio.0050304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wotton D, Lo RS, Lee S, Massague J. A Smad transcriptional corepressor. Cell. 1999;97:29–39. doi: 10.1016/s0092-8674(00)80712-6. [DOI] [PubMed] [Google Scholar]
- Zhang W, Kang JS, Cole F, Yi MJ, Krauss RS. Cdo functions at multiple points in the Sonic Hedgehog pathway, and Cdo-deficient mice accurately model human holoprosencephaly. Dev Cell. 2006;10:657–665. doi: 10.1016/j.devcel.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Zhang XM, Ramalho-Santos M, McMahon AP. Smoothened mutants reveal redundant roles for Shh and Ihh signaling including regulation of L/R asymmetry in the mouse node. Cell. 2001;105:781–792. [PubMed] [Google Scholar]
- Zhu CC, Dyer MA, Uchikawa M, Kondoh H, Lagutin OV, Oliver G. Six3-mediated auto repression and eye development requires its interaction with members of the Groucho-related family of co-repressors. Development. 2002;129:2835–2849. doi: 10.1242/dev.129.12.2835. [DOI] [PubMed] [Google Scholar]