

Abstract
The neuropathology of the primary dystonias is not well understood. We examined brains from identical twins with DYT1-negative, dopa-unresponsive dystonia. The twins exhibited mild developmental delays until age 12 years when they began developing rapidly progressive generalized dystonia. Genetic, metabolic, and imaging studies ruled out known causes of dystonia. Cognition was subnormal but stable until the last few years. Death occurred at ages 21 and 22 years. The brains were macroscopically unremarkable. Microscopic examination showed unusual glial fibrillary acidic protein–immunoreactive astrocytes in multiple regions and iron accumulation in pallidal and nigral neurons. However, the most striking findings were 1) eosinophilic, rod-like cytoplasmic inclusions in neocortical and thalamic neurons that were actin depolymerizing factor/cofilin-immunoreactive but only rarely actin-positive; and 2) abundant eosinophilic spherical structures in the striatum that were strongly actin- and actin depolymerizing factor/cofilin-positive. Electron microscopy suggested that these structures represent degenerating neurons and processes; the accumulating filaments had the same dimensions as actin microfilaments. To our knowledge, aggregation of actin has not been reported previously as the predominant feature in any neurodegenerative disease. Thus, our findings may shed light on a novel neuropathological change associated with dystonia that may represent a new degenerative mechanism involving actin, a ubiquitous constituent of the cytoskeletal system.
Dystonic syndromes are characterized by co-contractures of agonist-antagonist muscles leading to abnormal postures. The dystonias can be classified by age of onset, by regional distribution, and, more recently, by etiology.1 Using an etiologic approach, the primary dystonias (DYT1 and others) are disorders in which the clinical phenotype is dominated by dystonia.
Pathologically, early reports from patients dying with primary generalized dystonia described striatal lipid accumulation that could not be corroborated in later studies.2–5 Other cases exhibited striatal neuronal loss and a mosaic pattern of astrocytosis.6 Most cases of primary generalized dystonia examined to date show no significant cerebral pathological or consistent radiological abnormalities (for reviews, see Zeman and Dyken,5 McGeer and McGeer,7 and Zeman8). In some cases of adult-onset primary regional dystonia such as cranio-cervical dystonia or Meige disease, investigators have reported varying degrees of neuronal loss and either neurofibrillary tangle or Lewy body formation in the brainstem and cerebellum.9–11
Secondary dystonias share, but are not limited to, the dystonic phenotype. Common accompaniments of these dystonic states include parkinsonism, myoclonus, and tremor.1 The secondary dystonias can be further subdivided into cases without defined pathology (eg, drug induced, occupational) and cases associated with known pathological processes such as trauma or developmental, metabolic, and vascular disorders in which the basal ganglia often are involved (for reviews, see Fahn and colleagues,1 Bhatia and Marsden,12 Marsden and colleagues,13 and Obeso and colleagues14).
Hereditodegenerative disorders with dystonic manifestations represent yet another category of secondary dystonia that usually exhibits a clearly demonstrable and progressive neuropathology. Well-known examples include Wilson’s disease, Hallervorden–Spatz disease, and Huntington’s disease; rarer conditions include Lubag (DYT3),15,16 Leber’s hereditary optic neuropathy with dystonia,17,18 and Machado–Joseph disease (SCA-3).19 Here, we report male twins with the onset of rapidly progressive, dopa-unresponsive generalized dystonia at age 12 years. The extensive neuropathological involvement of the cortex and basal ganglia most likely places these cases into the latter category of dystonic syndromes, and the unique findings in their brains are the main subject of this report.
Patients and Methods
Clinical Evaluations
The twins reported here were followed at Emory University hospitals and clinics from age 14 years until their deaths. J.L.J. treated the patients during the last 6 years, examined members of the immediate family, and reviewed all available external and internal medical records.
Neuropathological Evaluation
After macroscopic external examination, the brains were sliced coronally, and the slices were examined carefully for abnormalities. The slices were then subdivided for fixation in 10% neutral buffered formalin; fixation in periodate-lysine-paraformaldehyde (PLP; 2% paraformaldehyde in phosphate-buffered 0.01M periodate, 0.067M lysine, pH 7.4) or rapid freezing. Small blocks of formalin-fixed tissue collected from multiple cortical and subcortical regions were embedded in paraffin.
Eight-micrometer-thick paraffin-embedded sections were stained with hematoxylin and eosin, silver stains (Bielschowsky and Sevier–Munger), thioflavine S, myelin stains (Woelcke and Luxol fast blue/periodic acid–Schiff [PAS]), iron, PAS with and without diastase pretreatment, and Masson’s trichrome. PLP-fixed, frozen sections were stained with Oil Red O. Additional paraffin-embedded sections were immunohistochemically labeled with a variety of markers (see Table for list of markers and staining parameters). Appropriate positive controls were included in each immunohistochemical experiment. For most antibodies, negative controls consisted of sections incubated without primary antibody. For the actin immunohistochemistry, preadsorption experiments also were conducted.
Table.
Immunohistochemical Markers Examined, Including Antibody Sources and Staining Parameters
| Marker | Antibody Source | Species | Primary Antibody Dilution | Incubation Temperature | Incubation Time | Pretreatments |
|---|---|---|---|---|---|---|
| Tau | Accurate Chemical & Scientific | Rabbit | 1:100 | 40°C | 30 min | None |
| Neurofilaments | Dako (clone 2F11) | Mouse | 1:100 | 40°C | 30 min | Pepsin (Biomeda) |
| α-Synuclein | Gift of Dr Bernardino Ghetti | Rabbit | 1:400 | 4°C | 24 hr | 90% formic acid |
| α-Synuclein | Zymed | Mouse | 1:100 | 4°C | 24 hr | 90% formic acid |
| GFAP | Dako (clone 6F2) | Mouse | 1:100 | 40°C | 30 min | None |
| GFAP | Gift of Dr Virginia Lee | Rabbit | 1:750 | 40°C | 30 min | None |
| Ubiquitin | Dako | Rabbit | 1:100 | 40°C | 30 min | None |
| Ubiquitin | East Acres Biologicals | Rabbit | 1:500 | 40°C | 30 min | None |
| β-Amyloid | Senetek (4G8) | Mouse | 1:5,000 | 4°C | 24 hr | Pepsin, 90% formic acid |
| Apolipoprotein E | Medix | Goat | 1:250 | 40°C | 30 min | Pepsin, 90% formic acid |
| Apolipoprotein E | Chemicon | Goat | 1:250 | 40°C | 30 min | Pepsin, 90% formic acid |
| Actin | Santa Cruz | Mouse | 1:40 | 4°C | 48 hr | Microwave in 0.01M citrate, pH 6.0, Autozyme (Biomeda) |
| Actin | Sigma | Mouse | 1:100 | 4°C | 48 hr | Microwave in citrate, Autozyme |
| Actin | Sigma | Rabbit | 1:2,000 | 4°C | 48 hr | Microwave in citrate, Autozyme |
| HAM-56 | Cell Marque | Mouse | 1:10 | 40°C | 30 min | Microwave in citrate |
| HLA-DR α-chain | Dako (clone TAL.1B5) | Mouse | 1:100 | 40°C | 30 min | Microwave in citrate |
| CD68 | Dako (clone PG-M1) | Mouse | 1:100 | 40°C | 30 min | Microwave in citrate |
| Amyloid precursor protein | Roche (clone 22C11) | Mouse | 1:200 | 4°C | 24 hr | Microwave in citrate |
| ADF/cofilin | Gift of Dr Hiroshi Abe | Mouse | 1:40 | 4°C | 24 hr | Microwave in citrate |
| ADF/cofilin | Cytoskeleton | Rabbit | 1:125 | 4°C | 24 hr | Microwave in citrate |
| Gelsolin | Sigma | Mouse | 1:500 | 4°C | 24 hr | None |
| MAP1b | Gift of Dr Lester Binder | Mouse | 1:300 | 4°C | 24 hr | Microwave in citrate |
| Tubulin | Gift of Dr Lester Binder | Mouse | 1:500 | 4°C | 24 hr | Microwave in citrate |
| Polyglutamine repeats | Gift of Dr J.-L. Mandel (1C2) | Mouse | 1:500 | 4°C | 24 hr | None |
| Met-enkephalin | INCSTAR | Rabbit | 1:1,000 | 4°C | 24 hr | None |
| NG2 | Chemicon | Rabbit | 1:50 | 4°C | 24 hr | Microwave in citrate |
| Synaptophysin | Biogenics (clone 5Y38) | Mouse | 1:320 | Room temperature | 25 min | 5 min in pressure cooker 15lb/in2 |
GFAP = glial fibrillary acidic protein; ADF = actin depolymerizing factor.
Additional Control Case Materials
Formalin-fixed, paraffin-embedded sections of basal ganglia from six cases of primary dystonia, including one DYT1 case, were provided by the Brain and Tissue Bank for Developmental Disorders at the University of Maryland for comparison with our cases. The six cases included five female patients and one male patient, age range 63 to 91 years, and none showed specific neuropathological findings at autopsy that could account for their clinical dystonia. In addition to hematoxylin and eosin stains, glial fibrillary acidic protein (GFAP), actin, and actin depolymerizing factor (ADF)/cofilin immunohistochemistry was performed on the sections from these six cases.
Electron Microscopy
One-millimeter cubes of PLP-fixed frontal cortex were fixed in 4% cacodylate-buffered glutaraldehyde overnight, post-fixed in 1% osmium tetroxide for 1 hour, and embedded in EMbed812 epoxy resin (Electron Microscopy Sciences, Fort Washington, PA). One-half-micrometer-thick sections were stained with toluidine blue. Ultrathin (approximately 800 angstroms thick) sections of areas containing cells with inclusions were contrasted with uranyl acetate and lead citrate and examined with a Philips EM201 electron microscope.
Immunoelectron microscopy was conducted using pre-embedding immunogold labeling for actin. Sections of PLP-fixed basal ganglia were rinsed in phosphate-buffered saline and processed according to the manufacturer’s instructions. Ultrasmall colloidal gold-conjugated secondary antibody (Aurion, Wageningen, the Netherlands) was used to bind to the primary antibody. After postfixation with 2.5% glutaraldehyde, gold particles in sections were intensified using R-gent Se-EM silver enhancement kit (Aurion). Sections were further fixed with 0.5% osmium tetroxide in 0.1M phosphate buffer for 15 minutes and processed for electron microscopy as described elsewhere.20 Selected sections were placed in 0.5% osmium tetroxide in phosphate buffer for 30 minutes and then flat embedded in Epon between sheets of Aclar and cured at 60°C for 2 to 3 days. Thin sections were cut using a Leica Ultracut S microtome and examined on a Hitachi H-7500 electron microscope.
Results
Clinical Phenotype
The twins were the low-birth-weight monozygotic products of a 32-week gestation complicated by near miscarriage in a 35-year-old gravida 5 para 4 white woman with a history of a previous twin miscarriage involving a different male partner. Unless otherwise indicated, the twins are discussed together, because their phenotypes were similar.
The twins were born with cleft lip and palate requiring multiple repairs. They were small for their age, and their limbs were small in relation to the rest of their bodies. Skeletal abnormalities included high foreheads, hypoplastic scapulas, and externally rotated hips. By age 10 years, they began developing kyphoscoliosis and severe antecolis. Achalasia presented at age 2 years; twin A initially stabilized on medical therapy, whereas surgical repair was required in twin B. Vision was normal at birth, and retinal exams before age 10 years were normal. By age 3 years, twin B developed spontaneous cataracts, aggravated later by trauma; by age 10 years, he was blind in one eye and had limited vision in the other. Vision in twin A was not affected. Sensory-neural hearing loss in both twins resulted in functional deafness by age 4 years, significantly affecting speech development.
Motor development was delayed and characterized initially as “mild cerebral palsy” with early “soft” signs suggestive of cortical motor dysfunction. The subsequent motor disorder began at age 12 years with the postural abnormalities noted above. Leg dystonia developed by age 14 years, progressing over 5 years from clumsy gait to inability to walk. As the dystonia generalized, the twins developed spontaneous bouts of oculogyric and opisthotonic crises. Cranial (spontaneous grimacing, dysarthria) and bulbar (forceful tongue protrusion, dysphagia) involvement began by age 14 years, further impairing speech and swallowing and necessitating gastrostomies by age 17 years. During that time, muscle tone evolved from normal to severe painful generalized dystonia. Hand and arm dystonia began around age 15 years with loss of fine and later gross motor skills. There was no tremor, parkinsonism, myoclonus, or autonomic dysfunction. Deep tendon reflexes were initially normal but later became difficult to obtain in the legs. Plantar responses remained flexor. There were no cerebellar signs, and the sensory exam was normal.
The twins’ early nonverbal intellectual function was in the just-below-average range. This mild impairment consisted of language acquisition deficits and other learning disabilities complicated by the hearing loss. By age 14 years, communication was further impaired by speech dysarthria and later pseudobulbar palsy. By age 17 years, intellectual function was increasingly difficult to test due to the multiple deficits. Evidence of progressive intellectual impairment and functional dementia consisted of decreasing ability to use American Sign Language and to direct or assist caregivers, as well as decreasing responsiveness to family and novelty.
Family History and Review of Systems
The mother is of Irish-English extraction with Chero-kee Indian and Jewish ancestry and has always been in good health. She smoked during the pregnancy but otherwise was not exposed to toxins or drugs. Little is known about the history of the estranged father, but when last seen 15 years ago, he had no obvious medical problems. Two half brothers are normal. There is no history of a similar illness or any other medical or neurological problem on the mother’s side of the family. At no point was there evidence of another illness not attributable to recurrent aspiration, to bowel dysmotility with pseudoobstruction, and, late in life, to recurrent urinary tract infections.
Treatments
Anticholinergics and antispasmodics were at best minimally effective at relieving the pain associated with the dystonic contractions; the deformities themselves were never helped. Focal injections of botulinum toxin also were used. Dopamine agonists were not effective. The intellectual dysfunction and apathy did not respond to the empiric use of antidepressants.
Diagnostic Tests
Genetic studies showed a normal karyotype, and sequencing of the entire mitochondrial genome in brain tissue showed no abnormalities. Additional tests showed that the twins were negative for the DYT1 mutation (courtesy of Drs Laurie Ozelius and Katherine Simms at Massachusetts General Hospital) and for the DDP1 (deafness/dystonia peptide 1) mutation. Extensive laboratory evaluations including neuroimaging, electroencephalography, cerebrospinal fluid analysis, radiology, muscle biopsy, routine laboratory tests, endocrine studies, and a wide variety of metabolic studies including mitochondrial oxidative phosphorylation enzymology in muscle were all negative for systemic illness or known metabolic disorders.
Neuropathological Findings
The fresh weights of the brains were 1,212gm (twin 1) and 1,290gm (twin 2). Both brains were macroscopically unremarkable with the exception of mild pallor of the substantia nigra. There was no significant cortical atrophy, and, on coronal sections, the basal ganglia showed no macroscopic changes. For the purpose of this report, the neuropathological findings were identical in both twins unless otherwise specified.
Microscopically, hematoxylin and eosin stains showed no frank reactive astrocytosis or neuronal loss in the neocortex, basal ganglia, or other subcortical structures. Throughout the brain, however, occasional neurons exhibited a pale cytoplasm suggesting early degeneration. GFAP immunohistochemistry showed numerous unusual GFAP-positive astrocytes with dense processes resembling strings of beads throughout the neocortex and basal ganglia (Fig 1). Within the neocortex, these GFAP-positive glia were most prominent in the white matter and at the gray–white matter junction. Within the basal ganglia, immunoreactive glia were most prominent in the striatum and internal capsule; there was no obvious clustering of GFAP-positive glia in “islands” as has been reported in some cases of primary dystonia and in Lubag.6,9,16 Minor findings included hypopigmentation of neurons in the substantia nigra, corresponding to the mild pallor noted grossly and reported previously in a case of dopa-responsive dystonia21 and iron deposition within neurons and glia of the globus pallidus and substantia nigra, also reported elsewhere and most likely representing a nonspecific finding.5 However, severe degeneration and extensive iron deposition in the pallidum and nigra, the hallmarks of Hallervorden–Spatz disease, were not present. Furthermore, Oil Red O did not show any significant lipid accumulation, nor did silver stains show any neurofibrillary tangles. Myelin stains were also unremarkable.
Fig 1.
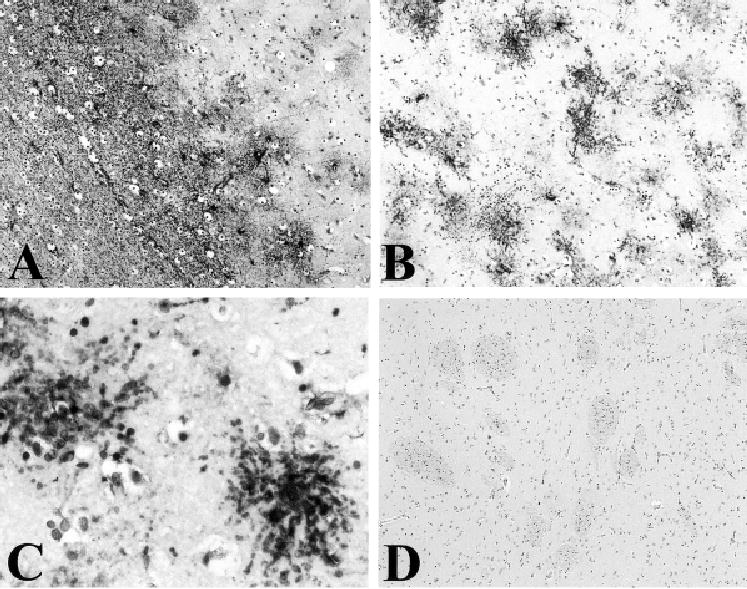
(A) Glial fibrillary acidic protein (GFAP) immunolabel of neocortex in Case 1 showing unusual-appearing, GFAP-positive glia, most prominent at the junction of the gray and white matter. (B, C) GFAP immunolabel in the striatum of Case 1; unusual-appearing, GFAP-positive glia are prominent but show no clustering in “islands.” (D) GFAP immunolabel is not present in the striatum of a patient with DYT1-type dystonia. Magnification: A, B, ×10; C, ×40; D, ×5.
A striking finding was extensive eosinophilic, rodlike cytoplasmic inclusions in neurons in the thalamus and throughout the neocortex (Fig 2); these inclusions resembled those previously reported in neurons of the basal ganglia and thalamus in alcoholics with and without liver damage, in individuals with various neurological diseases, in patients with myotonic dystrophy, and in nondiseased individuals.22–27 No rod-like inclusions were seen in neuronal nuclei, as was reported in neurons of the tectum in a case of Meige disease.10 The cytoplasmic inclusions in the neocortex and thalamus in our cases, like those previously described, stained red on trichrome stain and were PAS- and thioflavine S–negative. Unlike the striatal and thalamic inclusions described by others,28 most of the neocortical and thalamic inclusions in our cases were actin-negative with all antibodies tested (see Table); however, they immunolabeled strongly with antibodies to the actin-associated proteins, ADF/cofilin, which regulate actin turnover.
Fig 2.
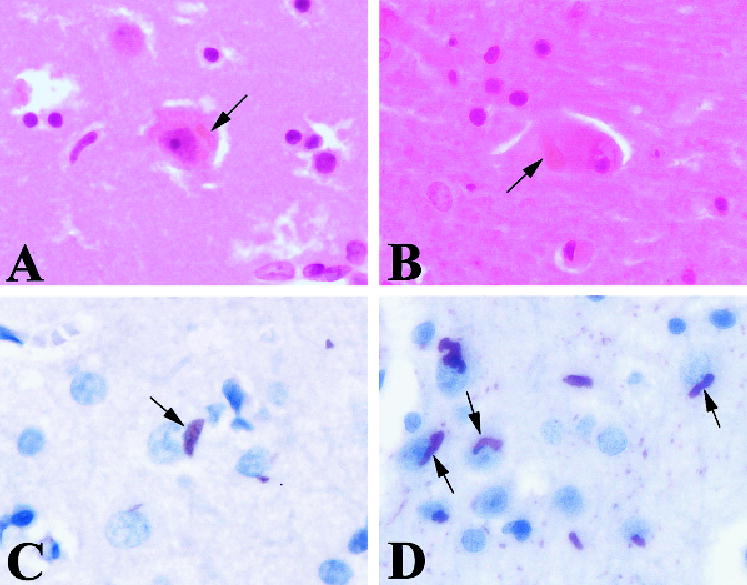
Eosinophilic rod-like inclusions (arrows) in the neocortex (A) and thalamus (B). These inclusions were strongly immunoreactive for the actin-associated proteins ADF/cofilin (D); only occasional inclusions were actin-positive (C). Magnification: ×40.
Another striking microscopic finding in our cases was abundant eosinophilic spherical structures were also seen in the striatum (Fig 3). These structures also were seen at lower densities in the globus pallidus and substantia nigra. They ranged in size from approximately 1 to 20μm and stained red on trichrome stain but did not stain with PAS, thioflavine S, or silver stains. Although it was not clear whether these structures were neuronal or glial in origin, electron microscopic studies suggest that they most likely represent degenerating neurons and processes. However, they did not appear to represent axonal spheroids, because they were immunonegative for neurofilaments, and ultrastructural studies failed to show the dense bodies and vesicular profiles typical of axonal spheroids.
Fig 3.
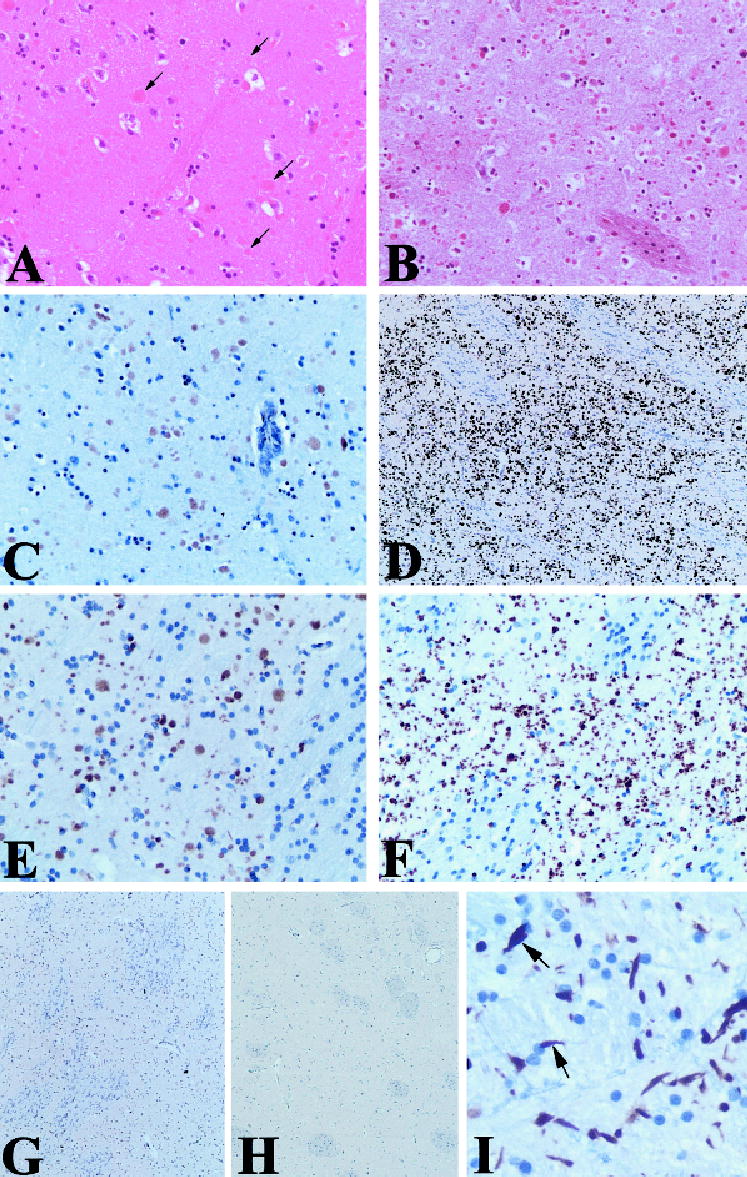
Unusual spherical structures of varying sizes (arrows) were seen on hematoxylin and eosin (A) and trichrome (B) stains in the striatum in Case 1. These structures were moderately immunoreactive with antibodies to ubiquitin (C) and strongly positive for actin (D, E) and ADF/cofilin (not shown). In Case 2 (F), the actin-immunoreactive structures were smaller and more irregularly shaped than in Case 1. Preadsorption of the actin antibody with peptide greatly diminished immunolabel (G). No actin immunore-activity was seen in the striatum of a DYT1-type dystonia case (H). Actin aggregates within the globus pallidus (I) were more elongate than those in the striatum, and some of these (arrows) were reminiscent of the glial inclusions seen in neurodegenerative diseases such as the synucleinopathies and tauopathies. Similar elongated aggregates were observed within the substantia nigra in both cases and within the subthalamic nucleus in Case 1 (not shown). Magnification: A to C, E, F, ×20; D, G, H, ×5; I, ×40.
Immunohistochemically, these spherical structures were negative for tau, neurofilaments, GFAP, α-synuclein, various macrophage and microglial markers, apolipo-protein E, β-amyloid, the amyloid precursor protein, and a variety of other markers (see Table for complete list of markers tested). They were lightly to moderately ubiquitin-positive in twin 1 but not twin 2. They were strongly positive for actin and ADF/cofilin in both twins (see Fig 3); in twin 1, these structures were predominantly spherical, whereas in twin 2 they tended to be more irregularly shaped. No such structures were seen in the basal ganglia of six other dystonia patients, one of whom was known to bear the DYT1 mutation. Additional actin and ADF/cofilin immunoreactivity was noted in the globus pallidus and substantia nigra in both twins and in the subthalamic nucleus in twin 1. In these regions, the aggregates were elongated and more process-like rather than spherical, and some had shapes reminiscent of the glial inclusions seen in multisystem atrophy and tauopathy (see Fig 3).
Electron Microscopic Findings
Ultrastructural evaluation of neocortical tissue showed irregularly shaped to oblong or needle-like aggregations of filaments within cells and processes (Figs 4A and B and 5A). The filaments, which measured 4.36nm in diameter, were arranged in parallel arrays with a periodicity of 11.6nm. Occasional aggregates also were identified within the neuropil. Axonal abnormalities, particularly swelling and disruption of the myelin sheath, also were observed (see Fig 5B).
Fig 4.
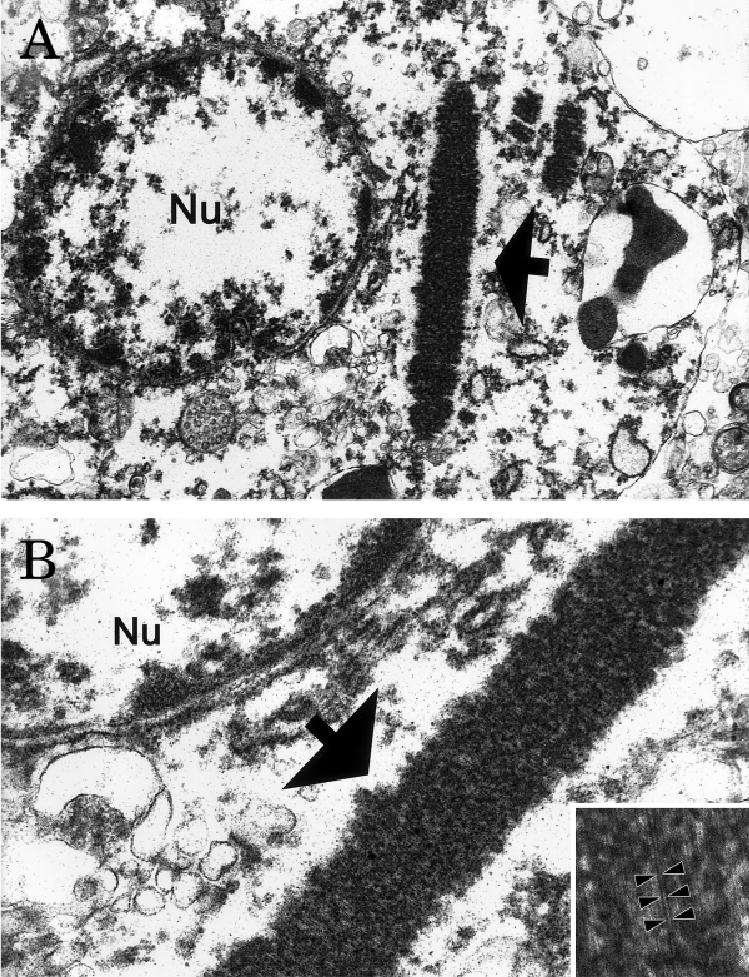
Ultrastructure of an inclusion (arrow) within a neocortical neuron seen at low (A) and intermediate (B) power. These aggregates of filaments were irregularly shaped to oblong or needle-shaped and formed parallel arrays (inset) with a periodicity of 11.6nm (arrowheads); the 4.36nm diameter of the filaments places them in the size range of actin microfilaments. Original magnification: A, ×26,760; B, ×76,210; inset, ×171,975.
Fig 5.
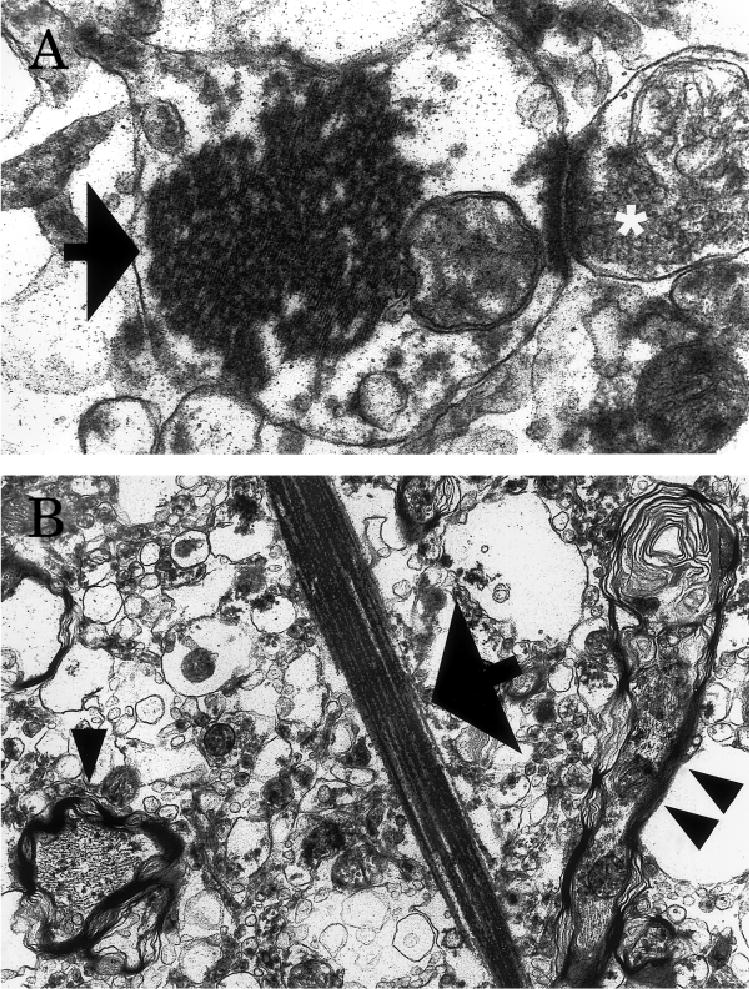
Electron micrographs of a filamentous inclusion (arrow) within a postsynaptic profile (the presynaptic bouton is indicated with a white asterisk) in the neocortex (A) and a needle-shaped inclusion (arrow) in the neuropil (B). Note the swelling and disruption of myelin in nearby axons (arrowheads, B) seen in longitudinal and cross-sections. Original magnification: A×26,760; B, ×18,800.
Electron microscopic evaluation of the spherical structures in the striatum showed variable sized membrane-bound accumulations of filaments approximately 5nm in diameter, placing them in the size range of actin microfilaments (Fig 6). Postembedding immunohistochemistry confirmed that these filamentous aggregates were actin-positive. Neither the spherical actin- and ADF/cofilin-positive structures in the striatum nor the rod-like inclusions in the neocortex and thalamus exhibited the “herringbone” appearance typical of Hirano bodies.
Fig 6.
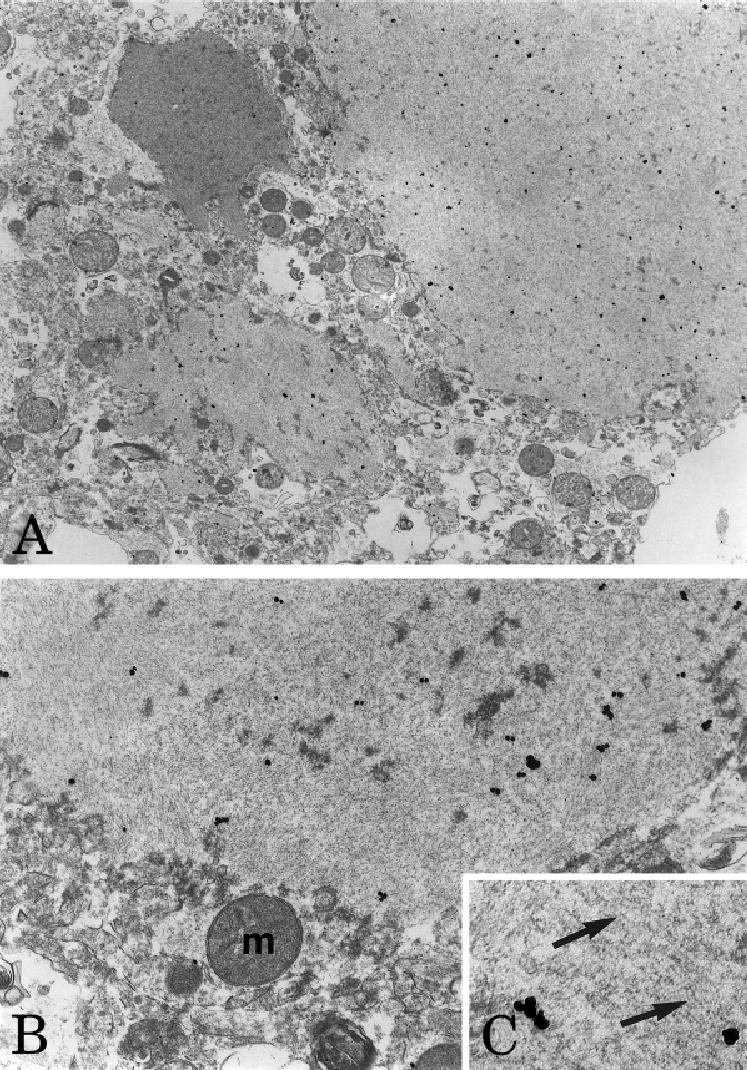
Ultrastructure of actin-immunoreactive spherical structures in the striatum. Low (A) and intermediate (B) power electron micrographs illustrating membrane-bound aggregates of filaments that were actin-immunoreactive, as shown by the accumulation of colloidal gold particles over these structures. The filaments (arrows, C) measured approximately 5nm in diameter, consistent with the diameter of actin microfilaments. Magnification: A, ×9,059; B, ×25,633; C, ×51,266.
Discussion
The twins presented here represent a unique progressive neurodegenerative disorder with a primarily dystonic phenotype of juvenile onset. This entity is distinct from the primary and many of the secondary dystonias described above, in that it has a well-defined and progressive neuropathology. The dystonic manifestations in adolescence were heralded by early bulbar signs that suggested a more widespread developmental disorder. For instance, the coincidence of signs like cleft lip and palate, early cataracts, sensory-neural hearing loss, mild dysmorphism (short limbs and high forehead) and achalasia may suggest a more systemic disorder of cytoskeletal proteins or embryogenesis. Early stable learning disabilities were later compounded by progressive cognitive decline.
The fact that the twins exhibited a virtually identical clinical and pathological phenotype suggests, but does not prove, that this is a genetically mediated disorder. Although there are many forms of genetically mediated dystonia, the disorder presented here cannot be explained by any of the known primary dystonias. The twins’ clinical phenotype also differs from that seen in any of the previously reported hereditodegenerative disorders with dystonic manifestations.
Moreover, the neuropathological findings differ dramatically from those reported previously in other neurodegenerative disorders with dystonic manifestations. The most striking finding in the brains of both twins was extensive aggregation of actin and/or the actin-associated protein ADF/cofilin in rod-like structures throughout the neocortex; in spherical structures probably representing degenerating neurons and processes in the striatum; and in elongated, neurite-like structures in the globus pallidus, subthalamic nucleus, and substantia nigra. Although eosinophilic, rod-like structures have been reported in normal individuals, alcoholics, myotonic dystrophy patients, and aging mice and are thought to be a nonspecific finding, they are typically localized to the striatum, thalamus, and substantia nigra in these individuals, although inclusions have been seen in a small proportion of cells in the frontal and temporal cortex in cases of myotonic dystrophy.22–24,26,27 To our knowledge, the extensive distribution of rod-like structures throughout the neocortex, as observed in this study, has not been reported elsewhere. Our observation that more cortical rods immunolabel with antibodies to cofilin than actin is surprising, given that cofilin is not known to aggregate or polymerize independently of actin.29,30 However, binding of cofilin to filamentous actin hinders phalloidin binding; thus, we cannot rule out the possibility that the actin epitopes recognized by the antibodies used in our study were masked by the presence of cofilin in the cortical rods.
The aggregation of actin and actin-related proteins in our cases is particularly intriguing in light of the involvement of other cytoskeletal proteins in a variety of developmental and neurodegenerative diseases. For example, aggregates of the microtubule-associated protein τ are a prominent feature in corticobasal degeneration, progressive supranuclear palsy, Pick’s disease, frontotemporal dementia and parkinsonism linked to chromosome 17, and Alzheimer’s disease.31–33 Aggregates of neurofilament, the neuronal intermediate filament protein, have been reported in motor neurons in amyotrophic lateral sclerosis, in the Lewy bodies of Parkinson’s disease and Lewy body dementia, in Pick bodies and Pick cells in Pick’s disease, and in ballooned neurons in corticobasal degeneration.31,32,34 Hirano bodies, which also contain actin and ADF/cofilin35,36 but are ultrastructurally different from the inclusions identified in these cases, are seen in the hippocampus in normal aging and as a minor finding in a variety of neurodegenerative diseases,31 but their significance is not clear, given their widespread occurrence. Minamide and colleagues36 reported ADF/cofilin immunoreactivity in plaque neurites in Alzheimer’s disease and also described the formation of actin- and ADF/cofilin-positive rods in cultured hippocampal neurons exposed to mediators of neurodegeneration including ischemia, oxidative stress, and excitotoxicity; ultrastructural studies of these rods have not been published. To our knowledge, our findings represent the first report of actin microfilament and ADF/cofilin aggregation as the predominant pathology in a neurodegenerative disease in vivo.
The colocalization of ADF/cofilin with actin in the aggregates in our cases suggests that the dysfunction in the twins was not limited to the actin protein but rather extended to include the actin regulatory system. ADF/cofilins are members of a complex system of proteins regulating turnover of actin filaments.29,30 De-phosphorylated ADF/cofilins increase actin filament turnover; phosphorylation of ADF/cofilins by Lim kinase 1 decreases their activity. Both hyperactivity and hypoactivity of the ADF/cofilins can result in the formation of actin aggregates.37–39 We are currently investigating whether the phosphorylation status and thus the activity of ADF/cofilins is altered in the brains of the twins, and, if so, whether the abnormality lies with the ADF/cofilins or with an upstream regulator of these cytoskeletal proteins.
In summary, we report novel neuropathological findings in identical twin brothers with a degenerative disorder presenting with multiple developmental abnormalities culminating in juvenile-onset generalized dystonia with particularly severe bulbar involvement. The extensive aggregation of actin and the actin regulatory proteins ADF/cofilin observed in these two cases suggests a dysfunction of the system regulating turnover of actin filaments in the cell. Given that dysregulation of cytoskeletal elements is a common theme in neurodegenerative diseases, our findings may have important implications not only in the field of dystonia, but also for the study of neurodegenerative disease, in general.
Acknowledgments
This work was supported by the Emory-Morehouse Alzheimer’s Disease Center (NIH AG10130, M.G., B.H.W.), National Institutes of Health (R01-AT00612-01, J.L.J.; N521328, AG13154, and N537167, D.C.W.), the Fauver Family Foundation, the Emory American Parkinson’s Disease Association Center of Excellence, the National Science Foundation (MCB-0110464, S.O.; IBN9983078, C.-A.G.), and the University Research Committee of Emory University and by a J. Worley Brown fellowship (V.P.) and an Ellison Foundation senior scientist grant (D.C.W.).
We thank L. Roback, R. Baul, and H. Yi for excellent technical assistance. We are also deeply grateful to the family of the twins for donating the twins’ brains and for their ongoing interest and involvement in this work, and to the Brain and Tissue Bank for Developmental Disorders at the University of Maryland for providing tissues from additional dystonia cases.
References
- 1.Fahn S, Bressman SB, Marsden CD. Classification of dystonia. In: Fahn S, Marsden CD, DeLong MR, eds. Advances in neurology (dystonia 3). Vol 78. New York: Lippincott-Raven, 1998:1–10. [PubMed]
- 2.Davison C, Goodhart SP. Dystonia musculorum deformans: clinicopathologic study. Arch Neurol Psychiatr (Chic) 1933;29:1108–1124. [Google Scholar]
- 3.Marinesco G, Nicolesco M. Un cas anatomo-clinique de dystonie contorsive spasmodique avec lésions du striatum et des centres sousthalamiques. Rev Neurol. 1929;1:973–980. [Google Scholar]
- 4.Richter H. Beiträge zur Klinik und pathologischen Anatomie der extrapyramidalen Bewegungsstörungen. Arch Psychiat Nervenkr. 1923;67:226–294. [Google Scholar]
- 5.Zeman W, Dyken P. Dystonia musculorum deformans. Clinical, genetic and pathoanatomical studies. Psychiatr Neurol Neurochir. 1967;70:77–121. [PubMed] [Google Scholar]
- 6.Gibb WRG, Kilford L, Marsden CD. Severe generalised dystonia associated with a mosaic pattern of striatal gliosis. Movement Disord. 1992;7:217–223. doi: 10.1002/mds.870070305. [DOI] [PubMed] [Google Scholar]
- 7.McGeer EG, McGeer PL. Pathology of the dystonias. In: Tsui JKC, Calne DB, eds. Handbook of dystonia. New York: Marcel Dekker, 1995:77–102.
- 8.Zeman W. Pathology of the torsion dystonias (dystonia musculorum deformans) Neurology. 1970;20:79–88. doi: 10.1212/wnl.20.11_part_2.79. [DOI] [PubMed] [Google Scholar]
- 9.Altrocchi PH, Forno LS. Spontaneous oral-facial dyskinesias: neuropathology of a case. Neurology. 1983;33:802–805. doi: 10.1212/wnl.33.6.802. [DOI] [PubMed] [Google Scholar]
- 10.Kulisevsky J, Marti MJ, Ferrer I, Tolosa E. Meige syndrome: neuropathology of a case. Movement Disord. 1988;3:170–175. doi: 10.1002/mds.870030209. [DOI] [PubMed] [Google Scholar]
- 11.Zweig RM, Hedreen JC, Jankel WR, et al. Pathology in brainstem regions of individuals with primary dystonia. Neurology. 1988;38:702–706. doi: 10.1212/wnl.38.5.702. [DOI] [PubMed] [Google Scholar]
- 12.Bhatia KP, Marsden CD. The behavioural and motor consequences of focal lesions of the basal ganglia in man. Brain. 1994;117:859–876. doi: 10.1093/brain/117.4.859. [DOI] [PubMed] [Google Scholar]
- 13.Marsden CD, Obeso JA, Zarranz JJ, Lang AE. The anatomical basis of symptomatic hemidystonia. Brain. 1985;108:463–483. doi: 10.1093/brain/108.2.463. [DOI] [PubMed] [Google Scholar]
- 14.Obeso JA, Giménez-Roldán S. Clinicopathological correlation in symptomatic dystonia. Adv Neurol. 1988;50:113–122. [PubMed] [Google Scholar]
- 15.Graeber MB, Kupke KG, Müller U. Delineation of the dystonia-parkinsonism syndrome locus in Xq13. Proc Natl Acad Sci USA. 1992;89:8245–8248. doi: 10.1073/pnas.89.17.8245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Waters CH, Faust PL, Powers J, et al. Neuropathology of Lubag (X-linked dystonia parkinsonism) Movement Disord. 1993;8:387–390. doi: 10.1002/mds.870080328. [DOI] [PubMed] [Google Scholar]
- 17.Jun AS, Brown MD, Wallace DC. A mitochondrial DNA mutation at nucleotide pair 14459 of the NADH dehydrogenase subunit 6 gene associated with maternally inherited Leber hereditary optic neuropathy and dystonia. Proc Natl Acad Sci USA. 1994;91:6206–6210. doi: 10.1073/pnas.91.13.6206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shoffner JM, Brown MD, Stugard C, et al. Leber’s hereditary optic neuropathy plus dystonia is caused by a mitochondrial DNA point mutation. Ann Neurol. 1995;38:163–169. doi: 10.1002/ana.410380207. [DOI] [PubMed] [Google Scholar]
- 19.Sudarsky L, Coutinho P. Machado-Joseph disease. Clin Neurosci. 1995;3:17–22. [PubMed] [Google Scholar]
- 20.Gutekunst CA, Li SH, Yi H, et al. The cellular and subcellular localization of huntingtin-associated protein 1 (HAP1): comparison with huntingtin in rat and human. J Neurosci. 1998;18:7674–7686. doi: 10.1523/JNEUROSCI.18-19-07674.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rajput AH, Gibb WRG, Zhong XH, et al. Dopa-responsive dystonia: pathological and biochemical observations in a case. Ann Neurol. 1994;35:396–402. doi: 10.1002/ana.410350405. [DOI] [PubMed] [Google Scholar]
- 22.Culebras A, Feldman RG, Merk FB. Cytoplasmic inclusion bodies within neurons of the thalamus in myotonic dystrophy. A light and electron microscopic study. J Neurol Sci. 1973;19:319–329. doi: 10.1016/0022-510x(73)90095-6. [DOI] [PubMed] [Google Scholar]
- 23.Fraser H. Eosinophilic bodies in some neurones in the thalamus of ageing mice. J Pathol. 1969;98:201–204. doi: 10.1002/path.1710980307. [DOI] [PubMed] [Google Scholar]
- 24.Kawano N, Horoupian DS. Intracytoplasmic rod-like inclusions in caudate nucleus. Neuropathol Appl Neurobiol. 1981;7:307–314. doi: 10.1111/j.1365-2990.1981.tb00102.x. [DOI] [PubMed] [Google Scholar]
- 25.Kojima K, Ogawa H. Alcoholic hyaline-like bodies of nerve cells. No To Shinkei. 1974;26:1000–1001. [PubMed] [Google Scholar]
- 26.Ono S, Inoue K, Mannen T, et al. Neuropathological changes of the brain in myotonic dystrophy—some new observations. J Neurol Sci. 1987;81:301–320. doi: 10.1016/0022-510x(87)90105-5. [DOI] [PubMed] [Google Scholar]
- 27.Peña CE, Katoh A. Intracytoplasmic eosinophilic inclusions in the neurons of the central nervous system. Acta Neuropathol. 1989;79:73–77. doi: 10.1007/BF00308960. [DOI] [PubMed] [Google Scholar]
- 28.Goldman JE, Horoupian DS. An immunocytochemical study of intraneuronal inclusions of the caudate and substantia nigra. Reaction with an anti-actin antiserum. Acta Neuropathol. 1982;58:300–302. doi: 10.1007/BF00688613. [DOI] [PubMed] [Google Scholar]
- 29.Bamburg JR, McGough A, Ono S. Putting a new twist on actin: ADF/cofilins modulate actin dynamics. Trends Cell Biol. 1999;9:364–370. doi: 10.1016/s0962-8924(99)01619-0. [DOI] [PubMed] [Google Scholar]
- 30.Bamburg JR. Proteins of the ADF/cofilin family: essential regulators of actin dynamics. Annu Rev Cell Dev Biol. 1999;15:185–230. doi: 10.1146/annurev.cellbio.15.1.185. [DOI] [PubMed] [Google Scholar]
- 31.Esiri MM, Hyman BT, Beyreuther K, Masters CL. Ageing and dementia. In: Graham DI, Lantos PL, eds. Greenfield’s neuropathology. Vol II. New York: Oxford University Press, 1997:153–233.
- 32.Lowe J, Lennox G, Leigh PN. Disorders of movement and system degenerations. In: Graham DI, Lantos PL, eds. Greenfield’s neuropathology. Vol II. New York: Oxford University Press, 1997:281–366.
- 33.Spillantini MG, Bird TD, Ghetti B. Frontotemporal dementia and parkinsonism linked to chromosome 17: a new group of tauopathies. Brain Pathol. 1998;8:387–402. doi: 10.1111/j.1750-3639.1998.tb00162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weller RO, Cumming WJK, Mahon M. Diseases of muscle. In: Graham DI, Lantos PL, eds. Greenfield’s neuropathology. Vol II. New York: Oxford University Press, 1997:489–581.
- 35.Maciver SK, Harrington CR. Two actin binding proteins, actin depolymerizing factor and cofilin, are associated with Hirano bodies. Neuroreport. 1995;6:1985–1988. doi: 10.1097/00001756-199510010-00008. [DOI] [PubMed] [Google Scholar]
- 36.Minamide LS, Striegl AM, Boyle JA, et al. Neurodegenerative stimuli induce persistent ADF/cofilin-actin rods that disrupt distal neurite function. Nat Cell Biol. 2000;2:628–636. doi: 10.1038/35023579. [DOI] [PubMed] [Google Scholar]
- 37.Nagaoka R, Kusano K, Abe H, Obinata T. Effects of cofilin on actin filamentous structures in cultured muscle cells. Intracellular regulation of cofilin action. J Cell Sci. 1995;108:581–593. doi: 10.1242/jcs.108.2.581. [DOI] [PubMed] [Google Scholar]
- 38.Ono S, Abe H, Obinata T. Stimulus-dependent disorganization of actin filaments induced by overexpression of cofilin in C2 myoblasts. Cell Struct Funct. 1996;21:491–499. doi: 10.1247/csf.21.491. [DOI] [PubMed] [Google Scholar]
- 39.Ono S, Baillie DL, Benian GM. UNC-60B, an ADF/cofilin family protein, is required for proper assembly of actin into myofibrils in Caenorhabditis elegans body wall muscle. J Cell Biol. 1999;145:491–502. doi: 10.1083/jcb.145.3.491. [DOI] [PMC free article] [PubMed] [Google Scholar]