

Abstract
Peptide sequence identification using tandem mass spectroscopy remains a major challenge for complex proteomic studies. Peptide matching algorithms require the accurate determination of both the mass and charge of the precursor ion and accommodate uncertainties in these properties by using a wide precursor mass tolerance and by testing, for each spectrum, several possible candidate charges. Using a data acquisition strategy that includes obtaining narrow mass-range MS1 “zoom” scans, we describe here a post-acquisition algorithm dubbed MAZIE, that accurately determines the charge and monoisotopic mass of precursor ions on a low-resolution Thermo LTQ-XL mass spectrometer. This is achieved by examining the isotopic distribution obtained in the preceding MS1 zoom spectrum and comparing to theoretical distributions for candidate charge states from +1 to +4. MAZIE then writes modified data files with the corrected monoisotopic mass and charge. We have validated MAZIE results by comparing the sequence search results obtained with the MAZIE-generated data files to results using the unmodified data files. Using two different search algorithms and a false discovery rate filter, we found that MAZIE-interpreted data resulted in 80% (using SEQUEST) and 30% (using OMSSA) more high-confidence sequence identifications. Analyses of these results indicate that the accurate determination of the precursor ion mass greatly facilitates the ability to differentiate between true and false positive matches, while the determination of the precursor ion charge reduces the overall search time but does not significantly reduce the ambiguity of interpreting the search results. MAZIE is distributed as an open-source PERL script.
Introduction
Mass spectrometry is a popular and powerful proteomics tool due to its ability to rapidly analyze proteins from complex biological samples. However, the acquisition of tens of thousands of MS2 scans during a typical mass spectral analysis necessitates automating data analysis. Though a variety of robust search algorithms have been developed to identify the peptide sequence corresponding to an MS2 spectrum, these results still present the problem of being able to distinguish a true positive sequence match from a false positive match.
The uncertainty of the precursor ion mass measurement dictates that search algorithms must employ wide mass window tolerances to ensure that the correct peptide candidate is included in the list of peptides to be examined, thus increasing the number of peptide candidates considered. However, by lengthening this list of candidate sequences, the chances that an incorrect sequence will receive a relatively high score increases and, thereby, makes it more difficult to identify the true positive match. Some statistical methods have been described with the goal of distinguishing the true-positive matches from the erroneous identifications(1-4). Despite these ongoing efforts, distinguishing between correct and incorrect matches is still among the greatest current problems of proteomics using mass spectroscopy.
Much of the uncertainty of spectrum-sequence matching is due to the fact that the most elemental physical properties of a peptide ion, to wit its charge and monoisotopic mass, are not definitively specified within MS2 spectra. The accuracy and resolution of the precursor mass associated with a particular MS2 is dependent upon the instrument and methodology used to acquire the previous MS1 “normal” scan from which the MS2 was derived. For Linear Ion Trap spectrometers, as a consequence of both its inherent resolution and binning effects generated by acquiring data in centroid mode, the mass accuracy of the MS1 “normal” scan is on the order of 0.5 Da and the isotopic distribution of the ion is converted to an ionic signal that is more representative of its average, instead of its monoisotopic, mass. Furthermore, even if the isotopic distribution can be somewhat resolved, the search algorithms must take into account whether or not the instrument acquisition happened to choose a higher isotopic peak of the ion and, thereby, misinterpret the mass of the peptide by as much as one or more Daltons.
Current search algorithms accommodate the loss of information of the monoisotopic mass and charge state of the precursor ion by employing more promiscuous search parameters. Typically, the algorithms match scans to database entries by assuming an average mass for the precursor ion and using a wide precursor mass tolerance windows of 1 to 2 Da. Furthermore, because the charge state is undetermined, the algorithms usually search the same MS2 scan using several potential charge states. This redundancy and over-searching requires more search time and, because more peptide sequences are included in the potential candidate list for a particular scan, makes it more difficult to distinguish the one true positive match from the growing population of false positive matches.
Recognizing the potential advantage of knowing the charge state, much effort has been put into accurate charge determination of peptide spectra through the examination of complementary fragment peak pairs (5-7), the distribution of the fragment ions (8), machine learning approaches (9,10), or the Fourier transform of isotopically-resolved mass spectra (11). These different efforts vary in their efficiency in distinguishing multiply charged ions from singly charged ions and in further separating doubly from triply charged states, but none of them address the issue of mass accuracy of the precursor ion.
High-resolution mass spectrometry instruments, such as the Fourier transform ion cyclotron resonance detector (FT-ICR) and the Orbitrap, provide greater mass resolution and, thereby, preserve the isotopic ion distribution. Deconvolution approaches (12,13), including THRASH (14) and the related algorithm MasSPIKE (15), have been applied to high-resolution data to extract charge states and monoisotopic masses. However, these efforts were primarily focused on macromolecule identifications and were not optimized for proteolytic digests of protein mixtures.
The value of improved charge state and monoisotopic mass information has been recognized, particularly the value of reducing the precursor mass tolerance window (16); identification of unique peptides was increased in one study by more than 20% as a direct result of the higher mass precision and unambiguous charge state (17). The recently-described DeconMSn algorithm is designed specifically for data acquired on the LTQ-FT and LTQ-Orbitrap instruments of Thermo Fisher Scientific (18). Primarily a modification of THRASH (14), DeconMSn can determine both the monoisotopic precursor mass and charge state from high-resolution MS1 scans and, though it cannot determine the monoisotopic mass of the precursor ion, it employs a modified SVM-based approach (9) to assign a charge state for low-resolution data.
In this report, recognizing that knowledge of both the monoisotopic mass and charge state of the precursor ion could potentially improve peptide matching efforts, we took advantage of the MS1 “zoom” scan rate available on the Thermo Fisher LTQ-XL to obtain MS1 mass data with greater precision. This MS1 zoom scan has sufficient mass resolution to preserve the isotopic distribution for ions with charge states up to +4, which is sufficient for standard tryptic digests that most commonly produce peptides with charge states of +2 or +3. We analyze this data with MAZIE, a “Mass and charge (Z) Inference Engine” that extracts both the charge state and monoisotopic mass of the precursor ion by examining the isotopic ion envelope of the MS1 “zoom” spectra. To quantify the effects for a relatively complex sample, we directly compare the results obtained by both the SEQUEST and OMSSA search algorithms for the MAZIE-modified and unmodified data. We conclude that MAZIE offers several significant advantages and offers advantages for routine pre-processing of data preceding database searching.
Experimental
Sample Preparation
Briefly, 7 mL of centrifuged human urine was reduced with 10 mM dithiothreitol, carboxyamidomethylated with 25 mM iodoacetamide, and then digested overnight at ambient temperature with 4 μg of trypsin. The peptides were fractionated using thin layer isoelectric focusing on an IPGphor rehydration tray using an Immobiline IPG Drystrip (13 cm, pH 3-10, GE Cat #17-6001-14) following manufacturer’s instructions. Peptides from one fraction (of 13) were extracted, purified using a C18 ZipTip (Millipore), dried, and re-dissolved in 20 μl of 3% acetic acid. Further details of this sample preparation will be presented more fully elsewhere.
Data Acquisition and Processing
LC-MS2 experiments were performed on an ETD-enabled LTQ-XL linear ion trap mass spectrometer (Thermo Fisher Scientific). Peptide samples were pressure loaded into a self-prepared 360 μm o.d. × 100 μm i.d. fused-silica column (Polymicro Technologies) packed with irregular (5-15 μm, 120 Ǻ) reverse-phase C18 beads (YMC). To retain the C18 beads on this “precolumn”, a ceramic frit had been created first by drawing up with capillary action a ~5 mm plug of a mixture of Kasil® (potassium silicate solution) (PQ Corporation) with formamide (Sigma) and then using ~100°C heat for 3-5 minutes to set the frit. After the peptide sample had been loaded on this “precolumn”, it was then washed with 20 column volumes of 0.1% acetic acid and then connected via a 0.012 in i.d. × 0.060 in o.d. PTFE Teflon® sleeve (Zeus) to a second fused-silica column, a self-packed PicoFrit® column (New Objective) with dimensions of 360 μm o.d. × 100 μm i.d. and a pre-fritted 10 μm tip. This analytical column was packed with regular (5 μm, 120Ǻ) reverse-phase C18 beads (YMC). Together, these two columns were mounted onto a Proxeon electrospray ionization sources that has been integrated with an Agilent 1100 series binary pump HPLC system. Chromatography used a flow rate of 60 nL/min with a 0–60% B gradient in 105 min followed by a 60-100% B gradient in 10 minutes. Solvent A was 0.1 M acetic acid and Solvent B was 80% acetonitrile with 0.1 M acetic acid.
The LTQ-XL mass spectrometer was operated in the data-dependent mode throughout the HPLC gradient. First, a full mass spectrum scan (300-2000 m/z) was acquired and the five ions with the highest intensity were selected for that chromatographic time point. For each of these five precursor ions, a MS1 zoom scan was first acquired in profile mode. The zoom scan, centered on the precursor m/z with a full width of 10.0 m/z, was then immediately followed by a MS2 CID spectrum of that same precursor using an isolation width of 2.0 m/z, an activation Q of 0.25, a normalized collision energy of 35%, and an activation time of 30 ms. After the zoom and MS2 scan for each of the top five precursor ions had been obtained, a new full mass spectrum scan was acquired and the process repeated. The duty cycle for this data acquisition cycle of 11 mass spectral scans was about 3 s. Dynamic exclusion was enabled with a repeat count of 3 over a 20 s period and with a 50 s exclusion duration.
For the SEQUEST searches, the “control” DTAs were those that were automatically generated by BioWorks/SEQUEST(19) software from the original LTQ RAW data file. Using the automatic charge state setting, SEQUEST constructed DTAs with potential charges of +1, +2, or +3 for most scans, with an occasional higher charge state for a few. For most scans, multiple DTAs were generated to account for multiple potential charge states. These DTAs were then searched by TurboSEQUEST v.28 (rev. 13) through the BioWorks Browser 3.3.1 SP1. The search parameters for all of the data files specified a tryptic digest that allowed for 2 missed cleavages with variable modifications of cysteine carbamidomethylation and methionine oxidation. The human FASTA database was acquired from through NCBI at ftp://ftp.ncbi.nih.gov/refseq. An in-house PERL script was used to generate a “forward-reverse” database from this original FASTA database by reversing each protein sequence and concatenating it immediately following the “forward” sequence with the text “REV” preceding the gi number.
All data sets are converted from the original Thermo RAW format to the .mzXML format using the program ReAdW.exe (http://tools.proteomecenter.org/ReAdW.php), provided by the Institute for Systems Biology. The MAZIE algorithm accesses the header information and scan data from mzXML files by calling an in-house modified version of the readmzXML.exe (http://tools.proteomecenter.org/readmzXML.php).
Strategy and implementation of the MAZIE algorithm
The MAZIE algorithm compares the isotopic ion distribution of the precursor ion, obtained from the MS1 ‘zoom’ scans, to theoretical distributions that are calculated using several simplifications that take advantage of the experimental conditions. First, because the analyzed samples are trypsinized, the vast majority of peptides will be less than 3 kDa (20). As a consequence, the calculation of the isotopic distribution can be limited to considering the first four isotopic peaks (k = 0,1,2,3) and a mass width of about 3.5 Da beginning near the monoisotopic mass (k = 0). Second, employing the concept of the “averagine” average amino acid (12), the peptide was treated as if a single average element was its sole constituent and that this average element had a single heavy isotope, instead of including in the calculation the explicit contribution for each of the elemental constituents (carbon, hydrogen, nitrogen, etc.). These simplifications greatly reduce the calculation complexity while still preserving the fundamental characteristics of the isotopic distribution that are required to determine the monoisotopic mass and charge state of the precursor ion.
With these simplifications, a binomial distribution was used to construct a probability mass function representing the theoretical ion distribution:
| (1) |
This model calculates the probability that a peptide ion with n total atoms has k heavy isotope atoms included within it, where p represents the relative probability of the heavy isotope. The approximation of the number of atoms, n is calculated as (precursor mass)×(charge)/18.4, where the constant is an approximation of the average atom mass. The heavy isotope probability, p was assigned as 1.07%, reflecting the average abundance of isotopic forms of H, C, O and N. These constants were adjusted empirically after comparing the calculated theoretical ion distributions with experimental distributions of known peptides with varied mass and charge. The relative peak intensities for the individual isotopic possibilities, k, are converted to a Gaussian shape with a line width, σ, that simulates the resolution of the instrument. The four theoretical peaks were then assembled into an isotopic distribution by spacing them 1/z relative to each other.
Outlining the overall flow of the algorithm, MAZIE steps through the scans of interest in the mzXML-formatted data file. For each MS2 scan, its corresponding high-resolution “zoom” MS1 scan is compared to the theoretical isotopic distribution for each charge, z = 1 to 4. To make the comparison for each charge state, the theoretical distribution is scanned along the mass axis and, at each mass step, normalized with respect to its monoisotopic peak and the observed ion intensity at that m/z. An overlap matrix is thus generated that quantifies the degree of overlap between the theoretical and experimental isotopic distributions as a function of both the monoisotopic mass of the precursor and its charge. Using empirically derived thresholds that were determined by examining overlap matrix values for manually verified scans from multiple independent data sets, MAZIE selects the charge and monoisotopic precursor mass with the greatest similarity to the experimental data. If the thresholds are not met, MAZIE associates multiple potential charge states for that scan. Finally, MAZIE writes the most probable mass and charge(s) for each MS2 scan in the header information of either individual DTA files (for SEQUEST searching) or concatenated DTA files (for OMSSA searching). MAZIE typically requires about an hour of processing time for data files that contain 10,000 MS2 scans when using a single 1.67 MHz Intel processor under the Mac/Unix platform.
MAZIE is a Perl script written using Perl 5.8.8 on the MacOS/BSD Unix platform and on Perl 5.8 installed in a Cygwin system under Windows XP. It contains dependencies of the Perl modules Math∷CDF, PDL, and PDL∷NiceSlice (http://www.CPAN.org) and an in-house modified version of readmzXML.exe (http://tools.proteomecenter.org/readmzXML.php) that is employed to read header and scan data from mzXML files. MAZIE is distributed under the Creative Commons License and is distributed, together with its dependencies, at http://faculty.virginia.edu/templeton.
Results and Discussion
We analyzed spectra from a complex urine proteomics sample derived from an isoelectric peptide fractionation. Using SEQUEST, we searched both the unmodified DTAs and the MAZIE-modified DTAs that had identical parameters except for the specified precursor mass and charge. To evaluate the effect of the MAZIE modification, we performed duplicate sets of searches for both data sets using either average or monoisotopic precursor mass parameters, at two different precursor mass tolerances as listed in Table 1. Because SEQUEST Xcorr scores tend to increase with peptide size, the best peptide match (highest Xcorr) for each scan was grouped according to its charge state. A False Discovery Rate (FDR) was calculated for each charge state group by tabulating the number of matches to “reverse” protein sequences obtained when searched simultaneously with the natural database sequences (4). The Xcorr value at the 3% FDR cutoff and the number of scans with Xcorr above this cutoff for each charge state are tabulated in Table 1 for each for the SEQUEST searches.
Table 1.
From SEQUEST search results, the cutoff Xcorr and the number of scans above that Xcorr as determined by a 3% FDR criterion that was applied to each individual charge state.
| Search Parameters | Charge | Unmodified DTAs | MAZIE-modified DTAs | ||
|---|---|---|---|---|---|
| Xcorr Cutoff | Num of Scans | Xcorr Cutoff | Num of Scans | ||
| ave/mono 0.7/0.5 | 1 | 3.39 | 14 | 3.02 | 3 |
| 2 | 3.73 | 85 | 3.07 | 15 | |
| 3 | 3.25 | 76 | 2.86 | 12 | |
| 4 | 3.26 | 2 | N/D | 0 | |
| Total | 177 | 30 | |||
| avg/mono 1.5/0.5 | 1 | 3.12 | 61 | 2.81 | 112 |
| 2 | 3.61 | 342 | 2.59 | 786 | |
| 3 | 2.91 | 369 | 2.60 | 452 | |
| 4 | 2.89 | 17 | 2.88 | 25 | |
| Total | 789 | 1375 | |||
| mono/mono 0.7/0.5 | 1 | 3.12 | 16 | 2.73 | 118 |
| 2 | 3.58 | 191 | 2.59 | 807 | |
| 3 | 3.33 | 248 | 2.60 | 498 | |
| 4 | 2.89 | 10 | 3.99 | 7 | |
| Total | 465 | 1430 | |||
| mono/mono 1.5/0.5 | 1 | 3.12 | 55 | 2.80 | 117 |
| 2 | 3.77 | 268 | 2.87 | 688 | |
| 3 | 3.33 | 285 | 2.73 | 465 | |
| 4 | 3.23 | 7 | 3.99 | 7 | |
| Total | 615 | 1277 | |||
Using unmodified DTAs, we found that searches conducted with an average precursor mass with a relatively wide tolerance of 1.5 Da gave optimal results. However, using MAZIE-modified DTAs, we found that the searches conducted using a monoisotopic mass and a relatively narrow 0.7 Da tolerance gave the highest confidence results. This reflects the accurate determination of the monoisotopic mass of the precursor ion using MAZIE. A relatively loose 3% FDR filter was employed to enable comparison of results in the small number of scans within the +1 and +4 charge state groups. The results in Table 1 reveal that the optimal SEQUEST search results for the MAZIE-modified DTA set showed an 80% improvement from the optimal search using the unmodified DTA set.
Figure 1 displays the data from these two optimal analyses, i.e. the 3% FDR filtered scans from the avg(1.5 Da) search results for the unmodified DTA set and the mono(0.7 Da) search results for the MAZIE-modified DTA set. Each data point in the two graphs of Figure 1 represents a single MS2 scan from the original data file. Data points lying along the diagonal indicate scans that received identical Xcorrs (Figure 1A) or ΔCn (Figure 1B) from both of these searches while the data points lying above the diagonal indicate scans that received an improved Xcorr (or ΔCn) from the MAZIE-modified DTA. We identified 6 abundant proteins present in the sample and plotted these in blue while all other points, including most incorrect assignments, are in red.
Figure 1.
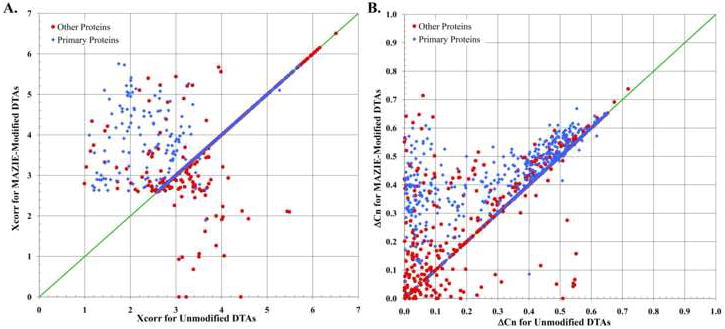
The change in the SEQUEST Xcorr and ΔCn parameters for DTAs generated from LTQ MS2 scans as a consequence of determining the monoisotopic mass and charge state of the original precursor mass of the scan. The sample represents an IEF fraction of a urine tryptic digest that contained six abundant proteins (◆) with lower levels of others (●). The Xcorr (A) and ΔCn (B) obtained from the unmodified DTAs are represented along the horizontal axis while the results obtained from the MAZIE-modified DTAs are along the vertical axis.
The figure reveals several important features of the SEQUEST search results. First, most of the data points lie along the diagonal in Figure 1A because the XCorr received by each scan is not dependent on either the mass difference between the experimental and theoretical mass nor on whether or not the precursor mass is considered as average or monoisotopic. The data points that lie above the diagonal have an improved Xcorr simply because the 1.5 Da precursor mass tolerance used by SEQUEST for the unmodified DTAs is too narrow to include the theoretical average mass of the correct peptide match. For the ΔCn parameter plotted in Figure 1B, many of the data points move above the diagonal, indicating that the difference between the best and second best identification has increased for much of the MAZIE-modified data. The improved ΔCn, indicating fewer false-discovery matches from the search results of the MAZIE-modified DTAs, is a direct consequence of the narrower precursor mass tolerance reducing the number of initial candidates that are searched and, thereby, reducing the number of incorrect matches.
Note that the SEQUEST search algorithm uses the same precursor mass tolerance window for each scan regardless of the charge state of the precursor ion. It could be argued, however, that the mass tolerance should instead reflect the uncertainty of the measured m/z of the ion and be linearly scaled according to the charge state of that precursor ion. This would enable tighter precursor mass tolerances to be employed for the scans with lower charge states and accommodate the propagation of instrument inaccuracy for the higher charge states. This approach is an option available with the OMSSA search algorithm.
To investigate the effect of mass tolerance scaling according to the charge, we tested the same trypsinized urine data with the OMSSA algorithm employing this option. For the MAZIE-modified DTAs, the OMSSA search considered only the charge state specified in the DTA header. For the unmodified data that does not have an accurate charge assignment, OMSSA considered all charge states from +1 to +4 for each scan. As before, both a monoisotopic and average precursor mass were considered with a range of mass tolerances. The results displayed in Table 2 illustrate again that the MAZIE-modified data resulted in more high-confidence matches than did the unmodified data when using their respective optimal search settings. Specifically, the search performed with mono(0.3 Da) precursor mass parameters, using MAZIE-modified data, resulted in about a 30% increase in the number of scans that pass a 1% FDR filter when compared to the search performed on the unmodified DTA set with avg(0.5 Da) precursor parameters.
Table 2.
The number of scans passing the 1% FDR filter for OMSSA search results, broken down by the charge state of the peptide identification.
| Search Parameters | Charge | Unmodified DTA s Number of Scans | MAZIE-modified DTA s Number of Scans |
|---|---|---|---|
| ave/mono 0.3/0.5 | 1 | 30 | 0 |
| 2 | 355 | 2 | |
| 3 | 365 | 14 | |
| 4 | 26 | 5 | |
| Total | 776 | 21 | |
| avg/mono 0.4/0.5 | 1 | 50 | 0 |
| 2 | 516 | 78 | |
| 3 | 418 | 242 | |
| 4 | 29 | 14 | |
| Total | 1013 | 334 | |
| avg/mono 0.5/0.5 | 1 | 52 | 0 |
| 2 | 615 | 336 | |
| 3 | 453 | 462 | |
| 4 | 39 | 36 | |
| Total | 1159 | 834 | |
| mono/mono 0.1/0.5 | 1 | 8 | 109 |
| 2 | 134 | 423 | |
| 3 | 38 | 413 | |
| 4 | 0 | 44 | |
| Total | 180 | 989 | |
| mono/mono 0.2/0.5 | 1 | 29 | 132 |
| 2 | 303 | 795 | |
| 3 | 103 | 533 | |
| 4 | 1 | 49 | |
| Total | 436 | 1509 | |
| mono/mono 0.3/0.5 | 1 | 50 | 132 |
| 2 | 454 | 800 | |
| 3 | 161 | 525 | |
| 4 | 7 | 49 | |
| Total | 672 | 1506 | |
Figure 2 plots the data points with OMSSA E-values better than the 1% FDR cutoff for these two particular searches with the MAZIE-modified results again located along the vertical axis. Similar to the SEQUEST results, most of the data points fall on or near the diagonal because they received similar E-values. An examination of the scans that received E-values that are significantly improved for the MAZIE-modified DTA show that these again mostly represent scans for which the relatively wide 0.5 Da tolerance for the average precursor mass of the unmodified data is not wide enough to include the theoretical average mass of the correct peptide.
Figure 2.
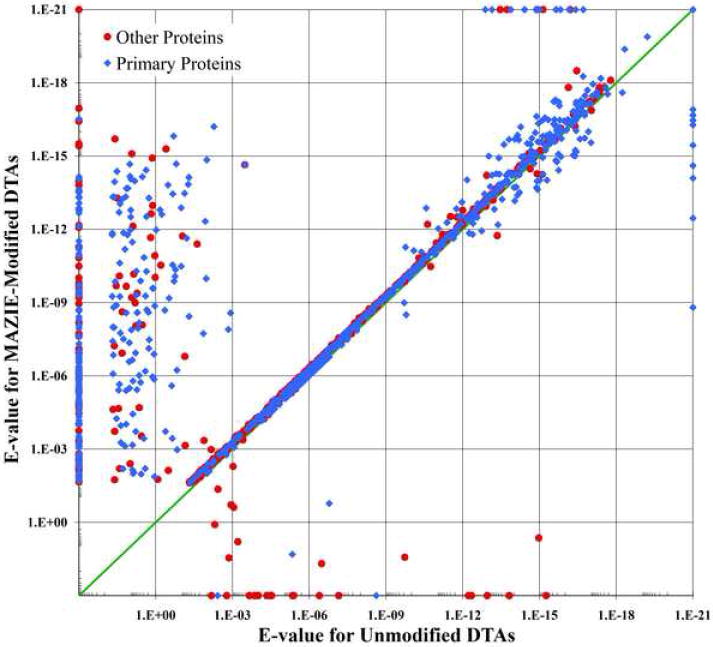
The change in the OMSSA E-value parameter for DTAs generated from LTQ MS2 scans as a consequence of determining the monoisotopic mass and charge state of the original precursor mass of the scan. The sample, data point symbols and data presentation are identical to Figure 1.
For both Figures 1A and 2, the small set of scan matches that scored worse for the MAZIE-modifed DTA (lie below the diagonal) represent either mixed/noisy spectra, for which MAZIE failed to determine the appropriate charge and/or monoisotopic mass, or false positive peptide matches from the search result on the unmodified DTA. Efforts are ongoing to adjust the MAZIE algorithm to better recognize the poor data conditions and to default to generating multiple DTAs for the charge state range of interest.
We then compared MAZIE to the previously-described DeconMSn algorithm obtained from http://omics.pnl.gov/software/DeconMSn.php (18) by using DeconMSn to analyze the same data set. Similar to MAZIE, DeconMSn generates DTA files that specify a corrected charge state determined by the algorithm or, if it is uncertain, defaults to generating two DTAs for a scan corresponding to a +2 and a +3 charge state. However, unlike the MAZIE algorithm, DeconMSn does not attempt to determine the monoisotopic mass associated with the precursor ion for mass spectral data acquired on low resolution spectrometers such as the LTQ. OMSSA searches were then conducted on DeconMSn DTAs, filtered by a 1% FDR cutoff, and then compared to the OMSSA mono(0.3 Da) results obtained for the MAZIE DTAs.
The comparison of the charge states determined by each of the algorithms to the charge state of the correct peptide match, based on the combined high-confidence OMSSA identifications of the three listed OMSSA searches, is displayed in the first column of Table 3. The MAZIE algorithm accurately determined the charge state of the precursor ion in 1187 out of 1558 total MS2 scans that passed the 1% FDR filter while DeconMSn unambiguously identified the precursor charge state for 814 scans. Table 3 also includes the sequence search results of the DeconMSn DTAs using the two best precursor parameters (identified in the analyses shown in Table 2), mono(0.3 Da) and ave(0.5 Da). Though the ave(0.5 Da) search setting for the precursor mass is the most appropriate because DeconMSn does not attempt to determine the monoisotopic mass of the precursor ion, the mono(0.3 Da) results of been included for completeness. The results illustrate that 1506 peptide matches passed the 1% FDR filter for the search result obtained with the MAZIE-modified DTAs while only 1171 matches did so for the DeconMSn DTAs, representing a 29% enhancement for the MAZIE-modified DTAs.
Table 3.
The number of scans for which MAZIE and/or DeconMSn accurately determined the charge state of its precursor ion and for which the subsequent OMSSA search of their corresponding DTAs obtained the correct peptide match.
| Charge | Accurate Charge State | MAZIE 0.3 mono vs DeconMSn 0.3 mono | MAZIE 0.3 mono vs DeconMSn 0.5 avg | |
|---|---|---|---|---|
| DeconMSn Only | 1 | 0 | 0 | 0 |
| 2 | 25 | 14 | 23 | |
| 3 | 76 | 9 | 18 | |
| 4 | 0 | 0 | 0 | |
| Totals | 101 | 23 | 41 | |
| MAZIE Only | 1 | 1 | 74 | 77 |
| 2 | 250 | 515 | 196 | |
| 3 | 179 | 310 | 57 | |
| 4 | 44 | 48 | 46 | |
| Totals | 474 | 947 | 376 | |
| Both | 1 | 131 | 58 | 55 |
| 2 | 537 | 285 | 604 | |
| 3 | 42 | 215 | 468 | |
| 4 | 3 | 1 | 3 | |
| Totals | 713 | 559 | 1130 | |
| Neither | 1 | 0 | 0 | 0 |
| 2 | 22 | 0 | 0 | |
| 3 | 246 | 0 | 0 | |
| 4 | 2 | 0 | 0 | |
| Totals | 270 | 0 | 0 | |
The above observations from these multiple search results illustrate that it is not the determination of the monoisotopic mass of the precursor ion, as opposed to its average mass, that improves the ability to distinguish the true positive from the false positive matches. Indeed, the mass error associated with the difference between the experimental and theoretical mass, regardless of whether it is the average or monoisotopic mass, does not significantly affect the subsequent peptide match score. This feature is illustrated graphically in Figures 1 and 2 by the majority of the scans lying along the diagonal. Instead, the improvement is a result of the MAZIE algorithm being able to more accurately determine the monoisotopic mass than the mass accuracy associated with the nominal MS1 scan of the LTQ mass spectrometer. Consequently, tighter precursor mass tolerances can be employed for the subsequent searches resulting in far fewer initial peptide candidates to be considered for each particular scan and, thereby, significantly reducing the likelihood that a relatively high-scoring, false positive match will be made. By reducing the number of false positive matches, more of the true positive matches can be distinguished. Furthermore, the determination of the precursor charge state does not result in increased search scores because the default data files generally include the correct ion charge state within the charge state range that is considered, typically from +1 to +4. However, by eliminating this redundancy MAZIE decreases the overall search time.
Because our data acquisition strategy collects an MS1 zoom scan before each MS2 spectrum, an obvious potential disadvantage is that a loss of information might be incurred as a direct result of fewer MS2 scans being acquired. Depending upon instrument and acquisition method settings, the acquisition time of the MS1 zoom scan is typically about half that required for the subsequent MS2 scan and, thus, reduces by a third the number of MS2 scans acquired for a fixed period of time. To examine this issue, we re-analyzed the same urine tryptic digest using the same instrumental setup except for the acquisition strategy. For this analysis, the “zoom” scans were eliminated and the acquisition duty cycle consisted of a full MS1 spectrum followed by five data-dependent MS2 spectra of the top five ions. The data from both of these acquisitions were processed in an identical manner as described except that the MAZIE algorithm was not used for either. The OMSSA searches for both datafiles were done with identical precursor parameters of ave(0.5 Da) over an equivalent 103 minute elution window. Without the zoom scans, 16485 MS2 scans were acquired and 165 unique peptides were identified that passed a 1% FDR. Using the zoom scan, 11213 MS2 scans were acquired and 161 unique peptides were identified that passed a 1% FDR, with 141 of these peptide identifications being common for both of the runs. Thus, for this relatively complex sample, the implementation of the zoom scan did not lead to a significant loss of useful information.
Conclusions
Our results indicate that MAZIE, using an acquisition plan on the Thermo LTQ-XL that includes MS1 ‘zoom’ scans, is an efficient and useful way of extracting more sequence information from complex proteomics samples. The MAZIE algorithm greatly facilitates the ability to distinguish true and false positive database search matches from complex proteomics data by accurately determining the monoisotopic mass of the precursor ion. It is the accurate determination of the precursor mass, as opposed to the whether or not it is the monoisotopic or average mass, that provides the major advantage. While the determination of the precursor ion charge state reduces the overall search time, it does not significantly reduce the ambiguity of interpreting the search results. Though the necessity of inserting an MS1 zoom scan before each MS2 scan reduces the number of MS2 scans collected, the improved accuracy of the data acquired can significantly increase the amount of useful information gained from the experiment.
Acknowledgments
Funding was provided through the National Cancer Institute, grant number CA126101.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tabb DL, McDonald WH, Yates JR. DTASelect and contrast: Tools for assembling and comparing protein identifications from shotgun proteomics. J Proteome Res. 2002;1:21–26. doi: 10.1021/pr015504q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.MacCoss MJ, Wu CC, Yates JR. Probability-based validation of protein identifications using a modified SEQUEST algorithm. Anal Chem. 2002;74:5593–5599. doi: 10.1021/ac025826t. [DOI] [PubMed] [Google Scholar]
- 3.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 4.Kall L, Storey JD, MacCoss MJ, Noble WS. Assigning significance to peptides identified by tandem mass spectrometry using decoy databases. J Proteome Res. 2008;7:29–34. doi: 10.1021/pr700600n. [DOI] [PubMed] [Google Scholar]
- 5.Dancik V, Addona TA, Clauser KR, Vath JE, Pevzner PA. De novo peptide sequencing via tandem mass spectrometry. J Comput Biol. 1999;6:327–342. doi: 10.1089/106652799318300. [DOI] [PubMed] [Google Scholar]
- 6.Sadygov RG, Eng J, Durr E, Saraf A, McDonald H, MacCoss MJ, Yates JR., 3rd Code developments to improve the efficiency of automated MS/MS spectra interpretation. J Proteome Res. 2002;1:211–215. doi: 10.1021/pr015514r. [DOI] [PubMed] [Google Scholar]
- 7.Hogan JM, Higdon R, Kolker N, Kolker E. Charge state estimation for tandem mass spectrometry proteomics. Omics. 2005;9:233–250. doi: 10.1089/omi.2005.9.233. [DOI] [PubMed] [Google Scholar]
- 8.Colinge J, Magnin J, Dessingy T, Giron M, Masselot A. Improved peptide charge state assignment. Proteomics. 2003;3:1434–1440. doi: 10.1002/pmic.200300489. [DOI] [PubMed] [Google Scholar]
- 9.Klammer AA, Wu CC, MacCoss MJ, Noble WS. Peptide charge state determination for low-resolution tandem mass spectra. Proc IEEE Comput Syst Bioinform Conf. 2005:175–185. doi: 10.1109/csb.2005.44. [DOI] [PubMed] [Google Scholar]
- 10.Na S, Paek E, Lee C. CIFTER: Automated charge-state determination for peptide tandem mass spectra. Anal Chem. 2008;80:1520–1528. doi: 10.1021/ac702038q. [DOI] [PubMed] [Google Scholar]
- 11.Tabb DL, Shah MB, Strader MB, Connelly HM, Hettich RL, Hurst GB. Determination of peptide and protein ion charge states by Fourier transformation of isotope-resolved mass spectra. J Am Soc Mass Spectrom. 2006;17:903–915. doi: 10.1016/j.jasms.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 12.Senko MW, Beu SC, Mclafferty FW. Determination of Monoisotopic Masses and Ion Populations for Large Biomolecules from Resolved Isotopic Distributions. J Am Soc Mass Spectr. 1995;6:229–233. doi: 10.1016/1044-0305(95)00017-8. [DOI] [PubMed] [Google Scholar]
- 13.Zhang ZQ, Marshall AG. A universal algorithm for fast and automated charge state deconvolution of electrospray mass-to-charge ratio spectra. J Am Soc Mass Spectr. 1998;9:225–233. doi: 10.1016/S1044-0305(97)00284-5. [DOI] [PubMed] [Google Scholar]
- 14.Horn DM, Zubarev RA, McLafferty FW. Automated reduction and interpretation of high resolution electrospray mass spectra of large molecules. J Am Soc Mass Spectr. 2000;11:320–332. doi: 10.1016/s1044-0305(99)00157-9. [DOI] [PubMed] [Google Scholar]
- 15.Kaur P, O’Connor PB. Algorithms for automatic interpretation of high resolution mass spectra. J Am Soc Mass Spectr. 2006;17:459–468. doi: 10.1016/j.jasms.2005.11.024. [DOI] [PubMed] [Google Scholar]
- 16.Beausoleil SA, Villen J, Gerber SA, Rush J, Gygi SP. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat Biotechnol. 2006;24:1285–1292. doi: 10.1038/nbt1240. [DOI] [PubMed] [Google Scholar]
- 17.Bakalarski CE, Haas W, Dephoure NE, Gygi SP. The effects of mass accuracy, data acquisition speed, and search algorithm choice on peptide identification rates in phosphoproteomics. Anal Bioanal Chem. 2007;389:1409–1419. doi: 10.1007/s00216-007-1563-x. [DOI] [PubMed] [Google Scholar]
- 18.Mayampurath AM, Jaitly N, Purvine SO, Monroe ME, Auberry KJ, Adkins JN, Smith RD. DeconMSn: a software tool for accurate parent ion monoisotopic mass determination for tandem mass spectra. Bioinformatics. 2008;24:1021–1023. doi: 10.1093/bioinformatics/btn063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eng JK, Mccormack AL, Yates JR. An Approach to Correlate Tandem Mass-Spectral Data of Peptides with Amino-Acid-Sequences in a Protein Database. J Am Soc Mass Spectr. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 20.Cagney G, Amiri S, Premawaradena T, Lindo M, Emili A. In silico proteome analysis to facilitate proteomics experiments using mass spectrometry. Proteome Sci. 2003;1:5. doi: 10.1186/1477-5956-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]