

Abstract
Glioblastomas (GBMs) are the most common and lethal primary brain tumor. Recent studies implicate an important role for a restricted population of neoplastic cells (glioma stem cells; GSCs) in glioma maintenance and recurrence. We now demonstrate that GSCs preferentially express the interleukin 6 (IL6) receptors interleukin 6 receptor alpha (IL6Rα) and glycoprotein 130 (gp130). Targeting IL6Rα or IL6 ligand expression in GSCs utilizing short hairpin RNAs (shRNAs) significantly reduces growth and neurosphere formation capacity while increasing apoptosis. Perturbation of IL6 signaling in GSCs attenuates signal transducers and activators of transcription 3 (STAT3) activation, and small molecule inhibitors of STAT3 potently induce GSC apoptosis. These data indicate that Stat3 is a downstream mediator of pro-survival IL6 signals in GSCs. Importantly, targeting IL6Rα or IL6 expression in GSCs increases the survival of mice bearing intracranial human glioma xenografts. IL6 is clinically significant as elevated IL6 ligand and receptor expression are associated with poor glioma patient survival. The potential utility of anti-IL6 therapies is demonstrated by decreased growth of subcutaneous human GSC derived xenografts treated with IL6 antibody. Together, our data indicate that IL6 signaling contributes to glioma malignancy through the promotion of GSC growth and survival, and that targeting IL6 may offer benefit for glioma patients.
Keywords: cancer stem cells, glioblastoma, interleukin 6, Stat3
INTRODUCTION
Glioblastoma (GBM, WHO grade IV astrocytoma) is the most common and aggressive primary brain tumor in adults [1]. Despite advances in cancer therapy, GBMs are incurable with an average survival of slightly more than one year past the initial diagnosis [1]. New GBM therapeutic strategies are desperately needed requiring insights into the biological and molecular mechanisms driving the tumor growth. GBMs are complex tumors that display cellular heterogeneity within the bulk tumor. Recent studies suggest that GBMs contain cellular subpopulations with potent tumorigenesis and some stem cell characteristics [2–9]. These glioma stem cells (GSCs) express neural stem cell markers (including the cell-surface antigen Prominin-1/CD133 and the transcription factor Olig2), self-renew as demonstrated by serial neurosphere formation, and differentiate into multiple nervous system lineages (neuronal, astrocytic, and oligodendroglial) [2–9]. GSCs drive tumor propagation in xenograft models, are highly angiogenic [6, 8], and are resistant to radio- and chemotherapies [5, 10]. These data strongly suggest GSCs are important for tumor maintenance and recurrence. Indeed, GSC markers may predict the survival of GBM patients [11, 12], strengthening the argument that GSC directed therapies may have important clinical applications.
Aberrant production and signaling of the circulated cytokine interleukin 6 (IL6) is tightly linked to tumor generation and poor disease outcome in many cancer types, including GBM [13–17]. GBM samples contain significantly higher levels of IL6 protein compared to those of control brains [15], and higher IL6 mRNA correlates with poor GBM patient survival [16]. Consistent with these data, loss of IL6 signaling prevents brain tumor development in a mouse model in which expression of the src oncogene is controlled by the promoter of the astrocyte marker glial fibrillary acidic protein (GFAP) [17]. Although IL6 may promote the growth of astrocytes [18], little is known about the specific biological mechanisms through which IL6 contributes to GBM initiation or progression. In other cancers, IL6 promotes chemoresistance, angiogenesis, and invasion [19–22], cellular behaviors which have all been linked to cancer stem cells. Breast cancer mammosphere survival and malignancy is promoted by IL6 [23], further suggesting a contribution of IL6 to cancer stem cell biology. Together, these data suggested that the role of IL6 signaling in GBM should be evaluated in the context of the GSC subpopulation.
The canonical IL6 signal transduction pathway is initiated by IL6 ligand binding to heteromeric plasma membrane receptor complexes formed from a specific IL6 binding receptor, IL6 receptor alpha (IL6Rα, gp80), and a common signal transducing receptor gp130 (glycoprotein 130). Upon receptor activation, intracellular signaling is propagated by Jak (Janus kinase) tyrosine kinase family members leading to the activation of transcription factors of the signal transducers and activators of transcription (STAT) family, particularly STAT3 [13]. STAT3 activation, as indicated by phosphorylation at tyrosine 705, is present in glioma patient samples and increases with tumor grade [17, 24, 25]. IL6 signals promote STAT3 activation in GBM cells in vitro, and targeting either STAT3 or IL6 decreases GBM cell survival [24, 26, 27]. Additional reports also link STAT3 to stem cell biology as STAT3 is required to maintain the propagation and pluripotency of normal embryonic stem cells and neural stem cells [14, 28, 29]. Together, these data led us to hypothesize that IL6 may activate STAT3 in GSCs to contribute to GBM progression. We have now examined the role of IL6 signaling in the specific context of cancer stem cells.
MATERIALS AND METHODS
Isolation of GSCs and Non-stem Glioma Cells and Cell Culture
Similar to our prior descriptions [4–8], matched cultures enriched or depleted for glioblastoma stem cells were isolated from the human glioblastoma xenografts (D456MG, D54MG) or fresh human surgical specimens either freshly derived (CCF1863) or immediately implanted in immunocompromised mice (T3359, T3691, T3832, T4105, T4121, and T4142), a method that has been described to preserve cancer stem cells in glioma models [30]. Patients provided informed consent under protocols approved by either the Cleveland Clinic Foundation or Duke University Institutional Review Boards. Briefly, viable tumors were disaggregated by Papain Dissociation System (Worthington Biochemical) and filtered by 70 μm cell strainer to remove tissue pieces according to the manufacturer’s instructions (Detailed Protocol: http://www.worthington-biochem.com/PDS/default.html). Cells were then cultured in stem cell culture medium supplemented as detailed below for at least four hours to recover surface antigens. Cells were then labeled with an allophycocyanin (APC)- or phycoerythrin (PE)-conjugated CD133 antibody (Miltenyi Biotec), and sorted by fluorescence-activated cell sorting (FACS). Alternatively, cells were separated microbead-conjugated CD133 antibodies and magnetic columns (Miltenyi Biotec). CD133 positive cells are enriched for glioma stem cells defined through functional assays of self renewal and tumor propagation whereas CD133 negative cells are depleted for non-stem glioma cells. GSCs were cultured in Neurobasal media supplemented with B27 without Vitamin A, L-glutamine, sodium pyruvate (Invitrogen), 10 ng/ml basic fibroblast growth factor (bFGF), and 10 ng/ml epidermal growth factor (EGF) (R&D Systems). Non-stem glioma cells were cultured for at least 12 hours in 10% serum containing DMEM to allow cell survival. After recovery, DMEM media was removed and the cells cultured in supplemented Neurobasal medium so experiments were performed in identical media. Non-stem glioma cells were cultured in Neurobasal media for at least 12 hours before experiments were performed. The cancer stem cell nature of the CD133 positive cells was confirmed by fluorescent in situ hybridization (Supplemental Table 1), serial neurosphere assays, and tumor formation assays, but cultures depleted of cancer stem cells did not self renew and or initiate tumors ([8] and data not shown)
Immunofluorescence Staining
Freshly frozen human glioma surgical biopsy samples were processed as previously described in accordance with a Duke University Medical Center Institutional Review Board approved protocol [5–8]. Slides were stained with polyclonal rabbit anti-IL6Rα (Abcam) with monoclonal mouse anti-CD133 (Miltenyi) or monoclonal mouse anti-gp130 with rabbit polyclonal anti-CD133 antibodies (Abcam). For sections of xenografts treated with IL6 antibody, slides were stained with polyclonal anti-Nestin (Abcam) or monoclonal PCAM (Abcam). Primary antibodies were incubated for 16 hours at 4°C followed by detection with Alexa 488 goat anti-mouse (Invitrogen) and Alexa 568 goat anti-rabbit (Invitrogen) secondary antibodies. Nuclei were stained with Hoechst 33342 (Invitrogen) and slides were mounted using Fluoromount (Calbiochem). Confocal z-stacks were taken by a 63× water immersion objective lens on a Leica SP5 confocal microscopre using sequential scans (blue, red, green).
Real Time PCR
Total RNA was prepared using the RNeasy kit (Qiagen), and reverse transcribed into cDNA using an iScript cDNA synthesis kit (BioRad). mRNA levels were measured using probes from SABiosciences with SYBR Green and a ABI-7900 system (Applied Biosystems).
Lentiviral Mediated shRNA Targeting
Lentiviral shRNA clones (Sigma Mission RNAi) targeting IL6Rα, IL6 and scramble control (SHC002) were purchased from Sigma (Supplemental Table 2). These vectors were co-transfected with the packaging vectors psPAX2 and pCI-VSVG (Addgene) into 293FT cells by lipofectamine 2000 (Invitrogen) to produce virus.
Small Molecule Inhibitors
Stattic (6-Nitrobenzo[b]thiophene-1,1-dioxide) and JSI-124 (Cucurbitacin I) were obtained from Calbiochem.
Cell Viability Assay
GSCs infected with lentivirus expressing the indicated shRNAs for 48 hours were plated in 96-well plates at 1000 cells per well. 24 hours after overnight recovery, plates were examined by the cell viability assay kit (Promega) at the indicated times. Results are reported from at least triplicate samples as the mean ± standard deviation.
Neurosphere Formation Assay
GSCs infected with lentivirus expressing the indicated shRNAs were plated in 24-well plates at 10 cells per well. After 7 days, the percentage of wells containing neurospheres was quantified and neurospheres imaged with an Olympus CK40 digital camera mounted to a light microscope.
Annexin V Staining
48 hrs after infection, GSCs were plated at 105 cells per well and allowed to recover for 24 hours prior to staining with an Annexin V kit (EMD Chemical) and FACS analysis. For small molecule inhibitor studies, GSCs were plated at 105 cells per well and allowed to recover for 24 hours prior to treatment with the indicated STAT3 inhibitors for 24 hours.
Caspase 3/7 Assay
GSCs infected with lentivirus expressing the indicated shRNAs for 48 hours were plated at 103 cells per well and assayed after 24 hours. For small molecule inhibitor studies, GSCs were plated at 103 cells per well and allowed to recover for 24 hours prior to treatment with the indicated STAT3 inhibitors for 24 hours. A caspase 3/7 assay kit (Promega) was used to determine caspase activity which is reported as the mean ± standard deviation.
Western Blotting and Antibodies
Western blots were performed as previously described [5–8]. Rabbit Polyclonal Antibodies for human IL6, IL6Rα (Abcam), STAT3, phospho-STAT3 (Cell Signaling), and actin (Millipore) were used according to the manufacturer’s instructions.
Intracranial Tumor Assays and IL6 Antibody Treatment
Intracranial or subcutaneous transplantation of GSCs into nude mice was performed as described [5–8] in accordance with a Duke University Institutional Animal Care and Use Committee approved protocol. Briefly, 48 hours after lentiviral infection, cells were counted and the indicated number of live cells implanted into the right frontal lobes of athymic nude mice. Mice were maintained until the development of neurological symptoms. Where indicated, animals were treated with 100 mg anti-IL6 antibody or phosphate buffered saline injected intraperotineally (IP) every two days until the termination of experiments when tumors were harvested, weighed, and examined.
TUNEL Assay
TUNEL apoptosis detection kit (Millipore) was utilized according to the manufacturer’s instructions.
Statistical Analysis
Results are reported as the mean ± standard deviation. Significance was tested by one-way ANOVA using GraphPad InStat 3.0 software (San Diego, CA) or MedCalc software (Belgium). For in vivo studies, Kaplan Meier curves and log-rank analysis were performed using MedCalc software.
RESULTS
GSCs Express IL6 Receptors and Ligand
To evaluate the potential contribution of IL6 signals to glioma biology in the context of the recently identified tumor subpopulations, we measured IL6 receptor expression in freshly isolated GSCs and non-stem glioma cells derived using our previously described methodology [5–8]. Enrichment or depletion of cancer stem cells was validated using functional assays, including propagation of tumors with characteristics of the parental sample and stem cell marker expression (Fig. 1A, Ref. 8, and data not shown). GSCs expressed elevated levels of IL6Rα and gp130 in comparison to non-stem glioma cells (Fig. 1A). Isolated GSCs cultured short term as neurospheres also showed co-expression of IL6Rα or gp130 with CD133 (Fig. 1B; Fig. S1A). We extended these studies to direct immunofluorescent staining of frozen sections of human glioma surgical biopsies that demonstrated co-localization of IL6 receptors (IL6Rα and gp130) and stem cell markers (including CD133) (Fig. 1C; Supplemental Fig. 1B). Consistent with these protein expression data, quantitative real-time PCR revealed that GSCs expressed higher IL6Rα, gp130, and Olig2 mRNA levels than matched non-stem glioma cells in four different glioblastoma samples and one primary human specimen (Fig. 2A–C; Supplemental Fig. 2). Although we detected IL6 in GSCs, IL6 mRNA levels were higher in non-stem glioma cells than matched GSCs in four out of five glioblastoma samples (Fig. 2D; Supplemental Fig. 2). Consistent with these data, secreted IL6 ligand levels were also higher in non-stem glioma cells as detected by an enzyme-linked immunosorbant assay (ELISA) (Supplemental Fig. 3). These data suggest the existence of both autocrine IL6 signaling in GSCs and paracrine signaling between non-stem glioma cells and GSCs. Taken together, these data demonstrated that the expression of IL6 receptors was elevated on GSCs in comparison to non-stem glioma cells.
Figure 1.
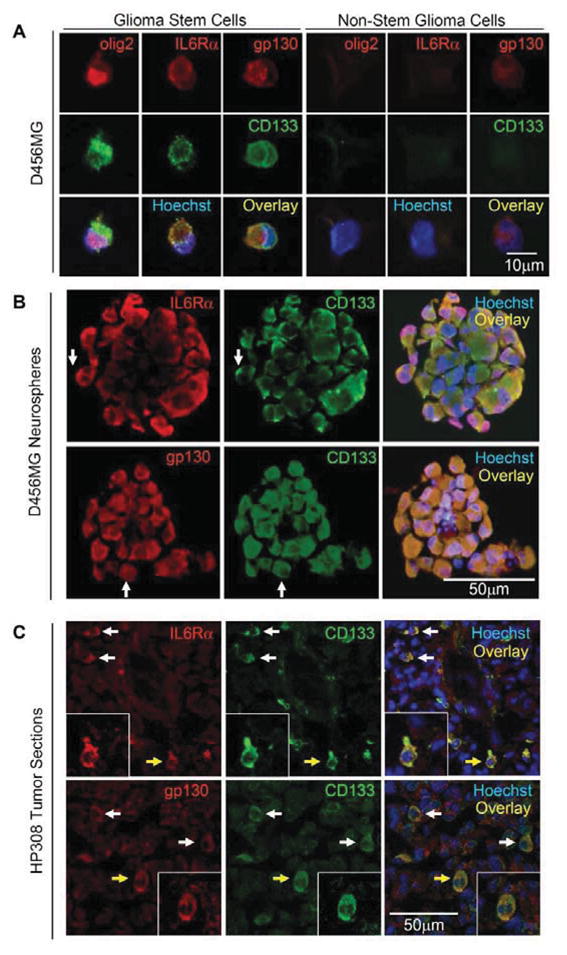
GSC markers co-expressed with IL6Rα and gp130. (A) The GSC marker CD133 co-localized with IL6Rα and gp130 in GSCs, but not non-stem glioma cells. (B) The GSC marker CD133 co-localized with IL6Rα and gp130 in neurospheres cultured in vitro from D456MG GSCs. Examples of areas with co-staining are highlighted with white arrows. (C) The GSC marker CD133 co-localized with IL6Rα and gp130 in the freshly frozen human glioma surgical biopsy specimen HP308 as demonstrated by immunofluorescent staining. Examples of cells with co-staining are highlighted with white arrows and the cell magnified in the inset is highlighted with a yellow arrow. Nuclei in all images were counterstained with Hoechst 33342.
Figure 2.

IL6 receptor and ligand mRNA levels indicated a potential paracrine loop between GSCs and non-stem glioma cells. Real-Time PCR was used to determine the relative mRNA levels of IL6Rα (A), gp130 (B), olig2 (C) and IL6 (D) in GSCs and non-stem glioma cells isolated from the long-term glioma xenografts D456MG and D54MG as well as from T3359 and T3832 patient specimens passaged short term in immunocompromised mice. The mRNA levels of IL6Rα (A) and gp130 (B) were generally higher in GSCs, whereas the mRNA level of IL6 was usually higher in non-stem glioma cells. (D) Olig2, a reported marker for GSCs, had consistently higher mRNA levels in isolated GSC populations. *, p<0.05 with comparison of non-stem glioma cells to matched GSCs.
Targeting IL6Rα in GSCs Decreases Growth and Survival
We assessed the functional significance of elevated IL6 receptors in GSCs by targeting IL6Rα using lentiviral transduced shRNA against IL6Rα (Sigma Mission RNAi). Two different sequences of shRNA directed against IL6Rα and a non-targeting shRNA were used for each experiment to control for potential off target shRNA effects (Supplemental Table 2). Both IL6Rα shRNA constructs led to a ~80% reduction in IL6Rα mRNA levels in GSCs in comparison to the non-targeting control (Fig. 3A). Loss of IL6Rα expression in GSCs significantly decreased cell growth over time associated with both decreased proliferation and increased cell death (Fig. 3B,C; Supplemental Fig. 4A,B). Targeting IL6Rα expression in GSCs decreased percentage of proliferating cells as demonstrated by a reduction in the number of cells in the S phase of the cell cycle as well as decreased thymidine incorporation (Supplemental Fig. 5). IL6Rα knockdown also increased apoptosis as demonstrated by elevated Annexin V positive cells (Fig. 3C) as well as increased caspase 3/7 activity (Fig. 3D; Supplemental Fig. 4B). Targeting IL6Rα expression also attenuated the ability to form neurospheres (Fig. 3C,D; Supplemental Fig. 4C,D) in cell culture. Of note, the neurospheres formed from the knockdown cells were smaller (potentially reflecting the decreased proliferation rate) and decreased in viability as shown by an inability to serially passage cells derived from neurospheres in the knockdown group (data not shown). As serial neurosphere formation is a key behavior of neural stem cells and GSCs that has been associated with self-renewal capacity [2–8], these data suggest that loss of IL6Rα impaired stem cell maintenance due in part to decreased cellular survival. Indeed, targeting IL6Rα, increased the expression of the differentiation markers S100β (astrocytes) and GalC (oligodendrocytes), demonstrating loss of IL6Rα signaling promoted differentiation (Supplemental Fig. 6A).
Figure 3.

Targeting IL6 receptor expression reduced GSC growth due to increased apoptosis. (A) Real-time PCR demonstrated IL6Rα expression in GSCs isolated from a D456MG human glioma xenograft was reduced via infection with lentivirus expressing two different shRNA constructs directed against IL6Rα (IL6Rα KD1 and IL6Rα KD2) when compared to lentivirus expressing a non-targeting control shRNA (NT). (B) Targeting IL6Rα via lentiviral shRNA significantly retarded the growth of D456MG GSCs as assessed with the Cell Titer Assay (Promega). Targeting IL6Rα decreases the survival of D456MG GSCs as demonstrated by Annexin V-FITC FACS analysis (C) and Caspase 3/7 activity (D). *, p < 0.01 with comparison to non-targeting shRNA. (E) Targeting IL6Rα expression attenuated the efficiency of D456MG GSCs to form neurospheres. The percentage of wells with neurospheres is indicated when infected cells were plated with ten cells per well in twenty four-well plates. (F) Representative images of neurospheres in E are shown.
Targeting IL6 Ligand in GSCs Decreases Growth and Survival
To determine if IL6 autocrine signaling in GSCs contributed to the phenotype exhibited with decreased IL6Rα expression, we utilized a similar lentiviral shRNA based targeting approach. Two different sequences of shRNA directed against IL6 were identified that reduced IL6 mRNA expression with an intermediate (IL6 KD1) and high efficiency (IL6 KD2) in GSCs (Supplemental Table 2; Fig. 4A). Targeting IL6 significantly inhibited GSC cell growth (Fig. 4B; Supplemental Fig. 4A) with a graded effect as IL6 KD2 reduced growth more rapidly and potently than IL6 KD1 (Fig. 4B), consistent with the relative knockdown efficiency. The reduced growth of IL6 knockdown cells was due to a reduction in the percentage of proliferating cells (Supplemental Fig. 5) and increased apoptosis (Fig. 4C,D; Supplemental Fig. 4B). Apoptosis, as demonstrated by elevated Annexin V positive cells (Fig. 4C) and increased caspase 3/7 activity (Fig. 4D), also reflected a relationship with knockdown efficiency. Targeting IL6 in GSCs significantly attenuated neurosphere formation capacity (Fig. 4E,F; Supplemental Fig. 4C,D) and the neurospheres that developed from the knockdown cells were smaller and could not be serially passaged (data not shown). These neurosphere formation data suggest that IL6 signals regulate stem cell maintenance, and we found that loss of IL6 increased the expression of differentiation markers (Supplemental Fig. 6B). Together with the similar results derived from IL6Rα targeting, these data support a pivotal role for autocrine IL6 signals in maintaining the survival of GSCs.
Figure 4.

Targeting IL6 ligand expression decreased GSC growth due to increased apoptosis. (A) Western blotting demonstrated IL6 expression in GSCs isolated from a D456MG human glioma xenograft was reduced via infection with lentivirus expressing IL6 shRNA constructs (IL6 KD1 and IL6 KD2) when compared to non-targeting control shRNA (NT). (B) Targeting IL6 via lentiviral shRNA significantly retarded the growth of D456MG GSCs as assessed with the Cell Titer Assay (Promega). (C,D) Targeting IL6 decreases the survival of D456MG GSCs as demonstrated by Annexin V-FITC FACS analysis (C) or Caspase 3/7 activity (D). *, p < 0.01 with comparison to non-targeting shRNA. (E) Targeting IL6 expression attenuated the efficiency of T3832 GSCs to form neurospheres. (F) Representative images of neurospheres in C are shown.
IL6 Signaling Promotes GSC Survival Through Stat3 Activation
As STAT3 is a downstream mediator of IL6 signaling and has important roles in embryonic and adult stem cells as well as glioma cell lines [24–29, 31], we explored STAT3 activation in GSCs with modulation of IL6 signaling. GSCs display an elevated level of basal phosphorylated STAT3 (Supplemental Fig. 7A) that was further induced upon the addition of exogenous IL6 (Supplemental Fig. 7B). Targeting IL6 signaling at the level of the receptor (IL6Rα) or ligand using shRNA inhibited levels of phosphorylated and total STAT3 (Fig. 5A). To further interrogate the role of STAT3 in mediating the effects of IL6 on GSC survival, we utilized small molecule inhibitors that decrease STAT3 activity by targeting STAT3 directly (Stattic) or Janus kinase (JSI-124) [27, 31]. Both STAT3 inhibitors reduced the activating phosphorylation of STAT3 in GSCs (Fig. 5B, Supplemental Fig. 8A). GSC cell proliferation and survival (Fig. 5C,D; Supplemental Fig. 8B–D) was dependent on STAT3 activity. STAT3 inhibitors reduced thymidine incorporation (Supplemental Fig. 8B) and induced apoptosis as measured by Annexin V staining (Fig. 5C; Supplemental Fig. 8C) and caspase 3/7 activity (Fig. 5D; Supplemental Fig. 8D). Taken together, our results support an essential role for IL6-mediated Stat3 activation in GSC growth and survival.
Figure 5.

STAT3 is a downstream mediator of IL6 in GSCs and mediates GSC survival. (A) STAT3 phosphorylation is decreased with IL6 or IL6Rα knockdown. 48 hours after T3691 and T3359 GSCs were infected with IL6 or IL6Rα shRNA expressing lentivirus, levels of phosphorylated STAT3 (at Tyr 705) and total Stat3 were reduced when examined by Western. Image J software was used to quantify the relative levels of phosphorylated STAT3 to total STAT3. (B) STAT3 inhibitors decreased the phosphorylation of of Stat3 in GSCs. T3691 and T3359 GSCs treated with vehicle control or 5 μM Stattic or JSI-124 for 4 hours have reduced Stat3 phosphorylation assessed via Western. (C) STAT3 inhibitors increase the percentage of Annexin V positive cells in GSCs. T3691 and T3359 GSCs treated with 5 μM Sttattic or 5 μM JSI-124 for 24 hours demonstrated an increased percentage of apoptotic cells as measured by FACS analysis with Annexin V-FITC and PI staining compared to DMSO control. (D) STAT3 inhibitors increase caspase 3/7 activity in GSCs. T3691 and T3359 GSCs treated with 5 μM Sttattic or 5 μM JSI-124 for 24 hours demonstrated increased activity in the caspase 3/7 assay (Promega) compared with DMSO control. *, p < 0.01 with ANOVA comparison to vehicle treated control.
IL6 Signaling Promotes Tumor Growth and Decreases Patient Survival
We next evaluated whether the critical effects of IL6 signals in vitro translate to in vivo survival difference by targeting IL6 receptor or ligand in intracranial tumor propagation. IL6Rα knockdown with two different shRNA constructs in GSCs prior to intracranial implantation into immunocompromised mice significantly increased survival compared to non-targeting control (Fig. 6A; Supplemental Fig. 9A). Similarly, targeting IL6 ligand expression in GSCs significantly increased survival of mice bearing human intracranial glioblastoma xenografts (Fig. 6B,C,D; Supplemental Fig. 9B). To determine if IL6Rα or IL6 expression could also impact glioma patient survival, we utilized the National Cancer Institute’s Repository for Molecular Brain Neoplasia Data (REMBRANDT) database. We found that upregulation of IL6Rα mRNA greater than two fold correlated with a significant decrease in survival (Fig. 6E). Similarly, upregulation of gp130 (the accessory receptor) was associated with decreased survival, although the number of patients expressing elevated gp130 was limited (Supplemental Fig. 10A). Consistent with a prior report linking IL6 to poor GBM prognosis [16], we also determined that glioma patients with an upregulation of IL6 mRNA greater than two fold have a decreased probability of survival compared to patients with reduced IL6 expression (Fig. 6F). When evaluating other IL6 family members which can also activate gp130, we found that leukemia inhibitory factor (LIF; Supplemental Fig. 10B) but not ciliary neurotrophic factor (CNTF; Supplemental Fig. 10C) expression was associated with poor patient survival, although there was no consistent elevation of LIF or its receptor in GSCs (Supplemental Fig. 11). These data demonstrate that IL6 signals promote the tumor initiating capacity of GSCs and strongly suggest that elevated IL6 signaling in GSCs contribute to poor patient outcome.
Figure 6.

Targeting either IL6Rα or IL6 suppressed tumor growth and increased the survival of mice bearing intracranial xenografts. (A) Kaplan-Meier curves demonstrate increased survival with knockdown of IL6Rα in T3359 GSCs. 5000 GSCs infected for 24 hours with lentivirus expressing non-targeting control shRNA (NT) or two different shRNAs directed against IL6Rα (IL6Rα KD1 and IL6Rα KD2) were injected into the right frontal lobes of immunocompromised mice. *, p < 0.001 with comparison to non-targeting control. (B) Kaplan-Meier curves demonstrated increased survival with knockdown of IL6 (IL6 KD1 and IL6 KD2) compared to non-targeting control shRNA (NT) in T3359 GSCs injected as in A. *, p < 0.01 with comparison to non-targeting control. (C) Kaplan-Meier curves demonstrated increased survival with knockdown of IL6 when 10000 D456MG GSCs were treated an injected as in B. *, p < 0.001 with comparison to non-targeting control. (D) Kaplan-Meier curves demonstrated increased survival with knockdown of IL6 in T3691 GSCs injected as in B. p < 0.001 with comparison to non-targeting control. (E) Clinical data from the National Cancer Institute’s Repository for Molecular Brain Neoplasia Data (REMBRANDT) database indicates that higher IL6Rα mRNA levels in gliomas correlate with poor patient survival. *, p = 0.0076 with comparison of survival probabilities for patients with up-regulated IL6R expression to those with down-regulated IL6Rα expression. (F) Clinical data from REMBRANDT indicates that higher IL6 mRNA levels in gliomas correlate with poor patient survival. *, p = 0.0342 with comparison of survival probabilities for patients with up-regulated IL6 expression to those with down-regulated IL6 expression.
IL6 Antibody Treatment Decreases the Growth of GSC Derived Tumors
As inhibition of IL6 signals could increase tumor latency in our animal models, we performed proof-of-principle studies targeting IL6 with a humanized antibody. Although large molecules like antibodies may have limited brain penetration due to restriction by the neurovascular unit, the recent clinical success of bevacizumab, a humanized neutralizing antibody against another ligand (vascular endothelial growth factor, VEGF), suggests that systemically administered antibodies may be useful as anti-glioma therapies. To evaluate the potential benefit of IL6 antibodies against gliomas in the absence of a brain-specific delivery restriction, we utilized a subcutaneous human glioma xenograft model and found that humanized IL6 antibody treatment reduced GSC tumor growth (Fig. 7; Supplemental Fig. 12). After GSC injection, treatment with IL6 antibody through intraperitoneal injection significantly reduced the volume of resulting tumors (Fig. 7A; Supplemental Fig. 12A). At the termination of experiments, the weight of tumors treated with IL6 antibody was significantly less than that of control (Fig. 7B; Fig. S12B). Histological analysis of the resulting xenografts demonstrated highly vascular and proliferative astrocytic tumors with pseudo-palisading necrosis characteristic of glioblastoma (Supplemental Fig. 13A). IL6 antibody treated tumors displayed a significantly lower percentage of proliferating cells (Fig. 7D,E) and a higher number of apoptotic cells than control tumors (Fig. 7F,G). The average number of cells positive for the stem cell marker Nestin was also decreased in IL6 antibody treated tumors (Supplemental Fig. 13B). In contrast, the intraperitoneal administration of IL6 antibody to mice bearing intracranial GSC tumors did not improve survival (data not shown) supporting a need of intraparenchymal delivery of the IL6 antibody for efficacy. These studies demonstrate that pharmacologic targeting of IL6 signaling has the capacity to reduce the growth of glioma xenografts and may be beneficial for glioblastoma patients.
Figure 7.

Systemic treatment with an anti-IL6 antibody inhibited the growth of human glioma xenografts in vivo. (A) Subcutaneous tumor volume was significantly decreased with IL6 antibody treatment. Animals subcutaneously injected with T3359 GSCs were intraperotineally injected 24 hours later with IL6 antibody at 100 mg every two days or PBS as a vehicle control. (B) Total tumor burden was reduced with IL6 antibody treatment as measured by tumor weight. *, p < 0.05 with comparison non-targeting control. (C) Images of xenografts measured in B. (D–E) Proliferation is decreased in IL6 antibody treated human glioma xenografts. Representative images of sections of control and IL6 antibody treated tumors stained with PCNA antibody (green) and Hoechst (blue) (D) were quantified based on 5 fields per tumor with six tumors for the control group and two tumors for the IL6 antibody treated group. *, p = 0.0488 with comparison to control. (F–G) Apoptosis is increased in IL6 antibody treated human glioma xenografts. Representative images of sections of control and IL6 antibody treated tumors stained with TUNEL (green) and Hoechst (blue) (D) were quantified based on 5 fields per tumor with three tumors per group. *, p = 0.036 with comparison to control.
DISCUSSION
Together, our data demonstrate an important role for IL6 signaling in GSCs. The IL6 receptors IL6Rα and gp130 were elevated in GSCs in comparison to non-stem glioma cells in sections of human patient specimens and isolated cell preparations. Targeting either IL6Rα or IL6 in GSCs significantly impaired their growth and survival in vitro, suggesting the importance of IL6 autocrine signals for GSC maintenance. IL6 signals were mediated through activation of STAT3, which was also critical for GSC survival. Targeting IL6Rα with shRNA or IL6 with shRNA or antibody increased tumor latency in mice bearing human glioma xenografts, suggesting that IL6 may be a novel cancer stem cell directed therapeutic target.
As IL6 may function as an autocrine and/or paracrine factor, we explored signaling in GSC maintenance in vitro and noted at least an autocrine role. However, cancer development is not a cell intrinsic process driven only by a collection of genetic errors in transformed cells. Tumor growth depends on the interactions between cancer cells and surrounding stroma cells, suggesting that paracrine effects of IL6 on GSCs may be critical in vivo. GSCs usually compose a small population (from 0.5% to 5%) of bulk tumors as demonstrated by immunohistochemical staining of GBM specimens and xenografts that demonstrates sporadic localization of GSCs surrounded by non-stem glioma cells [6]. The physical location (niche) of GSCs certainly suggests potential interactions with non-stem glioma cells. The finding that IL6 ligand (but not receptor) mRNA levels were higher in most non-stem glioma cells in comparison to matched GSCs supports the hypothesis that IL6 secreted by non-stem glioma cells may support GSC maintenance. If this paradigm of elevated ligand secretion from non-stem glioma cells with higher receptor expression on GSCs proves more broadly applicable, then non-stem glioma cells may prove to be a critical factor in the cancer stem cell niche.
The effects of IL6 activation in GBM have been largely undefined, but we now demonstrate a specific role for IL6 in GSC survival and tumorigenic capacity. As GSCs promote tumor maintenance through many biological mechanisms (invasion, angiogenesis, chemoresistance) that have also been found to be IL6 regulated [5, 6, 10, 15, 18–23], the potential for IL6 to control additional GSC-mediated behaviors exists. In particular, IL6 may regulate angiogenesis [15], and we previously determined GSCs are highly pro-angiogenic [6]. We also identified IL6 as one gene among a set of genes that are specifically unregulated in GSCs in comparison to non-stem glioma cells under hypoxia [8], a known “angiogenic switch” [32]. Hypoxia also induces IL6 expression in breast cancer cells grown as mammospheres, and IL-6 antibody treatment increases mammosphere cell death under hypoxic conditions [22]. Furthermore, IL6 increases VEGF transcription in GBM through STAT3 [33], demonstrating the potential involvement of both IL6 and STAT3 in a broad range of angiogenic behaviors. Together, these data suggest that IL6 may be additionally important for GSC survival under hypoxia and further contribute to GSC driven angiogenesis.
Clinical and laboratory evidence demonstrates that anti-IL6 directed therapies are well tolerated in patients, indicating their potential utility for anti-cancer treatments [34]. Humanized anti-IL6 and IL6Rα monoclonal antibodies have been evaluated in clinical trials and the use of IL6 conjugated toxins has also been proposed [34]. These data in combination with our results of IL6 antibody treatments of GBM xenografts, suggest that IL6 antibody may be useful against GBM. While treatment of GBMs is often complicated by the necessity of systemic treatments to cross the blood brain barrier, antibody based therapies (such as bevacizumab) have been administered intravenously and proven effective for GBM [35]. Similarly, the ability of IL6 antibody to bind and inactivate this growth factor in the bloodstream may prove efficacious for GBM patients.
We determined that a novel molecular pathway, IL6 signaling, is linked to GSC growth and survival. The dramatic benefit of IL6Rα and IL6 knockdown on the survival of mice bearing intracranial tumors and the effect of IL6 antibody against GBM xenografts, strongly suggest that targeting IL6 signals may be useful as a cancer stem cell directed therapy. Our studies provide evidence that inhibiting IL6 pathways should be considered for further exploitation in therapeutic development.
Supplementary Material
Acknowledgments
Financial Support: Financial support was provided by the Childhood Brain Tumor Foundation, the Pediatric Brain Tumor Foundation of the United States (J.R., X-F.W.), Accelerate Brain Cancer Cure (J.R.), Alexander and Margaret Stewart Trust, Brain Tumor Society (A.H., J.R.), Goldhirsh Foundation (J.R.), Sidney Kimmel Foundation, Damon Runyon Cancer Research Foundation (J.R.), Southeastern Brain Tumor Foundation (Y.C.), American Brain Tumor Association (J.W), NIH grants NS047409 (J.R.), NS054276 (J.R.), CA129958 (J.R.), CA116659 (J.R.), CA122998 (X-F.W.). The Duke University Brain Tumor Tissue Bank is supported by the Duke University Brain Cancer SPORE.
We thank Z. Su, Y. H. Sun, S. Keir, D. Satterfield, D. Kendall, L. Ehinger, and J. Funkhouser for technical assistance; M. Cook, B. Harvat, C. Shemo, and S. O’Bryant for assistance with flow cytometry. We appreciate the humanized IL6 antibody provided by C. Counter. We are also grateful to R. Wechsler-Reya and C. Counter for helpful discussions.
Footnotes
Publisher's Disclaimer: Disclaimers: The authors have no potential conflicts of interest.
Author Contributions: Hui Wang: conception and design, collection and/or assembly of data, data analysis and interpretation, manuscript writing
Justin D. Lathia: financial support, conception and design, collection and/or assembly of data
Qiulian Wu: collection and/or assembly of data
Jialiang Wang: financial support, collection and/or assembly of data
Zhizhong Li: conception and design, collection and/or assembly of data
John M. Heddleston: collection and/or assembly of data
Christine E. Eyler: collection and/or assembly of data
Jennifer Elderbroom: collection and/or assembly of data
Joseph Gallagher: collection and/or assembly of data
Jesse Schuschu: collection and/or assembly of data
Jennifer MacSwords: collection and/or assembly of data
Yiting Cao: financial support, collection and/or assembly of data
Roger E. McLendon: provision of study materials or patients, collection and/or assembly of data
Xiao-Fan Wang: conception and design
Anita B. Hjelmeland: financial support, conception and design, collection and/or assembly of data, data analysis and interpretation, manuscript writing
Jeremy N. Rich: financial support, conception and design, manuscript writing, final approval of manuscript
References
- 1.CBTRUS. Statistical Report: Primary Brain Tumors in the United States, 1998–2002. CBTRUS; 2005. Published by the . [Google Scholar]
- 2.Hemmati HD, Nakano I, Lazareff JA, et al. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci U S A. 2003;100:15178–15183. doi: 10.1073/pnas.2036535100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 4.Xie Z, Chin LS. Molecular and cell biology of brain tumor stem cells: lessons from neural progenitor/stem cells. Neurosurg Focus. 2008;24:E25. doi: 10.3171/FOC/2008/24/3-4/E24. [DOI] [PubMed] [Google Scholar]
- 5.Bao S, Wu Q, McLendon RE, et al. GSCs promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 6.Bao S, Wu Q, Sathornsumetee S, et al. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006;66:7843–7848. doi: 10.1158/0008-5472.CAN-06-1010. [DOI] [PubMed] [Google Scholar]
- 7.Bao S, Wu Q, Li Z, et al. Targeting cancer stem cells through L1CAM suppresses glioma growth. Cancer Res. 2008;68:6043–6048. doi: 10.1158/0008-5472.CAN-08-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li Z, Bao S, Wu Q, et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell. 2009;15:501–513. doi: 10.1016/j.ccr.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ligon KL, Huillard E, Mehta S, et al. Olig2-regulated lineage-restricted pathway controls replication competence in neural stem cells and malignant glioma. Neuron. 2007;53:503–517. doi: 10.1016/j.neuron.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eramo A, Ricci-Vitiani L, Zeuner A, et al. Chemotherapy resistance of glioblastoma stem cells. Cell Death Differ. 2006;13:1238–1241. doi: 10.1038/sj.cdd.4401872. [DOI] [PubMed] [Google Scholar]
- 11.Pallini R, Ricci-Vitiani L, Banna GL, et al. Cancer stem cell analysis and clinical outcome in patients with glioblastoma multiforme. Clin Cancer Res. 2008;14:8205–8212. doi: 10.1158/1078-0432.CCR-08-0644. [DOI] [PubMed] [Google Scholar]
- 12.Zeppernick F, Ahmadi R, Campos B, et al. Stem cell marker CD133 affects clinical outcome in glioma patients. Clin Cancer Res. 2008;14:123–9. doi: 10.1158/1078-0432.CCR-07-0932. [DOI] [PubMed] [Google Scholar]
- 13.Kishimoto T. Interleukin-6: from basic science to medicine--40 years in immunology. Annu Rev Immunol. 2005;23:1–21. doi: 10.1146/annurev.immunol.23.021704.115806. [DOI] [PubMed] [Google Scholar]
- 14.Hodge DR, Hurt EM, Farrar WL. The role of IL6 and STAT3 in inflammation and cancer. Eur J Cancer. 2005;41:2502–2012. doi: 10.1016/j.ejca.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 15.Choi C, Gillespie GY, Van Wagoner NJ, et al. Fas engagement increases expression of interleukin-6 in human glioma cells. J Neurooncol. 2002;56:13–19. doi: 10.1023/a:1014467626314. [DOI] [PubMed] [Google Scholar]
- 16.Tchirkov A, Khalil T, Chautard E, et al. Interleukin-6 gene amplification and shortened survival in glioblastoma patients. British J of Cancer. 2007;96:474–476. doi: 10.1038/sj.bjc.6603586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weissenberger J, Loeffler S, Kappeler A, et al. IL6 is required for glioma development in a mouse model. Oncogene. 2004;23:3308–3316. doi: 10.1038/sj.onc.1207455. [DOI] [PubMed] [Google Scholar]
- 18.Selmaj KW, Faroog M, Norton WT, et al. Proliferation of astrocytes in vitro in response to cytokines. J of Immunology. 1990;144:129–135. [PubMed] [Google Scholar]
- 19.Ishioka S, van Haaften-Day C, Sagae S, et al. Interleukin-6 (IL6) does not change the expression of Bcl-2 protein in the prevention of cisplatin-induced apoptosis in ovarian cancer cell lines. J Obstet Gynaecol Res. 1999;25:23–27. doi: 10.1111/j.1447-0756.1999.tb01117.x. [DOI] [PubMed] [Google Scholar]
- 20.Ancrile B, Lim KH, Counter CM. Oncogenic Ras-induced secretion of IL6 is required for tumorigenesis. Genes Dev. 2007;21:1714–1719. doi: 10.1101/gad.1549407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Conze D, Weiss L, Regen PS, et al. Autocrine production of interleukin 6 causes multidrug resistance in breast cancer cells. Cancer Res. 2001;61:8851–8858. [PubMed] [Google Scholar]
- 22.Sehgal PB, Tamm I. Interleukin-6 enhances motility of breast carcinoma cells. EXS. 1991;59:178–193. doi: 10.1007/978-3-0348-7494-6_12. [DOI] [PubMed] [Google Scholar]
- 23.Sansone P, Storci G, Tavolari S, et al. IL6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. J Clin Invest. 2007;117:3988–4002. doi: 10.1172/JCI32533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rahaman SO, Harbor PC, Chernova O, et al. Inhibition of constitutively active Stat3 suppresses proliferation and induces apoptosis in glioblastoma multiforme cells. Oncogene. 2002;21:8404–8013. doi: 10.1038/sj.onc.1206047. [DOI] [PubMed] [Google Scholar]
- 25.Wang H, Wang H, Zhang W, et al. Analysis of the activation status of Akt, NFkappaB, and Stat3 in human diffuse gliomas. Lab Invest. 2004;84:941–951. doi: 10.1038/labinvest.3700123. [DOI] [PubMed] [Google Scholar]
- 26.Konnikova L, Kotecki M, Kruger MM, et al. Knockdown of STAT3 expression by RNAi induces apoptosis in astrocytoma cells. BMC Cancer. 2003;3:23. doi: 10.1186/1471-2407-3-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Su Y, Zhang X, Gu J, et al. JSI-124 inhibits glioblastoma multiforme cell proliferation through G(2)/M cell cycle arrest and apoptosis augment. Cancer Biol Ther. 2008;7:1243–1249. doi: 10.4161/cbt.7.8.6263. [DOI] [PubMed] [Google Scholar]
- 28.Matsuda T, Nakamura T, Nakao K, et al. STAT3 activation is sufficient to maintain an undifferentiated state of mouse embryonic stem cells. EMBO J. 1999;18:4261–4269. doi: 10.1093/emboj/18.15.4261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Niwa H, Burdon T, Chambers I, et al. Self-renewal of pluripotent embryonic stem cells is mediated via activation of STAT3. Genes Dev. 1998;12:2048–2060. doi: 10.1101/gad.12.13.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shu Q, Wong KK, Su JM, et al. Direct orthotopic transplantation of fresh surgical specimen preserves CD133+ tumor cells in clinically relevant mouse models of medulloblastoma and glioma. Stem Cells. 2008;26:1414–1424. doi: 10.1634/stemcells.2007-1009. [DOI] [PubMed] [Google Scholar]
- 31.Schust J, Sperl B, Hollis A, et al. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol. 2006;13:1235–1242. doi: 10.1016/j.chembiol.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 32.Harris AL. Hypoxia--a key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2:38–47. doi: 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- 33.Loeffler S, Fayard B, Weis J, et al. Interleukin-6 induces transcriptional activation of vascular endothelial growth factor (VEGF) in astrocytes in vivo and regulates VEGF promoter activity in glioblastoma cells via direct interaction between STAT3 and Sp1. Int J Cancer. 2005;115:202–213. doi: 10.1002/ijc.20871. [DOI] [PubMed] [Google Scholar]
- 34.Trikha M, Corringham R, Klein B, et al. Targeted anti-interleukin-6 monoclonal antibody therapy for cancer: a review of the rationale and clinical evidence. Clin Cancer Res. 2003;9:4653–4665. [PMC free article] [PubMed] [Google Scholar]
- 35.Vredenburgh JJ, Desjardins A, Herndon JE, 2nd, et al. Bevacizumab plus irinotecan in recurrent glioblastoma multiforme. J Clin Oncol. 2007;25:4722–4729. doi: 10.1200/JCO.2007.12.2440. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.