

Synopsis
The major pathologic β-adrenergic (βAR) subtype in heart failure is the β1AR. Our laboratory has thus pursued genetic variation of the β1AR gene at the molecular, cellular, physiologic and clinical levels as the potential basis for interindividual variability in the response to β-blocker treatment in heart failure. This chapter will review these findings, with an emphasis on mechanism of action and future directions.
Keywords: adrenergic, adenylyl cyclase, myocardium, β-blocker, transgenic
β1AR coding polymorphisms
In the coding block of the intronless β1AR gene there are two common nonsynonymous single nucleotide polymorphisms (SNPs) at nucleotides 145 and 1165 [1,2]. While there are papers in the literature suggesting many other SNPs, a careful examination will show that the frequencies of these are often <0.01 (1%), have been found in only a few individuals, or have never been found in the homozygous state. These all indicate that such variants are either spurious or, quite rare and thus unlikely to impact public health. The location within the 7-transmembrane spanning β1AR of these two SNPs and the approximate allele frequencies are shown in Fig. 1. Concerning the amino acid position 389 polymorphism, it should be noted that Gly at position 389 was the originally cloned receptor and has often been denoted as the “wild-type.” However, Arg is more common in Caucasians, while Arg and Gly are about equally common in African-Americans [3]. So, it seems best not to designate “wild-type” vs. “polymorphism” but to simply state the allele, in the case of the β1AR, Ser49 or Gly49, and Arg389 or Gly389.
1.
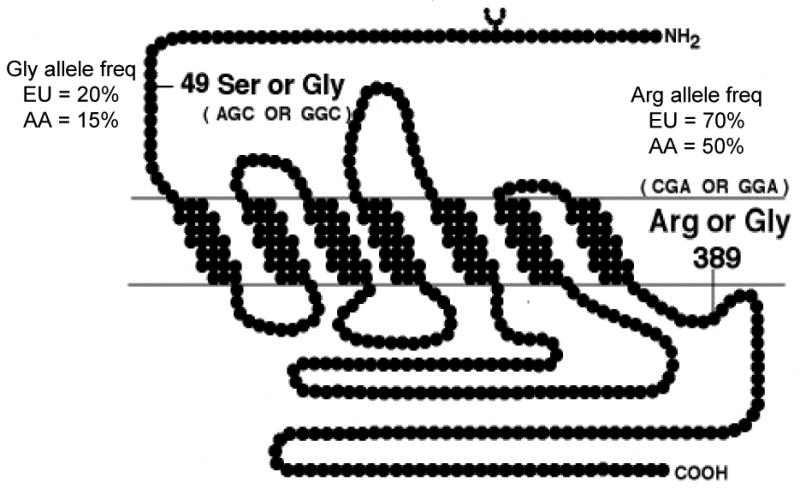
Localization of the common polymorphisms of the human β1AR. Shown is the predicted transmembrane topology of the β1AR, and the positions of the two common polymorphisms, with the triplet codons, nucleotide variations, amino acid variations, approximate allele frequencies in Americans from European (EU) or African (AA) descent.
Approach for studying β1AR polymorphisms
Fig. 2 indicates the approach that was taken for assessing the β1AR polymorphisms (and others that we have found in other genes [4-6]) in terms of signaling characteristics and clinical effect. As indicated, we initially concentrated on signal transduction in transfected cells so as to understand, in the most reductionist way, the potential differences between polymorphic receptors. In some cases this was followed by creation of myocyte-targeted transgenic mice which provide measurements of cellular and physiological signaling the cell-type of interest, under the conditions of the organ of interest. Small physiology-based clinical studies were also carried out, again building a wider base of knowledge about a given polymorphism within the context of the disease. When possible, we also utilize human tissue (in this case right ventricular trabeculae) from normal and failing hearts to further address phenotype under normal and diseased conditions. The pharmacogenetic clinical trials utilized all the results from these studies to formulate a hypothesis based on biologic plausibility. Finally, new drug or SNP targets can be derived from pathway analysis from ancillary studies, such as expression arrays performed on the human or transgenic mouse hearts [7].
2.
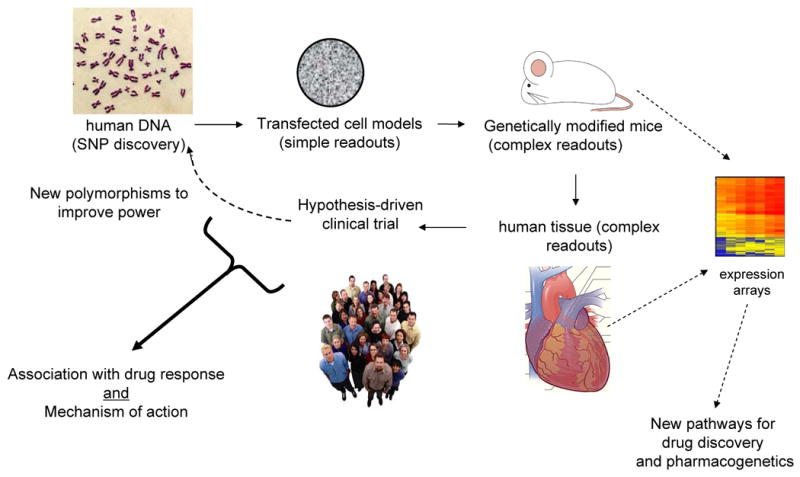
A progressive approach for polymorphism discovery, characterization, and clinical association. The dashed lines indicate ancillary studies derived from the primary study that may lead to additional pharmacogenetic loci or new drug targets.
The β1AR Arg389 and Gly389 signaling phenotype
The cDNAs for these two receptors were used to stably transfect Chinese hamster fibroblasts (CHW-1102 cells), and the major results that define the phenotype are shown in Fig. 3. Agonist completion binding studies showed no effects of the GTP analog GppNHp in Gly389 membranes (Fig. 3A) compared to Arg389 (Fig. 3B). In the former, regardless of the presence of GppNHp, Gly389 curves could be fit to a single site (Fig. 3B). In contrast, in the absence of GppNHp, Arg389 curves could be resolved into high-affinity and low-affinity binding sites, and in the presence of GppNHp only a low-affinity site [1]. The ratio of the low- to high-affinity sites, in essence, represents the potential change in Gibb's free energy that the agonist bound receptor conformation can convert, which represents the signal-transduction capacity. These studies suggested that Arg389 would have a greater agonist-promoted coupling to Gs/adenylyl cyclase compared to Gly389. As shown in Fig. 3C, this was the case. Isoproterenol-stimulated adenylyl cyclase activation was ∼3-fold greater for Arg as compared to Gly [1]. This was also the case for basal levels, consistent with the fact that receptors can “toggle” to the active state in the absence of agonist.
3.
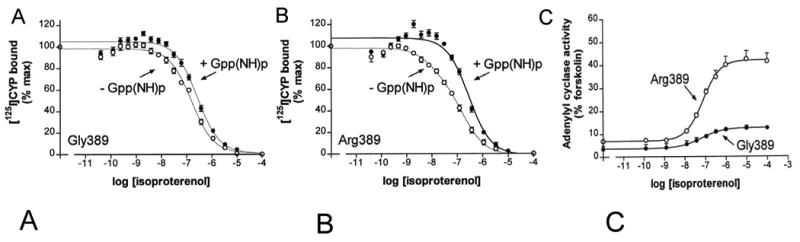
Pharmacologic properties of the β1AR variants at position 389 as assessed in transfected cells. A, B) agonist competition studies, C) agonist-stimulated adenylyl cyclase activities. Data are from ref [1].
Transgenic mice were made using the α-myosin heavy chain promoter, which targets the transgene (the human β1AR Arg389 or Gly389 cDNA) to the cardiomyocyte [8]. At equivalent expression levels, basal contractility (+dP/dt) was greater for 3-month-old Arg389 mice compared to Gly389 mice, as was maximal dobutamine stimulated contraction (Fig. 3A). Interestingly, at 6-months of age there was marked desensitization (essentially a total loss of responsiveness) of the Arg389 response, and ∼15% desensitization of the Gly389 response, while basal (no agonist) contractility remained greater for Arg vs. Gly389 (Fig. 3B). And, by 9-months of age, Arg389 mice had frank heart failure with depressed fractional shortening and increased mortality [8]. There are several interpretations of these results. One is that Arg389 is a risk factor for heart failure. We have shown though, that as a single variant, Arg389 and Gly389 allele frequencies in normal vs. heart failure patients are equivalent [9], indicating that alone this polymorphism does not represent a significant risk for human heart failure. Another interpretation is that there is a gradual change in the receptor phenotypes due to age, such that the Gly389 receptor becomes the hyperfunctional receptor near end-stage failure. An additional consideration is that the nature of the experiment (overexpression of both receptors) promotes long-standing enhanced contractility, eventually leading to enhanced desensitization of the Arg389 hearts, as well as heart failure, from such chronic stimulation. It turns out that there are elements of both of these latter two interpretations that were found in the human tissue (see below).
The first indication of a potential differential response to β-blockers based on the 389 genotype came from studies of 3-month old transgenic mice [8]. As shown in Fig. 4C, acute ex vivo administration of propranolol promoted a significant, and dose-dependent, decrease in +dP/dt in Arg389 hearts, but had minimal effect (and only at the highest concentration) for Gly389 hearts. In vivo, we examined heart rates by echocardiography in matched 3-month old mice treated with carrier or propranolol in the drinking water for 1-month. The Arg389 mice had a significant decrease in heart rate, while Gly389 mice did not (Fig. 4B).
4.
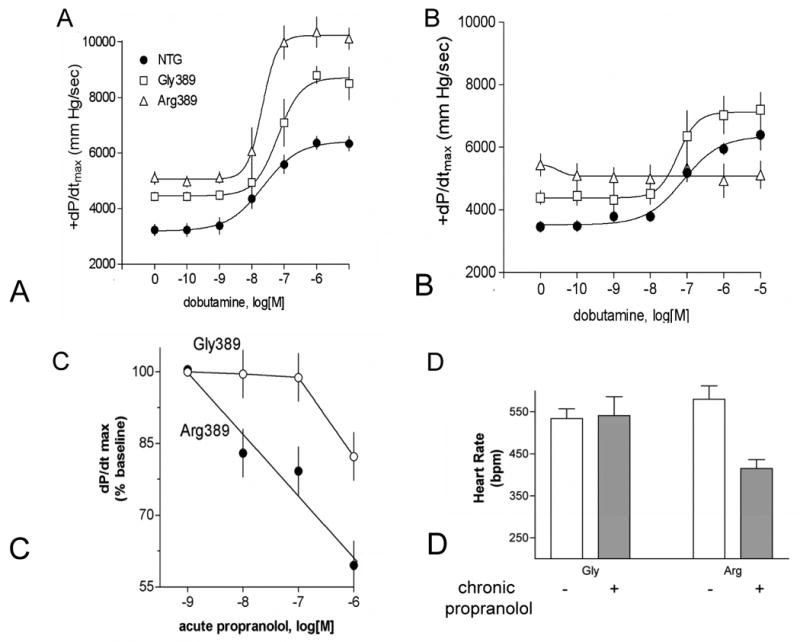
Physiologic characterization of the β1-Arg389 and -Gly389 receptors expressed in transgenic mice. A) contractile response to dobutamine in 3-month old mice, B) contractile response to dobutamine in 6-month old mice, C) response to acute ex vivo administration of propranolol, D) response to chronic administration of propranolol over a one-month time period in young mice. Data are from ref [8].
Ex vivo studies of non-failing and failing human right ventricular trabeculae [10] further defined the phenotype of Arg vs. Gly389. Fig. 5A shows that in normal hearts contractility in response to isoproterenol was greater for Arg389 homozygous hearts compared to Gly389 carrier hearts. These results were entirely consistent with the aforementioned studies of transfected cells and young transgenic mice. In the failing hearts (Fig. 5B) this same phenotype was seen, but note the scale difference. So, in comparison to the normal hearts, there was some “narrowing” of the phenotype, but in fact Arg389 hearts had greater contractility than Gly389 hearts even at end-stage heart failure. Additional studies were carried out with bucindolol (a β-blocker where DNA was available from a clinical trial) and carvedilol [10]. Carvedilol acted as a neutral antagonist, with no significant change noted in contraction with either genotype. In contrast, for the Arg389 trabeculae, bucindolol acted as an inverse agonist (i.e., decreased contractile force), while at Gly389 bucindolol was a neutral antagonist (Fig. 5C).
5.
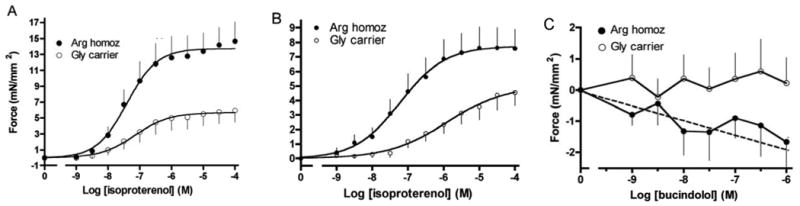
Contractile effects of β1-Arg389 and -Gly389 polymorphisms in isolated right ventricular trabeculae from normal end-stage failing human hearts. A) results from isoproterenol exposure to normal donor hearts, B) results from isoproterenol exposure to end-stage failing hearts obtained from cardiac transplant recipients, C) results from bucindolol exposure to failing hearts. Data are from ref [10].
The β1AR Ser49 Gly49 signaling phenotype
As indicated in Fig. 1, Gly is clearly the minor allele at position 49. This region of the receptor is in the extracellular amino terminus, and this region has not been subjected to extensive structure/function studies. Of note, there is a single N-linked glycosylation site on the human β1AR, at amino acid position 15. We expressed the Ser49 and Gly49 receptors in CHW-1102 cells and found no differences in basal or agonist-promoted adenylyl cyclase activities [2]. Binding affinities for isoproterenol, norepinephrine and metoprolol were not different between the two receptors. Another phenotype that we investigated was agonist-promoted downregulation, but we found no loss of β1AR expression after 24 hours of 10−5 M isoproterenol exposure in the culture medium. We then stably transfected HEK-293 cells. In these cells we actually found an upregulation of the Ser49 and a downregulation of Gly49. To eliminate new receptor synthesis, cells were treated with cycloheximide, and then vehicle or 10−5 M isoproterenol for 18 hours. Ser49 underwent ∼36% downregulation while Gly49 underwent ∼55% downregulation [2]. Short-term exposure (30 min) to agonist resulted in equivalent degrees of receptor internalization. Additional studies showed that there may be differences in glycosylation between Ser49 and Gly49. A high molecular weight band found with Ser49 solubilized membranes on SDS-PAGE was not observed with Gly49 membranes. The band was sensitive to in vivo exposure to the glycosylation inhibitor tunicamycin, and to N-glycosidase treatment in vitro. There was no effect of O-glycosidase treatment. It is unclear how N-glycosylation at position 15 could be affected by a polymorphism at position 49. Interestingly, we now know that β1ARs can form homodimers, so the high-molecular weight species could represent a minimally glycosylated dimer. Additional studies are required to understand the molecular basis of this phenotype. However, Levin et al. have also reported the enhanced agonist-promoted downregulation of the Gly49 receptor [11]. This group also reports, though, differences in basal and agonist-promoted stimulation of adenylyl cyclase, which we did not find. Nevertheless, a difference in agonist-promoted downregulation is a clinically relevant phenotype in heart failure, and thus there may be a role for the Gly49 variant in risk, progression, or therapeutic response to β-blockers in heart failure.
Association studies of β-blocker efficacy and β1AR polymorphisms
As discussed earlier, the most promising candidate polymorphism for distinguishing responses to β-blockers are the position 389 variants. The Arg389 receptor is “hyperfunctional,” which may be equated with a state that is more amenable to being antagonized by β-blockers. This appeared to be the case in the transgenic mice, where only Arg389 hearts responded acutely (decreasing contractility) or chronically (decreased heart rate) to propranolol. Thus a hypothesis that Arg389 was associated with improved outcomes in chronic heart failure treated with β-blockers was made. Of note, we also considered that in the absence of β-blockers, the SNP may have some effect on survival (as in the transgenic mice), so an ideal clinical trial would have a placebo arm so that the response could be stratified by genotype and treatment status, comparing Arg389 patients on placebo with those on β-blocker, and, Gly389 patients in the same manner. One study [12] that provided such groups was the Beta-blocker Extends Survival Trial (BEST). The DNA substudy of this trial consisted of archived DNA from 1040 patients who had been followed for up to ∼5 years: 525 on placebo and 515 on bucindolol. Recall that an additional genotype-dependent effect was noted with bucindolol (Fig. 5C) in the ex vivo failing heart studies, where this drug was an inverse agonist at Arg389 but not Gly389. Thus, particularly for bucindolol, the Arg389 genotype was hypothesized to be associated with a greater response compared to Gly389. We thus genotyped these samples at the 389 locus with the primary hypothesis being that compared to placebo, Arg389 homozygous patients would have an improvement in endpoints (survival, hospitalizations, or the combined endpoint) while Gly389 carrier patients would have shown no significant advantage of receiving bucindolol vs. placebo [10]. The subjects were well-matched in terms of ethnicity, etiology of heart failure, LVEF, blood pressure and other parameters (see Table 4 of Supporting Information from ref [10]). The salient results from these studies are shown in Fig. 6, which are Kaplan-Meyer curves of endpoints stratified by treatment and genotype. Cox-proportional hazards modeling was performed adjusting for age, sex and ethnicity [10]. The comparisons, adjusted for age, sex, and race, revealed that homozygous Arg bucindolol-treated patients had increased survival compared with Arg placebo-treated patients (HR = 0.62, 95% CI = 0.40–0.96, P = 0.03), an improvement of 38% in bucindolol over placebo. However, this same comparison in Gly carriers revealed no difference in survival curves (HR = 0.90, 95% CI = 0.62-1.30, P = 0.57), indicative of no statistically significant treatment response to bucindolol. There was also an apparent influence of β1AR genotype on the heart failure exacerbations during bucindolol treatment, as measured by time to first hospitalization due to heart failure: Arg389 HR = 0.64, 95% CI = 0.46–0.88, P = 0.006; Gly389 HR = 0.86, 95% CI = 0.64–1.15, P = 0.30. For the combined outcome of time to first heart failure hospitalization or death, a bucindolol associated favorable treatment effect was evident for Arg patients compared with placebo (HR = 0.66, 95% CI = 0.50–0.88, P = 0.004), but it was not apparent in bucindolol-treated Gly389 carriers vs. placebo (HR = 0.87, 95% CI = 0.67–1.11, P = 0.250).
6.
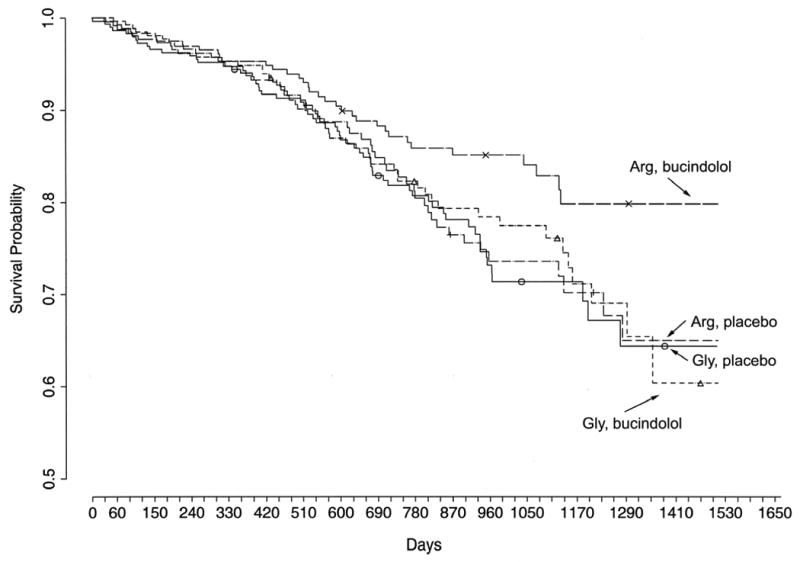
Kaplan-Meyer curves showing survival in patients stratified by genotype and drug-treatment in the BEST DNA substudy. The Arg389 patients receiving the active drug (bucindolol) had an improvement in survival compared to Arg389 patients receiving placebo. Both bucindolol and placebo treated Gly389 patients had equally poor outcomes. Data are from ref [10].
Taken together, these data point to the Arg389 locus as a predictor of the response to bucindolol [10]. Of note, in the original BEST, there was the suggestion that the drug may not have been as beneficial in African-Americans. Gly389 is more common in African-Americans compared to U.S. Caucasians (see Fig. 1); however, our findings with this polymorphism were not simply due to Gly being a marker of African ancestry. First, in the Cox-proportional hazards model, results were adjusted for race. Secondly, in an analysis of the white patients only, the beneficial effect of Arg389 in patients receiving bucindolol vs. placebo was observed, but was not found in Gly389 carriers [10], essentially mirroring what was found in the entire DNA substudy group.
We are aware of only one other double-blind placebo-controlled trial of a beta-blocker in heart failure where patient DNA was collected – the MERIT-HF DNA substudy [13]. However, the published information from this study showed a relatively high prevalence of AHA Class II patients compared to BEST, were followed for an average of one year, and had few events, so death or hospitalization was the primary endpoint. It is difficult to ascertain a pharmacogenetic effect from this report, though, because the metoprolol and placebo groups were combined. Recently Sehnert et al. [14] examined potential relationships with several polymorphisms including β1-389 in 637 patients in a retrospective study of subjects recruited from cardiac catheterization laboratories. The β-blockers were carvedilol or metoprolol, and there was not a placebo (or “no β-blocker”) group. The comparison was between outcomes in those with Arg389 or Gly389. No associations were found. This study is limited by its retrospective nature, the lack of uniform dosing, and a potential bias from recruiting patients who were receiving a cardiac catheterization. Nevertheless, the discrepancy between this study and ours with bucindolol, may be due to the pharmacologic differences between bucindolol, metoprolol and carvedilol. This brings to mind the question of whether the associations found in BEST are a class effect, or specific to bucindolol.
Future Directions
Many issues remain to be resolved regarding the β1AR polymorphisms and their potential influence on β-blocker response in heart failure. First, we now know that the promoter region of the β1AR gene is highly polymorphic, and that there are multiple haplotypes (specific combinations of polymorphisms) in the human population [15]. In whole-gene transfection studies, we have found that these alter β1AR expression and can be placed into at least three expression groups [15]. Thus these other polymorphisms, or the haplotypes, should be considered, as this approach may improve the predictive power. Secondly, it is well-recognized that there are other adrenergic receptors, and other non-receptor components, to the sympathetic signaling network of the heart. Studies with the α2CAR deletion polymorphism [4,9] and the G-protein coupled receptor kinase 5 polymorphism [6] highlight the necessity of considering multiple polymorphisms in multiple genes in the context of β-blocker pharmacogenomics. The statistical analysis of these potential complex interactions is not trivial, and may require some new approaches. And finally, the issue of specific drug vs. class effects needs to be resolved. We know that many of the β-blockers for heart failure treatment have unique (or different) properties. It is thus not entirely unexpected that they each may have a distinct group of polymorphisms that are most predictive of response. On the other hand, there may be some primary-effect polymorphisms that are useful for predicting response for all β-blockers.
Acknowledgments
This work was supported by Grants HL077101 and HL045967 from the National Institutes of Health
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mason DA, Moore JD, Green SA, et al. A gain-of-function polymorphism in a G-protein coupling domain of the human β1-adrenergic receptor. J Biol Chem. 1999;274(18):12670–74. doi: 10.1074/jbc.274.18.12670. [DOI] [PubMed] [Google Scholar]
- 2.Rathz DA, Brown KM, Kramer LA, et al. Amino acid 49 polymorphisms of the human Beta 1-adrenergic receptor affect agonist-promoted trafficking. J Cardiovasc Pharmacol. 2002;39:155–60. doi: 10.1097/00005344-200202000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Moore JD, Mason DA, Green SA, et al. Racial differences in the frequencies of cardiac β1-adrenergic receptor polymorphisms: analysis of c145A>G and c1165G>C. Hum Mutat. 1999;14(3):271. doi: 10.1002/(SICI)1098-1004(1999)14:3<271::AID-HUMU14>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 4.Small KM, Forbes SL, Rahman FF, et al. A four amino acid deletion polymorphism in the third intracellular loop of the human α2C-adrenergic receptor confers impaired coupling to multiple effectors. J Biol Chem. 2000;275(30):23059–64. doi: 10.1074/jbc.M000796200. [DOI] [PubMed] [Google Scholar]
- 5.Small KM, Mialet-Perez J, Seman CA, et al. Polymorphisms of the cardiac presynaptic α2Cadrenergic receptors: diverse intragenic variability with haplotype-specific functional effects. Proc Natl Acad Sci. 2004;101:13020–25. doi: 10.1073/pnas.0405074101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liggett SB, Cresci S, Kelly RJ, et al. A G-protein coupled receptor kinase-5 polymorphism that inhibits β-adrenergic receptor signaling is protective in heart failure. Nat Med. 2008;14(5):510–17. doi: 10.1038/nm1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Swift SM, Gaume BR, Small KM, et al. Differential coupling of Arg- and Gly389 polymorphic forms of the β1-adrenergic receptor leads to pathogenic cardiac gene regulatory programs. Physiol Genomics. 2008;35:123–31. doi: 10.1152/physiolgenomics.90225.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mialet-Perez J, Rathz DA, Petrashevskaya NN, et al. β1-adrenergic receptor polymorphisms confer differential function and predisposition to heart failure. Nat Med. 2003;9(10):1300–05. doi: 10.1038/nm930. [DOI] [PubMed] [Google Scholar]
- 9.Small KM, Wagoner LE, Levin AM, et al. Synergistic polymorphisms of β1- and α2C-adrenergic receptors and the risk of congestive heart failure. N Engl J Med. 2002;347:1135–42. doi: 10.1056/NEJMoa020803. [DOI] [PubMed] [Google Scholar]
- 10.Liggett SB, Mialet-Perez J, Thaneemit-Chen S, et al. A polymorphism within a conserved β1-adrenergic receptor motif alters cardiac function and β-blocker response in human heart failure. Proc Natl Acad Sci U S A. 2006;103(30):11288–93. doi: 10.1073/pnas.0509937103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Levin MC, Marullo S, Muntaner O, et al. The myocardium-protective Gly-49 variant of the beta 1-adrenergic receptor exhibits constitutive activity and increased desensitization and down-regulation. J Biol Chem. 2002;277(34):30429–35. doi: 10.1074/jbc.M200681200. [DOI] [PubMed] [Google Scholar]
- 12.BEST Trial Investigators. A trial of the beta-blocker bucindolol in patients with advanced chronic heart failure. N Engl J Med. 2001;344(22):1659–67. doi: 10.1056/NEJM200105313442202. [DOI] [PubMed] [Google Scholar]
- 13.White HL, De Boer RA, Maqbool A, et al. An evaluation of the β1-adrenergic receptor Arg389Gly polymorphism in individuals with heart failure: a MERIT-HF sub-study. Eur J Heart Failure. 2003;5:463–68. doi: 10.1016/s1388-9842(03)00044-8. [DOI] [PubMed] [Google Scholar]
- 14.Sehnert AJ, Daniels SE, Elashoff M, et al. Lack of association between adrenergic receptor genotypes and survival in heart failure patients treated with carvedilol and metoprolol. J Am Coll Cardiol. 2008;52(8):644–51. doi: 10.1016/j.jacc.2008.05.022. [DOI] [PubMed] [Google Scholar]
- 15.Small KM, Mialet-Perez J, Liggett SB. Genetic variation within the β1-adrenergic receptor gene results in haplotype-specific expression phenotypes. J Cardiovasc Pharmacol. 2008;51(1):106–10. doi: 10.1097/FJC.0b013e31815a958f. [DOI] [PubMed] [Google Scholar]