

Abstract
In addition to the proteolytic cleavages that give rise to amyloid-β (Aβ), the amyloid-β protein precursor (AβPP) is cleaved at Asp664 intracytoplasmically. This cleavage releases a cytotoxic peptide, APP-C31, removes AβPP-interaction motifs required for signaling and internalization, and is required for the generation of AD-like deficits in a mouse model of the disease. Although we and others had previously shown that Asp664 cleavage of AβPP is increased in AD brains, the distribution of the Asp664-cleaved forms of AβPP in non-diseased and AD brains at different ages had not been determined. Confirming previous reports, we found that Asp664-cleaved forms of AβPP were increased in neuronal cytoplasm and nuclei in early-stage AD brains but were absent in age-matched, non-diseased control brains and in late-stage AD brains. Remarkably, however, Asp664-cleaved AβPP was prominent in neuronal somata and in processes in entorhinal cortex and hippocampus of non-diseased human brains at ages <45 years. Our observations suggest that Asp664 cleavage of AβPP may be part of the normal proteolytic processing of AβPP in young (<45 years) human brain and that this cleavage is down-regulated with normal aging, but is aberrantly increased and altered in location in early AD.
Keywords: Alzheimer's disease, Amyloid-β protein precursor, APP-C31, APPNeo, Asp664, proteolytic processing
INTRODUCTION
Alzheimer's disease (AD) is the most common cause of dementia, affecting about 5.1 million people in the United States. Current treatment for AD is only marginally effective in most cases, consisting of drugs that may ameliorate cognitive and behavioral symptoms, but do not arrest or reverse the underlying neurodegenerative process. AD is characterized by senile plaques, neurofibrillary tangles, and loss of synapses and neurons in the brain. The predominant proteinaceous component in senile plaques is the amyloid-β peptide (Aβ), which has been proposed to mediate synaptic toxicity and functional deficits in AD [56]. Even though the processes involved in the generation of Aβ through cleavage of the amyloid-β protein precursor (AβPP) are being elucidated, the physiological function(s) of AβPP remain incompletely defined. Accumulating evidence now suggests that AβPP is involved in synapse formation and function [26,27,46] and that its carboxy (C)-terminus is involved in multiple cellular processes [10,19,24–27,51,52].
We [15,35] and others [17,61] have shown that, in addition to the proteolytic cleavages that give rise to Aβ, AβPP can be cleaved by caspases, enzymes required for the execution of programmed cell death, near its C-terminus. This cleavage releases a cytotoxic peptide, C31 [15,17,32,35,37,44,61], occurs in AD brains [2,12,17,35,63], and removes motifs important for the interaction of AβPP with cellular components [19,24–27,51,52]. Moreover, the C31 peptide released by C-terminal cleavage of AβPP can directly activate a classical pathway to apoptosis through an N-terminal Smac/DIABLO-like motif, which binds and antagonizes the family of inhibitor of apoptosis proteins (IAPs) [21].
Evidence for a crucial role of cleavage of AβPP at Asp664 in the pathogenesis of AD came from our studies that demonstrated that mice expressing a FAD-mutant human AβPP (hAβPP) transgene identical to that expressed in an established mouse model of AD (PDAPP mice), but carrying a mutation at the caspase cleavage site that prevents cleavage of hAβPP by caspases at Asp664 in vivo continued to produce Aβ and showed plaque deposition, but did not demonstrate anatomical, synaptic or behavioral deficits that characterize PDAPP mice [16,53]. These studies indicated that cleavage of AβPP at Asp664 has a crucial role in the generation of AD-like deficits in a transgenic mouse model of the disease. Considered together with recent in vitro evidence demonstrating the binding and multimerization of AβPP by Aβ [33,36,37,57], our observations strongly suggest that the generation of AD-like functional and anatomical deficits may be downstream of Aβ production, through a mechanism that involves AβPP processing at Asp664. One potential link between Aβ production and the generation of C31 has been described, with the demonstration that Aβ binds AβPP and induces AβPP multimerization, leading in turn to cleavage of the AβPP cytosolic tail and intracellular release of C31, followed by synaptic and neuronal damage [33,36,37,57].
In addition to generating a toxic peptide, C31, cleavage of AβPP at Asp664 removes protein-protein interaction motifs embedded in the C31 peptide. Thus, Asp664-cleaved AβPP lacks sequences that are required for its interaction with motor proteins, components of the stress response, and transcriptional transactivators. These motifs partially overlap with AβPP's YENPTY internalization signal [45] that is often found in cytoplasmic tails of membrane receptors [9]. The C31 peptide also contains a KF-FEQ sequence that allows chaperone-mediated autophagy [39]. The YENPTY motif acts as a binding site for several AβPP-interacting proteins such as kinesin [25], and phosphotyrosine-binding domain proteins such as Fe65 [14,52,62], Mint1/X11 [5,41], Disabled-1 [22], JIP-1 and JNK1 [40,54,59]. These interactions are thought to define,at least partially, a function for AβPP in axonal trafficking [19,24,25], in cellular motility [51,52], in vesicular transport [25], and in the regulation of synaptic function [26,27]. Cleavage of AβPP by transiently activated caspases or caspase-like proteases at neuronal terminals may therefore disrupt its interaction with several different protein complexes, and thus alter the normal signaling emanating from the molecule.
Consistent with the hypothesis that cleavage of AβPP at Asp664 may have a crucial role in AD pathogenesis, it was shown by us [15,35]; and others [2,17, 63] that fragments of AβPP cleaved at Asp664 are increased in AD brains compared to aged controls. The study of Asp664 cleavage of AβPP in intact or solubilized tissues and the positive identification of this C-terminal cleavage event was confirmed using a neoepitope antibody that was originally generated by Gervais et al. [17]. This antisera was raised against a synthetic peptide corresponding to the novel C-terminus of AβPP generated after cleavage at Asp664, and was extensively characterized by this group [17] by us [12, 15,16] and by others [2,63]. While our studies [15,35] had shown that Asp664-cleaved fragments of AβPP accumulate in AD brains as compared to age-matched controls, the studies of Zhao et al. demonstrated that Asp664 cleavage of AβPP may be an early event in the pathogenesis of the disease, closely associated with the appearance of active forms of caspase-3, and that Asp664-cleaved forms of accumulate in neuronal bodies as well as in neuronal terminals [63]. Moreover, Ayala-Grosso et al. demonstrated using the same cleavage-specific antibody that, in addition to being associated with plaques and dystrophic neurites in AD brains, Asp664-cleaved forms of AβPP were also found in several control cases that had a history of metabolic encephalopathy and stroke [2]. In both studies, immunoreactivity for the Asp664-cleaved forms of AβPP disappeared from older dense-core plaques, suggesting that these Asp664-cleaved fragments are cleared as the disease progresses.
Interestingly, our previous studies had shown that Asp664-cleaved fragments of AβPP were present in young, non-diseased mouse brains [16]. These observations raised the intriguing possibility that cleavage at Asp664 may occur in non-diseased conditions in human brain, at ages earlier than those of age-matched non-diseased subjects included in control groups in previous studies [2,15,17,63]. With the present study, we aimed to further our previous observations in aged AD and control brains and to define the temporal and spatial distribution of Asp664 cleavage of AβPP throughout life in non-diseased human brain. Confirming previous reports [2,63] and our previous studies [15,35] we showed that Asp664-cleavage of AβPP is pronounced in early AD but is not detectable in age-matched, non-diseased control brains, nor in late-stage AD. Consistent with previous data from other groups [2,15,35,63], a significant fraction of Asp664-cleaved AβPP fragments in AD brains colocalized with chromatin in neuronal nuclei. Remarkably, and in agreement with our previous observations in young, non-diseased mouse brains [16], Asp664-cleaved fragments of AβPP were prominent both in neuronal bodies and in processes of young, non-diseased human brains. The abundance of these Asp664-cleaved fragments diminished progressively with normal aging (defined here as the absence of development of AD) but was abnormally increased in subjects undergoing early AD changes. Our observations indicate that Asp664-cleaved forms of AβPP are abundant at neuronal terminals and in neuronal bodies in young, non-diseased human brain, and that these Asp664-cleaved AβPP fragments diminish in abundance with normal aging. However, and in agreement with previous reports [2,16,17,63], Asp664-cleaved AβPP fragments increased dramatically in AD. However, in contrast to previous reports [63], our data indicate that Asp664-cleaved AβPP fragments increase in early AD stages and ultimately decrease as the disease progresses. Taken together with the available experimental data [2,12,15–17,63], our results suggest that the C-terminal cleavage of AβPP at Asp664 may be part of physiological proteolytic processes that regulate AβPP signaling or turnover in young, non-diseased human brain, and that this putative regulatory event decreases with normal aging but is aberrantly activated in the early stages of AD.
MATERIALS AND METHODS
Antibodies
The APPNeo antibody recognizes the neoepitope generated after cleavage of AβPP at Asp664 (APP695 numbering) and has been extensively characterized by us and others [2,12,15–17,63]. Briefly, rabbits were immunized with the peptide 657CIHHGVVEVD664, which includes the nine amino acids immediately preceding the caspase cleavage site at position 664 in AβPP, coupled to KLH. Antisera were purified in three successive steps: 1. Antisera were affinity purified by binding to immobilized peptide antigen. Bound antibodies were eluted by a pH gradient. 2. The eluate from (1) was depleted of immunoglobulins recognizing the intact AβPP molecule by adsorption to a bridging peptide encompassing the cleavage site at Asp664 (TSIHHGVVEVDAAVTPEE) immobilized on an activated support. 3. The flowthrough from (2) was affinity-purified on immobilized immunogenic peptide. After washing, bound antibodies were eluted by a pH gradient, collected and stored in borate buffer.
The 5A3/1G7 monoclonal mixture and CT15 were described previously [16]. All HRP-conjugated secondary antibodies were purchased from Jackson ImmunoResearch Laboratories, West Grove, PA. Anti-calreticulin (SPA600) and anti-proline disulfide isomerase (PDI, SPA891) were purchased from Stressgen (Ann Arbor, Michigan). Alexa Fluor488-conjugated donkey anti-mouse, Alexa Fluor488 donkey anti-rabbit IgGs (H+L) were purchased from Molecular Probes, Carlsbad, CA. Nuclei were visualized using TOTO-3 iodide (642/660) nuclear counterstain (Molecular Probes).
Western blotting
Human brain tissue extracts were prepared from frozen autopsy material, kindly provided by the Harvard Brain Bank Tissue Resource Center, which is supported in part by PHS grant number MH/NS 31862. Both human and mouse extracts were solubilized in 0.15 M NaCl, 0.02 M Tris-Hcl pH = 7.5, 1.0% Triton X-100, 1 mM EDTA, 5 mM EGTA, 3 mM DTT containing a cocktail of protease inhibitors (Mini-Complete™, Invitrogen, Carlsbad, CA). Tissue extracts were maintained at 4°C. Homogenization of 50 mg/ml of tissue (wet weight) was carried out using an ultrasonic processor (Vibracell, Sonics) at 80% amplitude for ∼ 1.5 minutes, in 3 separate pulses. The extract was centrifuged for 10 min at 14,000 RPM and the pellet was resuspended in the same buffer to which sodium deoxycholate was added to a final concentration of 2%. The two supernatants were mixed at a 1:1 ratio and protein concentration was determined by a standard Bradford assay (Coomasie Plus, Pierce, Rockford, IL). Thirty mg of total protein per sample were disrupted in Laemmli buffer (Invitrogen) and resolved in 7% Tris-acetate gels (Invitrogen). The separated polypeptides were transferred to PVDF membranes (Schleicher and Schuell, Whatman, UK), blocked in 5% non-fat milk and immunoblotted with the indicated antibodies. Pharmacia, Pittsburgh, PA) and detected using Kodak Biomax MR or ML film (Kodak Eastman, New Haven, CT).
Immunohistochemistry
Mouse brain samples were from 2−4 month-old (mo) and 24 mo PDAPP transgenic mice, which express a human AβPP minigene carrying the Swedish and Indiana familial Alzheimer's disease (FAD) mutations [23,42] and from PDAPP(D664A) transgenic mice, which express the same FAD-hAβPP minigene but carry an additional D664A mutation that obliterates the Asp664 cleavage site at the C-terminus of hAβPP [16,53]. PDAPP mice were kindly provided by Lennart Mucke, Gladstone Institutes, University of California at San Francisco. Non-transgenic littermates were used as controls. Animals were bred and maintained at the Buck Institute Animal Facility under an approved IACUC protocol and in compliance with regulations in force for the use and care of animals in research. Human brain samples fixed in formalin were kindly provided by the Harvard Brain Bank Tissue Resource Center, which is supported in part by PHS grant number MH/NS 31862. Tissue was further processed for paraffin embedding. Mouse brains were fixed in 4% paraformaldehyde and also processed for paraffin embedding. Seven-micrometer microtome sections of paraffin sections from mouse and human brains were deparaffinized in xylene, rehydrated in 100, 95, 80 and 70% ethanol, and washed in 1 X TBS for 15 min at room temperature (RT). Microwave antigen retrieval was performed in 10mM citrate buffer (pH 6.0) for 5 min at 440 Watts. Slides were allowed to cool to RT for 20 min and were washed in 1 X TBS for 15 min. Samples were blocked in 10% normal donkey serum in 1 X TBS for 1 h at RT. APPNeo was applied at a dilution of 1:1000, α-calreticulin was applied at a 1:200 dilution and α-PDI at a 1:100 dilution in 1% bovine serum albumin (BSA) in 1 X TBS. Sections were incubated overnight at 4°C. Rabbit or rabbit and mouse pre-immune IgGs diluted to 1 mg/ml in 1% BSA in 1 X TBS were used as negative control. Sections were washed for 30 min in three changes of 1 X TBS. AlexaFluor488- and Alexa555-conjugated donkey α-rabbit and α-mouse secondary antibodies were applied at a 1:250 dilution. Sections were washed three times in TBS for 30 minutes, counterstained with TOTO-3 at a 1:500 dilution in PBS for 10 min, washed, and mounted with Prolong Gold antifade (Molecular Probes, Invitrogen). For DAB staining, tissue sections were treated for 15 min with 3% H2O2 to inactivate endogenous peroxidases. After primary antibody incubation, biotinylated goat α-rabbit IgG (Vector Laboratories, Burlingame, CA) was applied at a 1:200 dilution for 1 h at RT. Peroxidase-based ABC Elite kit (Vector Laboratories) was used according to the manufacturer's instructions followed by a 30 min wash in three changes of 1 X TBS. A liquid DAB kit (Vector Laboratories) was used for detection. Color development was monitored under the microscope. Sections were washed in 1 X TBS, briefly counterstained in aqueous hematoxylin, dehydrated, cleared, and mounted in Permount (Fisher Scientific, Pittsburgh, PA). Sections incubated without primary antibody or with preimmune rabbit IgG served as controls. Images were acquired using a Nikon Eclipse-8000 microscope and an Optronics Magnafire camera and software. Low magnification images were collected using a Nikon SMZ-U dissecting microscope and a CoolSnap camera and software. Single confocal images or z-stacks of images were acquired using a Nikon Eclipse-800 microscope and collected using Compix Simple PCI software. Where indicated, deconvolution was done using a maximum likelihood estimation algorithm with Huygens software and then processed by the Imaris imaging interphase (Bitplane AG, Zurich, Switzerland). All single confocal images and stacks of images were then processed in a SGI Octane R12 computer running Bitplane's Advanced Imaging Software suite. Analysis of colocalization of APPNeo and calreticulin were done using the Imaris Coloc algorithm [11].
Quantitation of APPNeo immunoreactivity
Selected anatomical locations were cornu ammonis 3 (CA3) or molecular layer (ML) regions of hippocampus, dorsal and medial entorhinal cortex, and parahippocampal gyrus. Three fields were randomly sampled and collected at 600X magnification within each region to represent the anatomical area of interest, avoiding overlap between fields. When relevant, areas examined in severe AD cases were selected in which the total number of nuclei were similar to those in non-diseased samples to minimize differences due to neuronal loss. Images were processed with Imaris imaging interphase (Bitplane AG). Total areas of APPNeo immunoreactivity were determined. Data were compiled using Excel (Microsoft, Redmond, WA) and significance of differences between groups were determined by Student's t test or one-way ANOVA using GraphPad Prism (GraphPad, San Diego, CA).
RESULTS
Asp664 cleavage of AβPP in mouse brains
The antibody that recognizes the neo-epitope generated by cleavage of AβPP at Asp664 in its intracellular, C-terminal domain has been extensively characterized in vitro by us and others [15,17], and in mouse [16] and human tissues [2,12,15,17,63]. This antibody was confirmed to be highly specific for the neo epitope generated after Asp664 cleavage by three criteria: (1) The ELISA titer for this preparation was < 1:142,000 (<5 ng/mL) against the immunizing peptide versus >1:70 (>10 mg/mL) against the bridging peptide that corresponds to the intact AβPP sequence across the caspase cleavage site at Asp664; (2) The antibody could immunoprecipitate or recognize in western blots AβPP that was truncated at Asp664 but not intact AβPP [12,15,17]; (3) The antibody could not recognize intact AβPP from healthy cells that were transfected with full-length AβPP but could readily react with AβPP fragments that were generated after treatment of these cells with staurosporine, in the absence of the caspase inhibitor BAF [15]. The same was true for NT2 cells [17]. Moreover, that APPNeo is highly specific for cleaved human (hAβPP) and mouse AβPP (mAβPP) was confirmed in various studies, both by us and by others [2,12,15,17,63].
Confirming our previous observations [16], cleavage of AβPP at Asp664 was increased in brain sections from mice expressing a hAPP transgene in which the D664 cleavage site was intact (PDAPPmice [23,42]), but not in sections from mice in which the Asp664 site had been mutated to Ala (PDAPP(D664A) mice [16, 53]. In agreement with our previous observations [16], APPNeo immunoreactivity in PDAPP(D664A) brains was indistinguishable from that in non-transgenic mouse brains, since in both genotypes all APPNeo epitopes available arise from cleavage of mAβPP given that transgenic hAβPP is ‘uncleavable’ due to the D to A substitution. Thus, and albeit at lower levels than in transgenic PDAPP brains, APPNeo immunoreactivity was detectable in non-transgenic mouse brains, indicating that endogenous mAβPP is cleaved at Asp664 in non-diseased conditions.
To determine Asp664 cleavage at different ages in non-transgenic, non-diseased mouse brains, we examined hippocampal and cortical sections from young (2 mo) and old (24 mo) non-transgenic mice and hAβPP transgenic mice by immunohistochemistry using APPNeo. As previously described [16], prominent APPNeo immunoreactivity indicative of AβPP cleavage at Asp664 was present in bodies of granular and pyramidal cells and in coarse deposits in the neuropil in the hippocampus and cortex of 2 mo hAβPP transgenic mice and also in non-transgenic littermates’, albeit at lower levels (Fig. 2a). Surprisingly, quantitative determinations of Asp664-cleaved forms of AβPP in young and aged transgenic and non-transgenic PDAPP mouse brains revealed that Asp664-cleaved AβPP decreased in transgenic PDAPP mouse brains while it increased in non-transgenic brains with increasing age (Fig. 2c, n = 12). To determine whether the observed changes in Asp664-cleaved AβPP were not a consequence of age-associated changes in hAβPP levels in mouse brains, we determined levels of hAβPP produced from the hAβPP transgene and of endogenous mAβPP in young (2−4 mo) and old (12−24 mo) transgenic and non-transgenic animals by western blot analysis. In contrast to the observed decrease in Asp664-cleaved forms of transgenic hAβPP in aged PDAPP transgenic brains, total levels of transgenic hAβPP increased with increasing age (Fig. 2d). Levels of endogenous mAβPP, on the other hand, remained constant in non-transgenic mouse brains with increasing age (Fig. 2e). Thus, the observed decrease in Asp664-cleaved forms of AβPP in aged PDAPP transgenic mouse brains was likely underestimated in our quantitative analyses (Fig. 2c) since levels of hAβPP, one of the substrates of cleavage in PDAPP transgenic mouse brains, increase in transgenic brains with increasing age (Fig. 2d). In addition, our data indicate that the increase in Asp664 cleavage of endogenous mAPP in aged animals (Fig. 1c) is not a consequence of increases in levels of expression of the protein.
Fig. 2.

Cleavage of transgenic hAPP and endogenous mAPP are differentially regulated by aging. a, Representative confocal images of young [2 months of age (mo)] and aged [2 years of age (2 y)] mouse brain sections immunostained with APPNeo b, APPNeo was replaced by preimmune rabbit sera. Sections were counterstained with TOTO-3 to visualize nuclei. The far red channel was pseudocolored blue. c, Total area of APPNeo immunoreactivity was quantified in hippocampal sections as described in Methods. d, Lysates from mouse brains as indicated were probed with an antibody that recognizes the extracellular domain of hAPP (5A3/1G7). e, Lysates from mouse brains as indicated were probed with an antibody that recognizes both hAPP and mAPP (CT15).
Fig. 1.
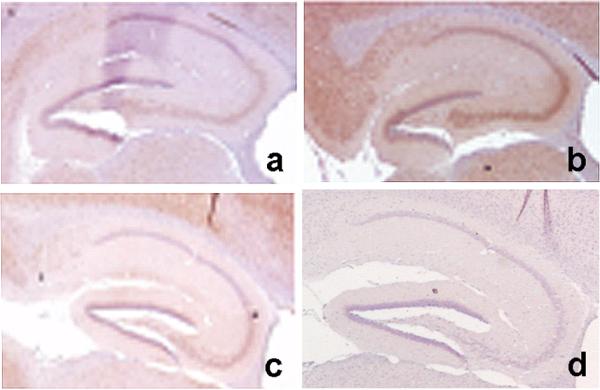
Cleavage of hAPP at Asp664 in vivo is abolished in PDAPP(D664A) transgenic mice. A rabbit antibody specific for the neoepitope generated after cleavage of AβPP at Asp664, APPNeo [2,12,15–17,63], was used to stain hippocampal sections of a, non-transgenic; b, PDAPP transgenic and c, PDAPP(D664A) transgenic mice. d, APPNeo antiserum was replaced by preimmune sera.
These results indicate that Asp664 cleavage of hAβPP, and possibly of endogenous mAβPP, is down-regulated with increasing age in PDAPP transgenic mice undergoing AD-like neurodegenerative changes. In contrast, Asp664 cleavage of endogenous mAβPP in non-diseased mouse brain increases with increasing age in the absence of neurodegenerative changes associated with overexpression of FAD-mutant hAβPP.
AβPP can be cleaved at Asp664 in association with the ER in mouse brain
AβPP is a membrane glycoprotein targeted to synapses [6,26,31,38,52] that is synthesized in association with the endoplasmic reticulum (ER) and is post-translationally processed in the secretory pathway. Since the distribution of APPNeo immunoreactivity in neuronal bodies in mouse brains was reminiscent of ER, we used an antibody specific for proline disulfide isomerase (PDI), an ER-resident protein, as a marker for ER together with APPNeo in immunohistochemical studies aimed to determine whether cleavage of AβPP occurs while in transit through this compartment. As shown in Fig. 3, Asp664-cleaved AβPP molecules in neuronal bodies in mouse cortex were associated with PDI-immunoreactive ER, suggesting that ER-associated cysteine protease(s) such as caspase-7 [47–49] may cleave hAβPP at Asp664 while in transit through the ER in PDAPP transgenic mouse brains. The APPNeo and PDI-immunoreactive signals prominently colocalized (Pearson's correlation coefficient (c) = 0.76). The large value of c for the colocalization of APPNeo and PDI-immunoreactive voxels indicated that the two immunoreactive signals were highly correlated, with 76% of the variability in the location of immunoreactive voxels for each channel predicted from the location of the immunoreactive voxels for the other channel.
Fig. 3.

Asp664-cleaved hAPP are associated with the ER. a, APPNeo (green channel) and α-PDI (red channel) antibodies were used in double immunofluorescence assays on transgenic mouse brain sections followed by Alexa488- and Alexa555-conjugated α-rabbit and α-mouse secondary antibodies respectively. TOTO-3 was used as counterstain to visualize nuclei. The far red channel was pseudocolored blue. Snapshots from maximum intensity projection Imaris Surpass volume images of a representative confocal z-stack for each channel (a, APPNeo, green; b, PDI, red; c, overlap; d, colocalization channel) are shown.
Cleavage of AβPP at Asp664 in human hippocampus
Pathogenic changes in AD involve predominantly association cortices, prominently the entorhinal cortex and hippocampus. It was shown previously that Asp664 cleavage of AβPP occurs in human brain and is increased in hippocampus and frontal cortex of AD patients [2,15,63] and in association with other brain pathologies [2]. To determine whether cleavage of AβPP at Asp664 may occur in young human brains, as suggested by our observations in young non-diseased mice, we examined hippocampus and entorhinal cortex sections of brains from AD patients and from either age-matched or young non-diseased controls using APPNeo. Immunoreactivity patterns and total areas of APPNeo immunoreactivity were determined in a cohort comprising 3 young (ages 23−45y) non-diseased patients, 13 AD cases (ages 68−92y) 5 of which were in the Braak II and III stages, 5 were in the Braak V and VI stages, 3 were AD brains without Braak classification, and in 5 age-matched controls (Table 1). As reported previously [2,15,17,63], quantitation of total APPNeo-immunoreactive areas in human brain sections revealed that Asp664-cleaved AβPP was significantly increased in AD hippocampus as compared with age-matched controls [2,15,63], Fig. 4a, c. Different Braak groups showed pronounced differences in APPNeo immunoreactivity, with earlier Braak stages (II and III) showing increased APPNeo staining whereas later stages (Braak V and VI) showed lower levels of APPNeo staining (Fig. 4a, d). The observed changes in levels of APPNeo immunoreactivity were confirmed by Western blot analyses of brain lysates (Fig. 4g). Thus, and in agreement with the studies of Ayala-Grosso et al. [2], we found a remarkable elevation in APPNeo immunoreactivity in early stages of AD and significantly decreased levels of reactivity in advanced Braak stages. Thus, our results suggest that cleavage of AβPP at Asp664 may be inversely related to the severity of pathology, and argue, in agreement with the observations by Zhao et al. [63], that accumulation of Asp664-cleaved forms of AβPP may represent an early neurodegenerative event in AD.
Table 1.
Patient demographics
| Group | N | Age (avg ± SE) | Sex (no. cases) | PMI |
|---|---|---|---|---|
| Young ND | 3 | 34 ± 5.4 | M(3) | U |
| Aged ND | 5 | 77.6 ± 5 | U(5) | U |
| Braak II and III | 5 | 80 ± 7 | M(2), F(3) | 19.9 ± 9.2 |
| Braak V and VI | 5 | 80.4 ± 2.5 | M(2), F(3) | 14.94 ± 6.19 |
| AD | 4 | 75.5 ± 10 | U(4) | 18.48 ± 2.39 |
N, number of cases; PMI, post-mortem interval; ND, non-diseased; AD, Alzheimer's disease.
Fig. 4.

Asp664 cleavage in human hippocampus. a, Representative low-magnification (100X) confocal images of hippocampal sections from patients at the indicated ages, stained with APPNeo and counterstained with TOTO-3 to visualize nuclei. Braak stages are denoted for AD cases. b and c, Total areas of APPNeo immunoreactivity in non-diseased and AD human samples were quantified as described in Methods. b, Left panel, averages ± SEM of young (20−45 Y) and aged (68−90 Y) are shown. Right panel, averages ± SEM of the indicated ages are shown. c, Total area of APPNeo immunoreactivity in control and AD groups. d, Total area of APPNeo immunoreactivity in different Braak stage groups within the AD group were compared to controls. Averages ± SEM are shown. AD, no break stage specified. e, Representative low-magnification confocal image of APPNeo-immunostained hippocampus in a 23 Y patient. TOTO-3 was used to counterstain nuclei. f, Representative high-magnification confocal images of CA3 hilus (a and b) and ML granular layer (c and d) in young (a and c) and aged (b and d) non-diseased human brains. g, Lysates from tissue samples obtained from the indicated groups were separated electrophoretically and reacted with the indicated antibodies. ND, non diseased.
To investigate whether cleavage of AβPP at Asp664 occurs in brains of younger non-diseased adults we stained brain sections from young (average 34 ± 5.4y) and older (68 and 90y) non-diseased and non-AD individuals respectively with APPNeo. Surprisingly, APPNeo immunoreactivity was prominent in bodies of granular cells (Fig. 4e) and in coarse deposits in the neuropil, likely representing mossy fiber terminal boutons (Fig. 4a), in CA3 hilus in young non-diseased adult brains (Fig. 4a and f [63]). Quantitative image analyses, however, revealed that areas of APPNeo immunoreactivity were pronouncedly decreased in older non-AD brains as compared to young non-diseased ones (Fig. 4b, left panel). Thus, in older non-AD brains, the distribution of Asp664-cleaved forms of AβPP was similar to that observed in younger patients, but their amount was markedly decreased (Fig. 4a and f). In Fig. 4f, compare insets a, c and b, d). Consistent with this observation, APPNeo immunoreactivity decreased monotonically with increasing age in the cohort examined (Fig. 4b, right panel). These decreases in Asp664-forms of AβPP in aged non-diseased brains were not associated with age-specific decreases in AβPP levels; while considerable inter-individual variability in total AβPP levels was observed among samples, these differences in AβPP levels were not correlated with age or with disease state (Fig. 4g, and data not shown). Whether a decrease in AβPP levels might account for the reduced levels of APPNeo staining observed in the advanced Braak stage group could not be determined since fresh frozen tissues were available only for a few samples analyzed. Thus, our results indicate that Asp664 cleavage of AβPP is prominent in young, non-diseased human hippocampus, and that this event declines with normal aging.
Cleavage of AβPP at Asp664 in human entorhinal cortex
The entorhinal cortex (EC), which has reciprocal connections with the hippocampus and various other cortical and subcortical structures, is particularly vulnerable to neuron loss and is thought to be an early site of AD pathology [13]. Since C-terminal fragments of AβPP accumulate at presynaptic terminals of entorhinal neurons [6], we sought to determine whether Asp664-cleaved forms of AβPP may be present in EC of human brain. Consistent with the high levels of Asp664-cleaved forms of AβPP detected in cell bodies of granular neurons and in mossy fiber terminals in hippocampus (Fig. 4a-b, e), APPNeo immunoreactivity was prominent in neuronal processes and cells showing glial morphology in the anterior parahippocampal gyrus of young non-diseased human brains (Fig. 5a, e). Projections and bodies of cells with stellate cell morphology in the superficial EC layers close to the pial surface, likely layer II, also showed prominent APPNeo immunoreactivity (Fig. 5d). APPNeo immunoreactivity was decreased in older, non-AD brains and was undetectable in AD brains (Figs 5a–c). Quantitative analyses revealed that, similar to our observations in hippocampus, APPNeo-immunoreactivity was higher in EC from young (20−45 Y) as compared to older (68−90 Y) non-AD adult brains (Fig. 5b). Total areas of APPNeo immunoreactivity in EC were highest between 30 Y and 45 Y and decreased with increasing age in non-diseased brains (Fig. 5b). In contrast to our observations in hippocampus, however, Asp664-cleaved AβPP was decreased in AD brains as compared to age-matched controls, although this difference did not reach significance due to high variability among non-diseased aged patients (Fig. 5a, c). Within the AD group, however, and consistent with our observations in hippocampus, total area of APPNeo staining was increased in early (Braak II and III) as compared to late (Braak V and VI) stages (Fig. 5c).
Fig. 5.

Asp664 cleavage in human entorhinal cortex. a, Representative low-magnification (100X) confocal images of hippocampal sections from patients at the indicated ages, stained with APPNeo and counterstained with TOTO-3 to visualize nuclei. Braak stage is denoted for the AD case. b and c, Total areas of APPNeo immunoreactivity in non-diseased and AD human samples were quantified as described in Methods. b, Left panel, averages ± SEM of young (20−45 Y) and aged (68−90 Y) samples are shown. Right panel, averages ± SEM of the indicated ages are shown. c, Left panel, total area of APPNeo immunoreactivity in control and AD groups. Right panel, total area of APPNeo immunoreactivity in different Braak stage groups within the AD group were compared to controls. Averages ± SEM are shown. d, Section views of a, green; b, red; c, overlay channels in stacks of confocal images collected from dorsal EC in brain sections of a 30 Y old non-diseased patient immunostained with APPNeo. f, Representative low-magnification (100X) confocal image of parahippocampal gyrus in brain sections of a 45 Y non-diseased patient immunostained with APPNeo. Sections were counterstained with TOTO-3 to visualize nucleicaption.
In addition to the prominent differences in the amount of Asp664-cleaved immunoreactivity of AβPP present in non-diseased young vs. older human hippocampi (Fig. 4) and EC (Fig. 5), the distribution of these Asp664-cleaved forms was also prominently different in the young vs. old tissues. While Asp664-cleaved AβPP was found in cell bodies, in coarse deposits in the neuropil in the hippocampus [63] and prominently in processes of young human entorhinal cortex (Fig. 5), it was present in coarse deposits (boutons) in the hilus of the dentate gyrus and in punctate deposits in the neuropil, and almost absent in entorhinal cortex (Fig. 5) in older non-diseased brains. Tissue distributions of APPNeo immunoreactivity in diseased vs. non-diseased human brain are summarized in Table 2.
Table 2.
Immunoreactivity patterns of Asp664-cleaved AβPP in non-diseased and AD human brains
| Localization of APPNeo immunoreactivity |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| ID | Region | Sex | Age (Y) | PMI (h) | Dx | Cell bodies | Processes | Boutons(hilus) | Punctate |
| R 380 | HC | NA | 23 | ND | NND | X | X | ||
| H-311 | HC | NA | 28 | ND | NND | X | X | ||
| H-356 | HC | NA | 30 | ND | NND | X | X | ||
| M-550 | Ant. HC | NA | 45 | 15 | NND | X | X | ||
| M-551 | HC | NA | 45 | 15 | NND | X | X | ||
| M-517 | Ant. HC | NA | 68 | 21.6 | ND | X | |||
| M-518 | HC | NA | 68 | 21.5 | ND | X | X | ||
| M-554 | HC | NA | 73 | 12 | ND | X | X | ||
| M-524 | HC | NA | 89 | 23.05 | ND | no ML | |||
| M-463 | HC | NA | 90 | 12.66 | ND | X | |||
| M-1669 | Ant. HC | M | 74 | 25 | Braak II | X | |||
| M-1671 | Ant. HC | F | 81 | 6.33 | Braak II | X | |||
| M-1678 | Ant. HC | F | 77 | 21.25 | Braak III | X | |||
| M-1680 | Ant. HC | M | 77 | 16.41 | Braak III | X | X | ||
| M-1682 | Ant. HC | F | 92 | 30.65 | Braak III | ||||
| M-1684 | Ant. HC s | M | 81 | 20.66 | Braak V | X | |||
| M-1675 | Ant. HC s | M | 77 | 14.32 | Braak VI | X | |||
| M-1673 | Ant. HC | F | 80 | 19.5 | Braak VI | X | |||
| M-1688 | Ant. HC | F | 80 | 15.25 | Braak VI | no ML | |||
| M-1686 | Ant. HC | F | 84 | 4.98 | Braak VI | X | |||
| M-520 | HC | NA | 66 | 15.58 | AD | no ML | |||
| M-1564 | HC | NA | 70 | 4.75 | AD | X | |||
| M-457 | HC | NA | 77 | 12.25 | AD | X | X | ||
| M-522 | HC | NA | 89 | 13.80 | AD | X | |||
HC, hippocampus; Ant. HC, anterior hippocampus; NA, not available; M, male; F, female; NND, no neurological disease; ND, non-diseased; AD, Alzheimer's disease; ML, molecular layer.
Asp664-cleaved AβPP fragments localize to nuclei in early AD
It was recently shown that the intracytoplasmic domain of AβPP generated after processing by β- or γ-secretases is involved in the activation of a transcriptional response either after its translocation to the nucleus in association with the adaptor molecule Fe65 and the histone acetyltransferase Tip60 [3,7,29,30], or indirectly by activation of Fe65 [8]. It was also previously reported that Asp664-cleaved forms of AβPP could be found in neuronal nuclei both in AD and in normal controls [63], although both the number of neurons displaying nuclear APPNeo reactivity and the intensity of staining were significantly higher in AD brains. Consistent with these observations, we found that APPNeo immunoreactivity was pronounced in nuclei of hippocampal neurons in AD but not in age-matched control brains (Fig. 6a). In non-AD brains, APPNeo staining was diffuse and cytoplasmic in bodies of granular cells or in the neuropil (Fig. 4 and Fig. 5a). Examination of stacks of high-magnification confocal images revealed that Asp664-cleaved fragments of AβPP colocalized prominently and specifically with chromatin (Fig. 6b). Similar to AβPP intracellular domain (AICD)-containing spherical nuclear spots previously described [60], we observed that APPNeo immunoreactivity delineated the perinuclear region and was found in speckle-like structures within the nucleus in some cells (Fig. 6c, panels a–c). In other cells, Asp664-cleaved AβPP accumulated in a compartment juxtaposed to the nucleus (Fig. 6c, panels e–f) or was abundantly and exclusively nuclear (Fig. 6c, panels d, g, h and i). Thus, Asp664-cleaved AβPP accumulate in nuclei of hippocampal neurons in AD brains. Although we could not identify the N-termini of the Asp664-cleaved fragments in AD nuclei, AβPP-immunoreactive fragments containing AβPP residues 649−664 (AβPP695 numbering) were detected in neuronal nuclei in vitro using antiserum I, which was raised against AβPP649−664 [55] (data not shown). In addition, it was shown previously that aminoterminally truncated species generated after processing by γ-secretases translocate to the nucleus [28,60]. Thus, APPNeo immunoreactive fragments in AD brain nuclei may represent presumptive secretase-cleaved fragments, including fragments 597−664 and 636/638−664 (AβPP695 numbering). Double immunostaining of human brain sections with antiserum I and APPNeo was precluded since both antibodies were generated in the same host species.
Fig. 6.

Asp664-cleaved AβPP fragments form punctate foci in nuclei of hippocampal neurons in AD brain. a, representative high-magnification (600X) confocal images of hippocampal brain sections from control and AD brains stained with APPNeo and counterstained with TOTO-3. Inset, digital magnification of a representative nucleus containing APPNeo-immunoreactive material. b, Section views of stacks of confocal images from AD brain stained with APPNeo (a) and counterstained with TOTO-3 (b). The regions of colocalization of the green and blue channels in (b) are shown in cyan. c, Representative high-magnification (2000X: 1000X magnification × 2X digital zoom) confocal images (a, b, c) or snapshots of maximum intensity projections Surpass volume images (d through i) of green and blue channels in stacks of confocal images of representative nuclei in non-diseased (a) or AD brain sections (b through i). APPNeo-immunoreactive material decorates the perinuclear region and discrete nuclear foci (b and c), in a region juxtaposed to the nucleus (e and f), or is associated with large regions of chromatin (d and g, h and i) in hippocampal neurons of AD brain.
Specificity of APPNeo for Asp664-cleaved forms of AβPP in human brain
To determine whether APPNeo staining in human brain tissues was specific as previously described [2, 15–17,63], we performed pre-adsorption studies using the immunizing peptide and a peptide that encompasses the epitope recognized by APPNeo plus five amino acid residues C-terminal to the Asp664 cleavage site (hence the first five N-terminal residues of the APP-C31 peptide (bridge peptide, [16,17], see Methods section)), which is identical to the continuous epitope in the intact form of full-length AβPP. Similar to our previous observations [16], we found that APPNeo staining in human brain sections was abolished if the antibody was pre-adsorbed with the immunizing peptide, but not if it was pre-adsorbed with a bridge peptide (Fig. 7a). Specific staining was differentiated with lipofuscin pigment autofluorescence in brains from older individuals by examination of signal overlap in the green, red and far red channels, and also in control sections incubated with immunogen-adsorbed APPNeo. In addition, all samples in the study were stained with an antibody specific for calreticulin, which is a calcium-binding protein located in storage compartments associated with the endoplasmic reticulum, to determine the integrity of the tissue. No difference in calreticulin-specific immunostaining, which delineated neuronal bodies in the hippocampus and entorhinal cortex, was observed between samples (Fig. 7b).
Fig. 7.

APPNeo specificity and integrity of human tissue. a, APPNeo staining is abolished if the APPNeo antiserum is preadsorbed with the immunogen peptide or if it is replaced by preimmune rabbit IgG. Immunostaining is unaffected when the APPNeo antiserum is preadsorbed with a peptide encompassing the Asp664 cleavage site and the first four amino acids of AβPP (bridge peptide, Methods). b, Calreticulin immunostaining reveals that the overall cellular structure and tissue integrity are preserved in AD samples. TOTO-3 was used to counterstain nuclei.
DISCUSSION
Consistent with previous reports [2,15,17,63], we found that Asp664-cleaved AβPP was increased in EC and in hippocampus in AD brains, and was minimal or undetectable in non-AD, age-matched, patients (Figs 4 and 5). Increased Asp664-cleaved forms of AβPP in hippocampus and EC were associated with the earlier stages during neurodegeneration, being prominent in early Braak stages but decreased with more advanced AD pathology (Figs 4 and 5). Interestingly, and similar to the findings of Zhao and colleagues [63] and Ayala-Grosso and colleagues [2],Asp664-cleaved AβPP fragments were detected in neuronal nuclei in AD brains (Fig. 6). Similar to Notch [1], it was recently shown that the AβPP intracellular domain (AICD) translocates to the nucleus together with the adaptor proteins Fe65 or Jip1b and the histone acetyltransferase Tip60, forming transcriptionally active complexes [3,7,28,30,60]. Since the Pro and Tyr residues in the NPTY sequence that is contained in the APP-C31 peptide released after Asp664 cleavage are required for Fe65 binding and transcriptional transactivation [7], translocation of AICD fragments must precede the formation of APPNeo-immunoreactive fragments in neuronal nuclei. Caspase-8 preferentially cleaves Asp664 in vitro [35,37], and it was recently shown that SUMO-modified caspases-8 [20] and −7 [4] are found in neuronal nuclei. SUMO-modified caspase-2 is also found in the nucleus [43,58]. Also, the caspase-9 proform can also be found in the nucleus, where it could be processed to its active form in a caspase-8- and caspase-3-dependent manner [50]. Thus, it is possible that caspase(s) present in neuronal nuclei cleave the 661VEVD664 motif in C-terminally intact AICD fragments translocated to the nucleus in association with Fe65 and/or Jip1b and Tip60. Consistent with this hypothesis, active caspases are preferentially found in AD brain [17,35,63]. Moreover, since the motif required for formation of the Fe65/TIP60 complex is contained in the APP-C31 peptide, it is conceivable that the APP-C31 peptide itself may be translocated to the nucleus as part of a transcriptionally active complex. Although our data do not provide any evidence to support or refute this hypothesis, in this scenario caspase cleavage of AβPP would precede translocation of an AβPP fragment to the nucleus.
Consistent with the presence of Asp664-cleaved forms of AβPP in non-diseased, young mouse brains [16], Fig. 1), we found that Asp664-cleaved AβPP was increased approximately ten-fold in young non-diseased human brains as compared to AD brains, and that the distribution of Asp664-cleaved AβPP was markedly different in young vs. older, non-diseased brain tissues. In young non-diseased brains, Asp664-cleaved forms of AβPP localized prominently to neuronal processes in the EC and were diffusely cytoplasmic in neuronal bodies and processes of stellate-like cells in EC (Fig. 5), and were also abundant in bodies of granular neurons and in mossy fiber terminals that formed synapses with CA3 hilus in hippocampus (Fig. 4). Both the abundance of Asp664-cleaved forms of AβPP (Fig. 4) and their distribution (Table 2) were markedly different in aged, non-diseased human brains, which showed a pronounced decrease in abundance and exclusively bouton-like and punctate distribution. To summarize, our data suggest that Asp664-cleaved forms of AβPP are present in cell bodies and projections in young non-diseased human brain and diminish significantly in both locations in association with normal aging, but are still present at lower levels and restricted to cell bodies and also in nuclei, in early AD.
The age-related changes in Asp664 cleavage of AβPP in non-transgenic mouse brains differed markedly from those in human non-diseased brain. In contrast to the age-associated decrease in Asp664 cleavage observed in non-diseased humans, a 2.5-fold increase was observed in aged vs. young non-transgenic mice (Fig. 2). Asp664 cleavage of AβPP in PDAPP transgenic mice modeling AD, however, mimicked the pattern observed in human AD brains. In PDAPP transgenic animals, Asp664-cleavage increases were observed in association with the earliest AD-like neurodegenerative changes, concomitant with the initial increase in amyloid-beta production at ∼2−3 mo. As in AD cases, Asp664 cleavage decreased thereafter, as AD-like degenerative processes progress in PDAPP transgenic mice (Fig. 2). Although our results can be interpreted as indicating differential cleavage susceptibility of mouse and human AβPP, no differences are present in primary sequence at or in proximity to the site of cleavage between hAβPP and mAβPP. Thus, it is more likely that Asp664 cleavage of AβPP (both human and mouse forms) may be downregulated in PDAPP transgenic mouse brains as a consequence of, or possibly as a response to, AD-like neurodegenerative processes taking place in the brains of transgenic animals. AD-like pathological processes in PDAPP mice arise not only from the overexpression of AβPP and the accumulation of Aβ, but also as a result of cleavage of AβPP at Asp664 [16,53]; it is thus conceivable that Asp664 cleavage may be downregulated with increasing age and pathology, perhaps as part of a protective response. Since accumulation of soluble amyloid from and Asp664 cleavage of endogenous mAβPP do not induce neurodegenerative-like processes in non-transgenic mice, it is conceivable that regulation of Asp664 cleavage is controlled or modulated through different mechanisms than those operating in PDAPP transgenic mice. Thus, as our results suggest, ongoing AD-like processes possibly result in the downregulation of Asp664 cleavage of both endogenous mAβPP and of transgenic hAPP in transgenic, but not in non-transgenic, PDAPP mouse brains (Fig. 2). Although both the increase in Asp664 cleavage of endogenous mAβPP in non-transgenic brains and the decrease of Asp664 cleavage of likely both mouse and hAβPP in transgenic mouse brains were significant, the magnitude of the change was significantly larger for the downregulation of Asp664 cleavage of mouse and/or hAβPP in transgenic brains (26-fold) as compared to the upregulation of Asp664 cleavage of endogenous mAβPP in non-transgenic brains (2.5-fold, Fig. 2), arguing for a stronger effect of age on the regulation of Asp664 cleavage in brains of mice modeling AD. Remarkably, Asp664 cleavage in aged transgenic mouse brains was significantly lower than that in both young and aged non-transgenic brains. This observation lends further support to the hypothesis that active processes may downregulate cleavage of both mouse and human AβPP in aged transgenic brains undergoing AD-like degeneration.
Consistent with our previous [16] and present (Fig. 2) observations in young non-transgenic mice, Asp664-cleaved forms of AβPP were prominent in neuronal bodies and in terminals of young, non-diseased human brain (Figs 4 and 5). Although the activity or activities responsible for Asp664 cleavage of AβPP in vivo are still unknown, it is conceivable that low levels of active caspases, which cleave Asp664 in vitro, may affect Asp664 cleavage in vivo. The activation of caspases at neuronal processes in healthy, non-diseased neurons has been demonstrated in several studies (reviewed by Gilman and Mattson [18]). Moreover, it was recently shown that caspases are present in synapses and dendrites of neurons, where they can be activated in response to glutamate receptor stimulation and calcium influx. Interestingly, it was recently reported that a pan-caspase inhibitor or a caspase-1-specific inhibitor significantly enhanced the AMPA-mediated component of LTP without affecting the NMDA-dependent component [34]. It is also conceivable that Asp664 cleavage is affected by proteases different from caspases, such as calpains. The questions of what are the proteolytic activities involved and what are the mechanisms responsible for the regulation of Asp664 cleavage in vivo, however, remain unanswered. Cleavage of AβPP at Asp664 removes the GYENPTY protein-protein interaction motif which acts as a binding site for several AβPP-interacting proteins and partially overlaps with AβPP's internalization signal [45]. Thus, Asp664 cleavage of AβPP by transiently activated caspases or caspase-like proteases during its processing in the secretory pathway, or in neuronal terminals, may serve to regulate signaling AβPP since only AβPP molecules that have not undergone Asp664 cleavage and contain the GYENPTY motif are competent for both protein-protein interactions and for retrieval from the cell surface through endocytosis. Thus, in agreement with previous reports [2,15–17,63], our results provide further support for a crucial role of Asp664 in the pathogenesis of AD [16] and suggest that cleavage at Asp664 may serve a regulatory function as part of the proteolytic processing of AβPP.
ACKNOWLEDGEMENTS
Human tissues were provided by the Harvard Brain Tissue Resource Center, which is supported in part by PHS grant number MH/NS31862. We thank Drs. George Tejada and Francine Benes at the HBTRC for their help and Dr. Lennart Mucke (U. California, San Francisco) for PDAPP transgenic mice. We also thank Dr. Meredith Halks-Miller for critically reading the manuscript. Animal work involved in the present study was carried out in accordance with regulations and guidelines of the Buck Institute Institutional Animal Care and Use Committee (IACUC). None of the authors has a financial interest in the results reported. This work was supported by National Institutes of Health Grants NS45093 and AG05131 to D.E.B., NIRG-04−1054 from the Alzheimer's Association and a grant from the S.D. Bechtel, Jr. Foundation to V.G. V.G. wishes to thank the John Douglas French Alzheimer's Foundation and Mrs. Eloise Goodhew Barnett for their support.
References
- 1.Artavanis-Tsakonas S, Matsuno K, Fortini ME. Notch signaling. Science. 1995;268:225–232. doi: 10.1126/science.7716513. [DOI] [PubMed] [Google Scholar]
- 2.Ayala-Grosso C, Ng G, Roy S, Robertson GS. Caspase-cleaved amyloid precursor protein in Alzheimer's disease. Brain Pathol. 2002;12:430–441. doi: 10.1111/j.1750-3639.2002.tb00460.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baek SH, Ohgi KA, Rose DW, Koo EH, Glass CK, Rosenfeld MG. Exchange of N-CoR corepressor and Tip60 coactivator complexes links gene expression by NF-kappaB and beta-amyloid precursor protein. Cell. 2002;110:55–67. doi: 10.1016/s0092-8674(02)00809-7. [DOI] [PubMed] [Google Scholar]
- 4.Besnault-Mascard L, Leprince C, Auffredou MT, Meunier B, Bourgeade MF, Camonis J, Lorenzo HK, Vazquez A. Caspase-8 sumoylation is associated with nuclear localization. Oncogene. 2005;24:3268–3273. doi: 10.1038/sj.onc.1208448. [DOI] [PubMed] [Google Scholar]
- 5.Biederer T, Cao X, Sudhof TC, Liu X. Regulation of APP-dependent transcription complexes by Mint/X11s: differential functions of Mint isoforms. J Neurosci. 2002;22:7340–7351. doi: 10.1523/JNEUROSCI.22-17-07340.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buxbaum JD, Thinakaran G, Koliatsos V, O'Callahan J, Slunt HH, Price DL, Sisodia SS. Alzheimer amyloid protein precursor in the rat hippocampus: transport and processing through the perforant path. J Neurosci. 1998;18:9629–9637. doi: 10.1523/JNEUROSCI.18-23-09629.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cao X, Sudhof TC. A transcriptionally correction of transcriptively active complex of APP with Fe65 and histone acetyltransferase Tip60. Science. 2001;293:115–120. doi: 10.1126/science.1058783. [DOI] [PubMed] [Google Scholar]
- 8.Cao X, Sudhof TC. Dissection of amyloid-beta precursor protein-dependent transcriptional transactivation. J Biol Chem. 2004;279:24601–24611. doi: 10.1074/jbc.M402248200. [DOI] [PubMed] [Google Scholar]
- 9.Chen WJ, Goldstein JL, Brown MS. NPXY, a sequence often found in cytoplasmic tails, is required for coated pit-mediated internalization of the low density lipoprotein receptor. J Biol Chem. 1990;265:3116–3123. [PubMed] [Google Scholar]
- 10.Cirrito JR, May PC, O'Dell MA, Taylor JW, Parsadanian M, Cramer JW, Audia JE, Nissen JS, Bales KR, Paul SM, DeMattos RB, Holtzman DM. In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changes in amyloid-beta metabolism and half-life. J Neurosci. 2003;23:8844–8853. doi: 10.1523/JNEUROSCI.23-26-08844.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Costes SV, Daelemans D, Cho EH, Dobbin Z, Pavlakis G, Lockett S. Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophys J. 2004;86:3993–4003. doi: 10.1529/biophysj.103.038422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cottrell BA, Galvan V, Banwait S, Gorostiza O, Lombardo CR, Williams T, Schilling B, Peel A, Gibson B, Koo EH, Link CD, Bredesen DE. A pilot proteomic study of amyloid precursor interactors in Alzheimer's disease. Ann Neurol. 2005;58:277–289. doi: 10.1002/ana.20554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Du AT, Schuff N, Zhu XP, Jagust WJ, Miller BL, Reed BR, Kramer JH, Mungas D, Yaffe K, Chui HC, Weiner MW. Atrophy rates of entorhinal cortex in AD and normal aging. Neurology. 2003;60:481–486. doi: 10.1212/01.wnl.0000044400.11317.ec. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fiore F, Zambrano N, Minopoli G, Donini V, Duilio A, Russo T. The regions of the Fe65 protein homologous to the phosphotyrosine interaction/phosphotyrosine binding domain of Shc bind the intracellular domain of the Alzheimer's amyloid precursor protein. J Biol Chem. 1995;270:30853–30856. doi: 10.1074/jbc.270.52.30853. [DOI] [PubMed] [Google Scholar]
- 15.Galvan V, Chen S, Lu D, Logvinova A, Goldsmith P, Koo EH, Bredesen DE. Caspase cleavage of members of the amyloid precursor family of proteins. J Neurochem. 2002;82:283–294. doi: 10.1046/j.1471-4159.2002.00970.x. [DOI] [PubMed] [Google Scholar]
- 16.Galvan V, Gorostiza OF, Banwait S, Ataie M, Logvinova AV, Sitaraman S, Carlson E, Sagi SA, Chevallier N, Jin K, Greenberg DA, Bredesen DE. Reversal of Alzheimer's-like pathology and behavior in human APP transgenic mice by mutation of Asp664. Proc Natl Acad Sci USA. 2006;103:7130–7135. doi: 10.1073/pnas.0509695103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gervais FG, Xu D, Robertson GS, Vaillancourt JP, Zhu Y, Huang J, LeBlanc A, Smith D, Rigby M, Shearman MS, Clarke EE, Zheng H, Van Der Ploeg LH, Ruffolo SC, Thornberry NA, Xanthoudakis S, Zamboni RJ, Roy S, Nicholson DW. Involvement of caspases in proteolytic cleavage of Alzheimer's amyloid-beta precursor protein and amyloidogenic A beta peptide formation. Cell. 1999;97:395–406. doi: 10.1016/s0092-8674(00)80748-5. [DOI] [PubMed] [Google Scholar]
- 18.Gilman CP, Mattson MP. Do apoptotic mechanisms regulate synaptic plasticity and growth-cone motility? Neuromolecular Med. 2002;2:197–214. doi: 10.1385/NMM:2:2:197. [DOI] [PubMed] [Google Scholar]
- 19.Gunawardena S, Goldstein LS. Disruption of axonal transport and neuronal viability by amyloid precursor protein mutations in Drosophila. Neuron. 2001;32:389–401. doi: 10.1016/s0896-6273(01)00496-2. [DOI] [PubMed] [Google Scholar]
- 20.Hayashi N, Shirakura H, Uehara T, Nomura Y. Relationship between SUMO-1 modification of caspase-7 and its nuclear localization in human neuronal cells. Neurosci Lett. 2006;397:5–9. doi: 10.1016/j.neulet.2005.11.057. [DOI] [PubMed] [Google Scholar]
- 21.Hell K, Saleh M, Crescenzo GD, O'Connor-McCourt MD, Nicholson DW. Substrate cleavage by caspases generates protein fragments with Smac/Diablo-like activities. Cell Death Differ. 2003;10:1234–1239. doi: 10.1038/sj.cdd.4401298. [DOI] [PubMed] [Google Scholar]
- 22.Homayouni R, Rice DS, Sheldon M, Curran T. Disabled-1 binds to the cytoplasmic domain of amyloid precursor-like protein 1. J Neurosci. 1999;19:7507–7515. doi: 10.1523/JNEUROSCI.19-17-07507.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L. Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc Natl Acad Sci USA. 1999;96:3228–3233. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kamal A, Stokin GB, Yang Z, Xia CH, Goldstein LS. Axonal transport of amyloid precursor protein is mediated by direct binding to the kinesin light chain subunit of kinesin-I. Neuron. 2000;28:449–459. doi: 10.1016/s0896-6273(00)00124-0. [DOI] [PubMed] [Google Scholar]
- 25.Kamal A, Almenar-Queralt A, LeBlanc JF, Roberts EA, Goldstein LS. Kinesin-mediated axonal transport of a membrane compartment containing beta-secretase and presenilin-1 requires APP. Nature. 2001;414:643–648. doi: 10.1038/414643a. [DOI] [PubMed] [Google Scholar]
- 26.Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- 27.Kim JH, Anwyl R, Suh YH, Djamgoz MB, Rowan MJ. Use-dependent effects of amyloidogenic fragments of (beta)-amyloid precursor protein on synaptic plasticity in rat hippocampus in vivo. J Neurosci. 2001;21:1327–1333. doi: 10.1523/JNEUROSCI.21-04-01327.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kimberly WT, Zheng JB, Guenette SY, Selkoe DJ. The intracellular domain of the beta-amyloid precursor protein is stabilized by Fe65 and translocates to the nucleus in a notch-like manner. J Biol Chem. 2001;276:40288–40292. doi: 10.1074/jbc.C100447200. [DOI] [PubMed] [Google Scholar]
- 29.Kimberly WT, Zheng JB, Guenette SY, Selkoe DJ. eThe intracellular domain of the beta-amyloid precursor protein is stabilized by Fe65 and translocates to the nucleus in a notch-like manner. J Biol Chem. 2001;276:40288–40292. doi: 10.1074/jbc.C100447200. [DOI] [PubMed] [Google Scholar]
- 30.Kogel D, Schomburg R, Copanaki E, Prehn JH. Regulation of gene expression by the amyloid precursor protein: inhibition of the JNK/c-Jun pathway. Cell Death Differ. 2005;12:1–9. doi: 10.1038/sj.cdd.4401495. [DOI] [PubMed] [Google Scholar]
- 31.Koo EH, Sisodia SS, Archer DR, Martin LJ, Weidemann A, Beyreuther K, Fischer P, Masters CL, Price DL. Precursor of amyloid protein in Alzheimer disease undergoes fast anterograde axonal transport. Proc Natl Acad Sci USA. 1990;87:1561–1565. doi: 10.1073/pnas.87.4.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.LeBlanc A, Liu H, Goodyer C, Bergeron C, Hammond J. Caspase-6 role in apoptosis of human neurons, amyloidogenesis, and Alzheimer's disease. J Biol Chem. 1999;274:23426–23436. doi: 10.1074/jbc.274.33.23426. [DOI] [PubMed] [Google Scholar]
- 33.Lorenzo A, Yuan M, Zhang Z, Paganetti PA, Sturchler-Pierrat C, Staufenbiel M, Mautino J, Vigo FS, Sommer B, Yankner BA. Amyloid beta interacts with the amyloid precursor protein: a potential toxic mechanism in Alzheimer's disease. Nat Neurosci. 2000;3:460–464. doi: 10.1038/74833. [DOI] [PubMed] [Google Scholar]
- 34.Lu C, Wang Y, Furukawa K, Fu W, Ouyang X, Mattson MP. Evidence that caspase-1 is a negative regulator of AMPA receptor-mediated long-term potentiation at hippocampal synapses. J Neurochem. 2006;97:1104–1110. doi: 10.1111/j.1471-4159.2006.03800.x. [DOI] [PubMed] [Google Scholar]
- 35.Lu DC, Rabizadeh S, Chandra S, Shayya RF, Ellerby LM, Ye X, Salvesen GS, Koo EH, Bredesen DE. A second cytotoxic proteolytic peptide derived from amyloid beta-protein precursor. Nat Med. 2000;6:397–404. doi: 10.1038/74656. [DOI] [PubMed] [Google Scholar]
- 36.Lu DC, Shaked GM, Masliah E, Bredesen DE, Koo EH. Amyloid beta protein toxicity mediated by the formation of amyloid-beta protein precursor complexes. Ann Neurol. 2003;54:781–789. doi: 10.1002/ana.10761. [DOI] [PubMed] [Google Scholar]
- 37.Lu DC, Soriano S, Bredesen DE, Koo EH. Caspase cleavage of the amyloid precursor protein modulates amyloid beta-protein toxicity. J Neurochem. 2003;87:733–741. doi: 10.1046/j.1471-4159.2003.02059.x. [DOI] [PubMed] [Google Scholar]
- 38.Lyckman AW, Confaloni AM, Thinakaran G, Sisodia SS, Moya KL. Post-translational processing and turnover kinetics of presynaptically targeted amyloid precursor super-family proteins in the central nervous system. J Biol Chem. 1998;273:11100–11106. doi: 10.1074/jbc.273.18.11100. [DOI] [PubMed] [Google Scholar]
- 39.Massey A, Kiffin R, Cuervo AM. Pathophysiology of chaperone-mediated autophagy. Int J Biochem Cell Biol. 2004;36:2420–2434. doi: 10.1016/j.biocel.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 40.Matsuda S, Yasukawa T, Homma Y, Ito Y, Niikura T, Hiraki T, Hirai S, Ohno S, Kita Y, Kawasumi M, Kouyama K, Yamamoto T, Kyriakis JM, Nishimoto IM. c-Jun N-terminal kinase (JNK)-interacting protein-1b/islet-brain-1 scaffolds Alzheimer's amyloid precursor protein with JNK. J Neurosci. 2001;21:6597–6607. doi: 10.1523/JNEUROSCI.21-17-06597.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McLoughlin DM, Irving NG, Miller CC. The Fe65 and X11 families of proteins: proteins that interact with the Alzheimer's disease amyloid precursor protein. Biochem Soc Trans. 1998;26:497–500. doi: 10.1042/bst0260497. [DOI] [PubMed] [Google Scholar]
- 42.Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of abeta 1−42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paroni G, Henderson C, Schneider C, Brancolini C. Caspase-2 can trigger cytochrome C release and apoptosis from the nucleus. J Biol Chem. 2002;277:15147–15161. doi: 10.1074/jbc.M112338200. [DOI] [PubMed] [Google Scholar]
- 44.Pellegrini L, Passer BJ, Tabaton M, Ganjei JK, D'Adamio L. Alternative, non-secretase processing of Alzheimer's beta-amyloid precursor protein during apoptosis by caspase-6 and -8. J Biol Chem. 1999;274:21011–21016. doi: 10.1074/jbc.274.30.21011. [DOI] [PubMed] [Google Scholar]
- 45.Perez RG, Soriano S, Hayes JD, Ostaszewski B, Xia W, Selkoe DJ, Chen X, Stokin GB, Koo EH. Mutagenesis identifies new signals for beta-amyloid precursor protein endocytosis, turnover, and the generation of secreted fragments, including Abeta42. J Biol Chem. 1999;274:18851–18856. doi: 10.1074/jbc.274.27.18851. [DOI] [PubMed] [Google Scholar]
- 46.Priller C, Bauer T, Mitteregger G, Krebs B, Kretzschmar HA, Herms J. Synapse formation and function is modulated by the amyloid precursor protein. J Neurosci. 2006;26:7212–7221. doi: 10.1523/JNEUROSCI.1450-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rao RV, Hermel E, Castro-Obregon S, del Rio G, Ellerby LM, Ellerby HM, Bredesen DE. Coupling endoplasmic reticulum stress to the cell death program. Mechanism of caspase activation. J Biol Chem. 2001;276:33869–33874. doi: 10.1074/jbc.M102225200. [DOI] [PubMed] [Google Scholar]
- 48.Rao RV, Peel A, Logvinova A, del Rio G, Hermel E, Yokota T, Goldsmith PC, Ellerby LM, Ellerby HM, Bredesen DE. Coupling endoplasmic reticulum stress to the cell death program: role of the ER chaperone GRP78. FEBS Lett. 2002;514:122–128. doi: 10.1016/s0014-5793(02)02289-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reddy RK, Mao C, Baumeister P, Austin RC, Kaufman RJ, Lee AS. Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors: role of ATP binding site in suppression of caspase-7 activation. J Biol Chem. 2003;278:20915–20924. doi: 10.1074/jbc.M212328200. [DOI] [PubMed] [Google Scholar]
- 50.Ritter PM, Marti A, Blanc C, Baltzer A, Krajewski S, Reed JC, Jaggi R. Nuclear localization of procaspase-9 and processing by a caspase-3-like activity in mammary epithelial cells. Eur J Cell Biol. 2000;79:358–364. doi: 10.1078/S0171-9335(04)70040-0. [DOI] [PubMed] [Google Scholar]
- 51.Sabo SL, Ikin AF, Buxbaum JD, Greengard P. The Alzheimer amyloid precursor protein (APP) and FE65, an APP-binding protein, regulate cell movement. J Cell Biol. 2001;153:1403–1414. doi: 10.1083/jcb.153.7.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sabo SL, Ikin AF, Buxbaum JD, Greengard P. The amyloid precursor protein and its regulatory protein, FE65, in growth cones and synapses in vitro and in vivo. J Neurosci. 2003;23:5407–5415. doi: 10.1523/JNEUROSCI.23-13-05407.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Saganich MJ, Schroeder BE, Galvan V, Bredesen DE, Koo EH, Heinemann SF. Deficits in synaptic transmission and learning in amyloid precursor protein (APP) transgenic mice require C-terminal cleavage of APP. J Neurosci. 2006;26:13428–13436. doi: 10.1523/JNEUROSCI.4180-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Scheinfeld MH, Roncarati R, Vito P, Lopez PA, Abdallah M, D'Adamio L. Jun NH2-terminal kinase (JNK) interacting protein 1 (JIP1) binds the cytoplasmic domain of the Alzheimer's beta-amyloid precursor protein (APP) J Biol Chem. 2002;277:3767–3775. doi: 10.1074/jbc.M108357200. [DOI] [PubMed] [Google Scholar]
- 55.Selkoe DJ, Podlisny MB, Joachim CL, Vickers EA, Lee G, Fritz LC, Oltersdorf T. Beta-amyloid precursor protein of Alzheimer disease occurs as 110- to 135-kilodalton membrane-associated proteins in neural and nonneural tissues. Proc Natl Acad Sci USA. 1988;85:7341–7345. doi: 10.1073/pnas.85.19.7341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- 57.Shaked GM, Kummer MP, Lu DC, Galvan V, Bredesen DE, Koo EH. Abeta induces cell death by direct interaction with its cognate extracellular domain on APP (APP 597−624) Faseb J. 2006;20:1254–1256. doi: 10.1096/fj.05-5032fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shirakura H, Hayashi N, Ogino S, Tsuruma K, Uehara T, Nomura Y. Caspase recruitment domain of procaspase-2 could be a target for SUMO-1 modification through Ubc9. Biochem Biophys Res Commun. 2005;331:1007–1015. doi: 10.1016/j.bbrc.2005.04.019. [DOI] [PubMed] [Google Scholar]
- 59.Taru H, Kirino Y, Suzuki T. Differential roles of JIP scaffold proteins in the modulation of amyloid precursor protein metabolism. J Biol Chem. 2002;277:27567–27574. doi: 10.1074/jbc.M203713200. [DOI] [PubMed] [Google Scholar]
- 60.von Rotz RC, Kohli BM, Bosset J, Meier M, Suzuki T, Nitsch RM, Konietzko U. The APP intracellular domain forms nuclear multiprotein complexes and regulates the transcription of its own precursor. J Cell Sci. 2004;117:4435–4448. doi: 10.1242/jcs.01323. [DOI] [PubMed] [Google Scholar]
- 61.Weidemann A, Paliga K, Durrwang U, Reinhard FB, Schuckert O, Evin G, Masters CL. Proteolytic processing of the Alzheimer's disease amyloid precursor protein within its cytoplasmic domain by caspase-like proteases. J Biol Chem. 1999;274:5823–5829. doi: 10.1074/jbc.274.9.5823. [DOI] [PubMed] [Google Scholar]
- 62.Zambrano N, Buxbaum JD, Minopoli G, Fiore F, De Candia P, De Renzis S, Faraonio R, Sabo S, Cheetham J, Sudol M, Russo T. Interaction of the phosphotyrosine interaction/phosphotyrosine binding-related domains of Fe65 with wild-type and mutant Alzheimer's beta-amyloid precursor proteins. J Biol Chem. 1997;272:6399–6405. doi: 10.1074/jbc.272.10.6399. [DOI] [PubMed] [Google Scholar]
- 63.Zhao M, Su J, Head E, Cotman CW. Accumulation of caspase cleaved amyloid precursor protein represents an early neurodegenerative event in aging and in Alzheimer's disease. Neurobiol Dis. 2003;14:391–403. doi: 10.1016/j.nbd.2003.07.006. [DOI] [PubMed] [Google Scholar]