

Abstract
A formal total synthesis of the antitubercular natural product was accomplished. This work was undertaken to address certain stereochemical problems in our initial synthesis. By using an ester group as a surrogate for a methyl group, we were able to intercept a key intermediate in our first synthesis with better selectivity and greater convergence than had previously been the case.
Introduction
The problem of tuberculosis has accompanied mankind for millennia.1 At present, approximately one third of the world’s population is infected with the causative organism, Mycobacterium tuberculosis. Of this group, 5–10% proceed to active disease. Over two million people a year die worldwide due to tuberculosis.
Though still a disease that can be treated, tuberculosis often requires chemotherapy on the order of months or years to achieve a cure. This long course often results in non-compliance on the part of patients and this has led to the emergence of moderately drug resistant (MDR) and extensively drug resistant (XDR) strains of the disease. The latter are resistant to the typical first line of drugs used for the treatment of TB as well as at least two drugs comprising the second line of defense
The persistence of this disease and in particular the rise of exceptionally resistant strains of the TB microorganism have made the discovery of new chemotherapies a priority. As is often the case, natural products can provide lead compounds for the development of new drugs, and a number of natural products with antitubercular properties have been identified.2
One such compound is pseudopteroxazole (1), which was isolated from the sea whip Pseudopterogorgia elisabethae by Rodriguez and co-workers.3 This compound displayed significant activity against M. tuberculosis H37Rv and thus is a potential lead for the development of antitubercular agents. Corey corrected the stereochemical assignment originally given to 1 and also produced the first total synthesis of the compound.4 We published a synthesis of 1 as well,5 further demonstrating the synthetic utility of enantiomerically pure benzothiazines in synthesis.
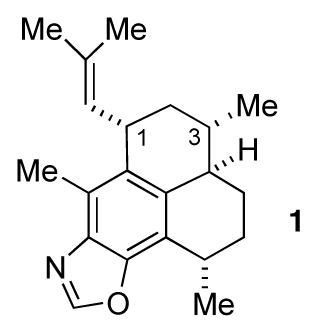
However, our first synthesis was not without some problems. A key step involved the intramolecular conjugate addition of the sulfoximine carbanion derived from 2 to afford 3 (Scheme 1). This proceeded in high yield and diastereoselectivity. Unfortunately, the diastereoselection was in a direction opposite to that desired and ultimately the stereochemistry at the methyl-bearing carbon atom, the one that would be carbon 3 of pseudopteroxazole, had to be corrected. Once this was done, the synthesis of diene 4 proved to be straightforward, as did the conversion of the latter to pseudopteroxazole. We thus set out to prepare 4 via a modified route, and this report details the results of our efforts.
SCHEME 1.

Summary of our first synthesis of pseudopteroxazole
Results and Discussion
Our plan was to bring in all of the elements needed to construct 4 at a very early stage of the synthesis and use the inherent diastereoselectivity of the benzothiazine formation to our advantage. First, we made phosphonate 8 from commercially available 3-methyl-2-butenal 5 and trimethyl phosphonoacetate 6 in 4 steps (Scheme 2).6 This reagent was then coupled with 2-bromo-3-methoxy-5-methylbenzaldehyde 97 to give 10 with high stereoselectivity using Ba(OH)2 as base (Scheme 3).8
Scheme 2.

Preparation of Phosphonate 8
Scheme 3.

Formal Synthesis of Pseudopteroxazole
A key step was the Buchwald-Hartwig coupling of triene 10 with sulfoximine (R)-11.9 We were pleased to find that the coupling product 12 could be obtained in 81% yield, with minimal complications due to Heck reactions, a side reaction that we feared might dash our hopes of using this strategy.10
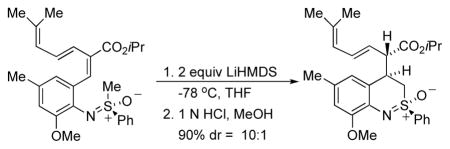
The next critical step was the intramolecular Michael addition.11 Treatment of 12 with LiHMDS followed by quenching with HCl in cold methanol afforded 13 with a diastereoselectivity that was as high as 10:1.12 The stereochemical outcome of the reaction could be rationalized on the basis of a model we presented earlier4 and, in fact, was the same stereochemical outcome observed for the formation of 3. However, the key difference is that we planned to convert the ester group in 13 to a methyl group en route to 4.
Thus, treatment of 13 with DIBAL afforded the corresponding alcohol 14 in 88% yield. Mesylation afforded the corresponding mesylate 15. Attempted reduction of this mesylate with various reducing agents did not proceed very successfully. After numerous attempts at reduction of this and related sulfonates, we ultimately used a procedure that presumably resulted in the in situ formation of the corresponding iodide with concomitant reduction. Thus, treatment of 15 with excess lithium iodide and excess lithium triethylborohydride at −30 °C for 26 h resulted in the formation of 4 in 79% yield, thereby completing a formal total synthesis of 1.
Conclusion
This approach to 4 required 6 steps from aldehyde 9 (conversion of 12 to 13 shows the workup as step 2). The old procedure (Scheme 1) required 7 steps from the same aldehyde, but suffered from the need to separate a significant amount of undesired stereoisomer en route to the synthesis of 4. Further, in this new approach, we have demonstrated that the palladium-catalyzed coupling of a sulfoximine to a highly unsaturated compound is possible in high yield, without loss of material due to other palladium-catalyzed processes. The reaction time for the coupling is significantly shorter than what has been typically reported and only a slight excess of 11 was needed to produce a very high yield of 12.
This work further establishes that benzothiazines are easily prepared templates that are useful for total synthesis. Further studies of their synthesis and applications will be reported in due course.
Experimental Section
(2E, 3E)-Isopropyl 2-(2-bromo-3-methoxy-5-methylbenzylidene)-6-methylhepta-3,5-dienoate (10)
To a solution of bromo aldehyde 9 (2.22 g, 9.7 mmol) and phosphonoacetate 8 (3.43 g, 11.8 mmol) in 120 mL THF and 6 mL of H2O, Ba(OH)2 (7.35 g, 43 mmol) was added in portions with vigorous stirring at 40 °C. After 10 min, the reaction was allowed to reach rt and was diluted with 200 mL CH2Cl2. It was washed with 1×100 mL saturated NaHCO3 and 1×100 mL brine. It was dried with MgSO4, filtered through Celite and concentrated in vacuo. After flash chromatography (1% TEA, 10% ethyl acetate in hexane), 3.2 g (84%) of the brom oester was obtained as a viscous oil. IR (neat): 2974, 2930, 1714, 1234, 1096 cm−1; 1H NMR (CDCl3, 300 MHz) δ 7.33(s, 1H), 7.16 (dd, 1H, J = 11.0, 15.6), 6.79 (s, 1H), 6.67 (s, 1H), 6.22 (d, 1H, J = 15.6 Hz), 5.82 (d, 1H, J = 11.0 Hz), 5.22 (septet, 1H, d = 6.0 Hz), 3.90 (s, 3H), 2.33 (s, 3H), 1.79 (s, 6H),1.38 (s, 3H), 1.36 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 167.0, 155.8, 138.3, 137.6, 137.4, 135.8, 132.2, 131.8, 126.2, 123.9, 122.2, 112.2, 110.1, 68.4, 56.2, 26.2, 21.8, 21.4, 18.6; HRMS calcd for C20H25O3BrNa [M+Na]+ 415.0879; Found: 415.0875.
Isopropyl (2E,3E)-2-(2-{[R-methylphenyl(oxido)-λ6-sulfanylidene]amino}benzylidene)-6-methylhepta-3,5-dienoate (12)
A 100 mL round bottom flask with condenser was charged with palladium acetate (15 mg, 0.065 mmol), rac-BINAP (60 mg, 0.1 mmol), in 35 mL toluene. The mixture was stirred for 15 min at rt. The bromoester 10 (510 mg, 0.5 mmol) and (R)-11 (220 mg, 0.77 mmol) in 5 mL toluene was added, followed by addition of Cs2CO3 (1.17 g, 2 mmol). It was refluxed at 110 °C for 12 h. Then it was diluted with 40 mL CH2Cl2, filtered through Celite, which was washed with 3×50 mL CH2Cl2, and concentrated in vacuo. After flash chromatography (25% ethyl acetate in hexanes), 491 mg (81%) of 12 was obtained as pale yellow semisolid. IR (film): 3064, 2974, 2925, 1703, 1560, 1454, 1336, 1270, 1233, 1094, 735 cm−1; 1H NMR (CDCl3, 500 MHz) δ 8.00 (dd, 2H, J = 1.5, 10.0 Hz), 7.77 (s, 1H), 7.56−7.50 (m, 3H), 7.20 (dd, 1H, J = 11.0, 15.5 Hz), 6.80 (s, 1H), 6.60 (s, 1H), 6.40 (d, 1H, J = 15.5 Hz), 5.87 (d, 1H, J = 11.0 Hz), 5.21 (m, 1H, J = 6.0 Hz), 3.59 (s, 3H), 3.10 (s, 3H), 2.28 (s, 3H), 1.81 (s, 6H), 1.36 (s, 3H), 1.34 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 167.6, 152.2, 142.4, 137.2, 136.9, 132.3, 132.0, 131.6, 130.7, 130.2, 129.8, 128.9, 127.5, 126.6, 123.4, 113.1, 67.8, 55.6, 46.0, 26.2, 22.0, 21.2, 18.6; HRMS calcd for C27H33NO4SNa [M+Na]+ 490.2022; Found: 490.2016; [α]25D = 77.975 (c 0.79, CHCl3).
Isopropyl (2S,3E)-6-methyl-2-[(2R. 4R)-2-methyl-2-oxido-3,4-dihydro-2 λ4,1-benzothiazin-4-yl]hepta-3,5-dienoate (13)
A 100 mL round-bottom flask was charged with bromo ester (1.58 g, 3.38 mmol) in 40 mL THF. LiHMDS 6 mL (1 M in toluene, 6 mmol) was added dropwise to it at −78 °C. After 10 min at −78 °C, the reaction was quenched with 1 N HCl in methanol at −78 −°C. It was poured into water, extracted with 3×20 mL CH2Cl2, dried with MgSO4, and concentrated in vacuo. After flash chromatography (30% ethyl acetate in hexanes), 1.28 g (81%) of 13 was obtained as the main isomer. IR (film): 2970, 2921, 2868, 1720, 1462, 1245, 1102 cm−1; 1H NMR (CDCl3, 500 MHz) δ 8.10–8.12 (m, 2H), 7.62–7.66 (m, 1H), 7.54–7.57 (m, 2H), 6.68 (s, 1H), 6.64 (s, 1H), 6.26 (dd, 1H, J = 11.0, 15.0 Hz), 5.77 (d, 1H, J = 11.0 Hz), 5.49 (dd, 1H, J = 7.5, 15.0 Hz), 4.89 (septet, 1H, J = 6.0 Hz), 3.96 (t, 1H, J = 7.0 Hz), 3.88 (s, 3H), 3.60–3.64 (m, 1H), 3.52–3.56 (m, 2H), 2.30 (s, 3H), 1.75 (s, 3H), 1.69 (s, 3H), 1.15 (d, 3H, J = 6.5 Hz), 1.05 (d, 3H, J = 6.0 Hz); 13C NMR (CDCl3, 125 MHz) δ 172.3, 152.6, 139.4, 136.8, 133.8, 132.1, 131.1, 129.9, 129.5, 129.4, 124.9, 124.9, 124.3, 119.4, 111.9, 68.4, 56.2, 51.1, 49.2, 38.4, 26.2, 21.8, 21.6, 18.6; HRMS calcd for C27H33NO4SNa [M+Na]+ 490.2022; Found: 490.2012; [α]25D = −60.48 (c 1.66, CHCl3).
(2 R,4R)-,4-[(1S,2E)-1,5-Dimethyl-2,4-hexadienyl]-3,4-dihydro-8-methoxy-6-methyl -2-phenyl-2γ4–2,1-benzothiazine-2-oxide (4)
To a solution of mesylate 15 (71 mg, 0.15 mmol) and LiI (201 mg, 1.5 mmol) in 7.5 mL dry THF at −30 °C, was added 1.5 mL of 1 M LiBHEt3 in THF slowly. After it was kept at −30 °C for 26 h, it was diluted with 15 mL DCM and quenched with 10 mL 10% NaOH, and 5 mL 30% H2O2. After it was stirred for 30 min at rt, it was washed with 10 mL saturated Na2S2O3 solution, followed by 30 mL brine. After drying with Na2SO4, it was concentrated under vacuum. Chromatography (20% ethyl acetate in hexanes) afforded 45 mg (79%) of 4 as a colorless oil. The NMR data matched the published data.4
Supplementary Material
Acknowledgments
This work was supported by the NIH (1R01-AI59000–01A1) to whom we are grateful.
SUPPORTING INFORMATION Characterization data for compound 16 and copies of proton and carbon spectra for previously unreported compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.See: Mitscher LA, Baker W. Med Res Rev. 1998;18:363–374. doi: 10.1002/(sici)1098-1128(199811)18:6<363::aid-med1>3.0.co;2-i.Friedman LN, editor. Tuberculosis: Current Concepts and Treatment. CRC; Boca Raton, FL: 2001. Kaufmann SHE, Rubin E, editors. Handbook of tuberculosis; molecular biology and biochemistry. Wiley-VCH; Weinheim: 2008. Kaufmann SHE, Britton WJ, editors. Handbook of tuberculosis; immunology and cell biology. Wiley-VCH; Weinheim: 2008. Kaufmann SHE, van Helden P, editors. Handbook of tuberculosis; clinics, diagnostics, therapy and epidemiology. Wiley-VCH; Weinheim: 2008.
- 2.Gutierrez-Lugo MT, Bewley CAJ. Med Chem. 2008;51:2606–2612. doi: 10.1021/jm070719i. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Copp BR, Pearce AN. Nat Prod Rep. 2007;24:278–297. doi: 10.1039/b513520f. [DOI] [PubMed] [Google Scholar]
- 3.Rodriguez AD, Ramirez C, Rodriguez II, Gonzalez E. Org Lett. 1999;1:527–530. doi: 10.1021/ol9907116. [DOI] [PubMed] [Google Scholar]
- 4.(a) Johnson TW, Corey EJ. J Am Chem Soc. 2001;123:4475–4479. doi: 10.1021/ja010221k. [DOI] [PubMed] [Google Scholar]; (b) Davidson JP, Corey EJ. J Am Chem Soc. 2003;125:13486–13489. doi: 10.1021/ja0378916. [DOI] [PubMed] [Google Scholar]
- 5.(a) Harmata M, Hong X. Org Lett. 2004;6:2201–2203. doi: 10.1021/ol049334+. [DOI] [PubMed] [Google Scholar]; (b) Harmata M, Hong X. Org Lett. 2005;7:3583–3583. doi: 10.1021/ol0515412. [DOI] [PubMed] [Google Scholar]
- 6.Minami T, Tokumasu S, Mimasu R, Hirao I. Chem Lett. 1985:1099–1102. [Google Scholar]
- 7.Koyama H, Kamikawa T. J Chem Soc Perkin Trans 1. 1998;1:203–209. [Google Scholar]
- 8.Nicolaou KC, Nold AL, Milburn RR, Schindler CS, Cole KP, Yamaguchi J. J Am Chem Soc. 2007;129:1760–1780. doi: 10.1021/ja068053p. [DOI] [PubMed] [Google Scholar]
- 9.(a) Harmata M, Pavri N. Angew Chem, Int Ed. 1999;38:2419–2422. doi: 10.1002/(sici)1521-3773(19990816)38:16<2419::aid-anie2419>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]; (b) Bolm C, Hildebrand JP. Tetrahedron Lett. 1998;39:5731–5734. [Google Scholar]; (c) Bolm C, Hildebrand JP. J Org Chem. 2000;65:169–175. doi: 10.1021/jo991342u. [DOI] [PubMed] [Google Scholar]
- 10.Only trace amounts of a compound assigned as an intramolecular Heck reaction product were isolated whenever this reaction was performed. This product 16 was characterized and details on it are reported in the supporting information. The enantiopurity of 12 is considered to be the same as that of (R)-11, which was established by both optical rotation and HPLC to be 100%, within experimental error.
- 11.Harmata M, Hong X. J Am Chem Soc. 2003;125:5754–5756. doi: 10.1021/ja034744z. [DOI] [PubMed] [Google Scholar]
- 12.Diastereomeric ratios were determined based on proton NMR analysis of crude reaction mixtures. The diastereoselectivity varied from 7–10:1. The minor isomer was not characterized but could be separated from the desired diastereomer by column chromatography.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.