

Abstract
Acid functionalized single walled carbon nanotubes were covalently grafted to chitosan by first reacting the oxidized carbon nanotubes with thionyl chloride to form acyl-chlorinated carbon nanotubes which are subsequently dispersed in chitosan and covalently grated to form composite material, CNT–chitosan, 1, which was washed several times to remove un-reacted materials. This composite has been characterized by FTIR, 13C NMR, TGA, SEM and TEM and has been shown to exhibit enhanced thermal stability. The reaction of 1, with poly lactic acid has also been accomplished to yield CNTchitosan–g-poly(LA), 2 and fully characterized by the above techniques. Results showed covalent attachment of chitosan and chitosan–poly lactic acid to the carbon nanotubes.
Keywords: Chitosan, Carbon nanotube, TEM, NMR, TGA
1. Introduction
Aqueous injectable, in situ gel-forming systems have received recent attention especially in tissue engineering because of the several advantages they offer when compared to scaffolds. First of all, the ease of introduction of in situ gel forming matrix through needle injection to defect sites, therefore removing the need for surgical implantation [1]. Second, the ease of conformity to the surrounding upon introduction, and ease of introduction of cells, therapeutic agents and growth factors by simple mixing, combined with not having to worry about residual solvents which are common in preformed scaffolds [2]. Chitosan, a N-deacetylated derivative of chitin, consists of 2-amino-2-deoxy (1-4)-β-D-glucopyranose residue (or D-glucosamine units) and is derived from the partial alkaline deacetylation of chitin. Chitins, chitosan’s precursor, are cellulosic type biopolymers which are widely distributed in nature, especially in the shell of crustaceans, the cell wall of fungi and exoskeleton of insects. The excellent biocompatibility, biodegradability, non toxicity and bioresorbability [3–6] have been widely used in many areas such as pharmaceutics, tissue engineering, and as food additive, and textiles [4,7–10].However, native chitosan is practically insoluble in organic solvents and is only soluble in aqueous acids, which therefore limits direct applications in tissue engineering. Although covalent attachment of functional groups for the modification of polymer properties (such as relative degree of hydrophobicity), and because of the polymer’s ability to form a semi rigid precipitate that can be processed into a variety of useful forms including gels, scaffolds, beads, fibers and films [11–13]. In spite of these favorable properties, the poor mechanical strength and the loss of structural integrity especially under wet conditions, limits chitosan’s application in bone tissue engineering. Carbon nanotubes possess high tensile strengths, and are ultra-light weight with excellent thermal and chemical stability. Many applications for carbon nanotubes have been proposed in the field of biomedical devices that include bio-sensors, drug and vaccine delivery [14,15]. Incorporation of super strong lightweight carbon nanotube structures into chitosan matrix offers a novel approach to the design of high performance composite materials with superior mechanical properties. However, the atomically smooth graphene surface of nanotubes can provide only limited load transfer from the matrix to nanotubes across the nanotube/polymer interface because of weak Van der Waals interfacial bonding through covalent grafting of carbon nanotubes to the chitosan to form the CNT–Chitosan nanocomposites.
The carbon nanotubes can provide improved mechanical strength and better structural integrity especially under physiological condition. Although multiwalled carbon nanotubes have been used as doping materials for three dimensional chitosan scaffolds to develop a highly conductive, porous composite materials, direct covalent attachment of carbon nanotube to the chitosan was not established [16] and in few cases, chitosan and carbon nanotubes were covalently grafted to composites using ultrasonic assisted acid oxidation of nanotubes followed by thionylation and dispersion in chitosan [17]. In this letter, we report a three-step methods to synthesize chitosan–carbon nanotubes composites with and without added polylactic acid as biocompatible materials especially for bone tissue engineering.
2. Experimental details
2.1. Materials
Low molecular weight chitosan, potassium persulfate, 85% lactic acid solution, sulfuric acid, nitric acid, and thionyl chloride were obtained from Sigma Aldrich. Bacterial protease was donated from Enzyme Development, Minneapolis, Minnesota. High purity single wall carbon nanotubes (SWCNTs) were purchased from Helix Materials Solutions.
2.2. Instruments and measurements
FTIR (IR-200 Thermo-Nicolet 2.2) (KBr) in the range of 400–4000 cm−1 was used to confirm the presence of functional groups in chitosan and its derivatives. Freeze-dried samples and KBr were used to prepare pellets. The thermal stability was analyzed in a Thermogravimetric analyzer (TGA) Q500 and analysis was performed using Universal V3.4C TA Instruments. The runs were performed in the temperature range of 30 °C to 800 °C and consisted of a ramp at a steady rate of 20 °C/min. The nitrogen flow rate was maintained at 60 mL/min. For gel permeation chromatography, the soluble chitosan derivatives were dissolved in 10% sodium acetate solution as the mobile phase. The mobile phase was pH 4 using ammonium hydroxide, 10% methanolic solution then, filtered using 0.2 μm nylon membrane syringe filter. Samples were injected into Viscotek GPCmax VE 2001 GPC Solvent/Sample Module before injection into the GPC. Resulting chromatograms were analyzed using Omni SEC 4.5.6 Administration software. Pullulan was used as the standard. The Scanning Electron Microscopy, SEM was performed using FEI Quanta 600 FE-SEM at 10 kV or using JEOL JSM at 10 kV. Specimens for high-resolution imaging were coated with 4 nm Pt/Pd layers using Cressington 208HR. Solid-state NMR experiments were performed with a Bruker Avance-400 spectrometer equipped with a standard 4 mm MAS probe head. The 13C NMR spectra were recorded at spinning rates of 11 kHz with a CP-pulse sequence. Chemical shifts were referred to TMS (external).
2.3. Preparation of degraded chitosan and grafted chitosan derivatives
2.3.1. Purification
Ten grams (10 g) of chitosan was dissolved in 200 mL of 1% acetic acid solution, then filtered, using glass wool to remove insoluble sample. Two hundred (200) mL of 2 M sodium hydroxide was added to the filtered sample with continuous stirring. After removal of excess NaOH solution, the filtrate was washed with distilled water until a pH 7.0 was reached.
2.3.2. Degradation
Initial studies were performed to determine the time needed for a desired molecular weight for chitosan derivatives. Ten grams (10 g) of purified chitosan was dissolved in 300 mL of 0.1 M sodium acetate/0.2 M acetic acid pH 5.4. Neutral bacterial protease (0.1 g) was added to the dissolved chitosan solution and was placed in a hot water bath at 60 °C at 24 h intervals. Aliquots were removed after 24, 48, 72, 96 and 120 h. Each aliquot was boiled for 10 min, allowed to cool at room temperature and the pH was adjusted to 9. Each aliquot was washed three times with deionized water before freeze drying. Molecular weights were determined using GPC as described above. The degree of deacetylation as determined using 1H NMR was 85%.
Degraded chitosan derivative at 48 h (0.5 g), hereafter called CH86K, was dissolved in 2% acetic acid solution and reacted with K2S2O8 (0.02 g) for 66 h under nitrogen purge. The reaction was stopped with 10 mL methanol. The pH of the reaction mixture was adjusted to 9 before washing three times with de-ionized water. The sample was freeze dried.
2.3.3. Preparation of grafted chitosan derivatives
CH86K (0.5 g) was reacted with K2S2O8 (0.02 g) for 20 h before adding 85% lactic acid solution (0.0555 mole) for an additional 2 h. Each reaction was quenched by adding 10 mL tetrahydrofuran before freeze-drying. The dried product was washed continuously with methanol to remove unreacted residue. The presence of lactic acid was confirmed using FTIR and solid state NMR.
2.4. Preparation of carbon nanotubes–chitosan composites
CNTs were oxidized with a 3:1 H2SO4:HNO3 for 24 h as to yield carboxylic acid functionalized SWCNTs, hereafter referred to as CNT–COOH. The carboxylic acid group was converted to formyl chloride via reaction with thionyl chloride for 24 h at 75 °C while refluxing, hereafter product is referred to as CNT–COCl. The CNT–COCl (400 mg) was reacted with CH86K (2 g) in 100 mL 2% acetic acid at 75 °C for 24 h while stirring. After reaction was stopped, sample was washed three times with 2% acetic acid to remove unreacted chitosan. The product, hereafter referred to as CNT–chitosan, 1 was dried as described before.
CNT–chitosan, 1 (0.1 g) was reacted with K2S2O8 (0.02 g) and 4 mL 85% lactic acid solution in 2% acetic acid solution at 75 °C for 2 h. Product was centrifuged at 20,000 rpm and washed twice with water before drying at 90 °C and 30 in Hg. The product hereafter will be referred to as CNT–chitosan–PLA. 2 Product was worked up as before.
3. Result and discussion
The CNT–Chitosan 1 and CNT–Chitosan–PLA, 2, were synthesized as summarized in Fig. 1.
Fig. 1.
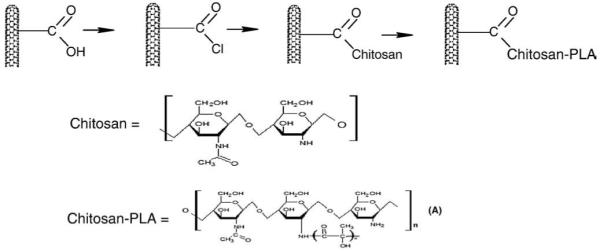
Summarizes the synthetic scheme for carbon nanotubes–chitosan and carbon nanotubes–chitosan–poly lactic acid composites.
3.1. FTIR
The FTIR peaks for pure show the broad peaks at 3450 cm−1 due to the stretching vibration –OH superimposed on –NH stretching band and broaden due to inter hydrogen bonds of polysaccharides. The 1656 cm−1 attributed to the presence of acetyl unit with –C=O stretching; 1579 cm−1 attributed to N–H bending, the 1376 cm−1 attributed to –CH3 symmetrical angular deformation; 1153 cm−1 attributed to β (1,4) glycosidic bonds; and 1091 cm−1 attributed to C–O–C stretching vibration. In the chitosan–PLA, the 3450 cm−1 peak becomes more broadened, due to extensive hydrogen bonding, and a slight low energy shift in the –C=O stretching to 1598 cm−1. The FTIR of SWCNTchitosan–g-poly(LA), 2, confirmed the presence of chitosan and lactic acid and specifically the presence of a strong peaks at 1401 cm−1 due to the presence of C=O of lactic acid. Additionally a new absorption peak at 1586 cm−1 indicates an overlap of the amide band and the amino group of the chitosan which is in agreement with the literature [18a–c]. Peaks due to β (1,4) glycosidic bonds and C–O–C stretching vibration were shifted to 1123 cm−1 and 1041 cm−1, respectively. In addition, the broad peak observed in Chitosan–PLA appears as two resolved peaks at 3450 cm−1 and 3120 cm−1, the latter peak was also observed in the CNT–COOH spectrum. Comparative FTIR of Chitosan, chitosan–PLA, CNT–COOH, CNT–Chitosan and CNT–Chitosan–PLA is shown in Fig. 2a.
Fig. 2.

a. Comparative FTIR of Chitosan, chitosan–PLA, CNT–COOH, CNT–Chitosan and CNT–Chitosan–PLA. 2. b. 13 C Solid State NMR of 1(A) and 2(B).
3.1.1. Solid state 13C NMR
Solid state 13C NMR was performed by Dr. Vladimir Bakhmoutov at NMR lab at Texas A&M University. In Fig. 2b, a comparison of solid state C-13 NMR of CNT–Chitosan, 1, and CNT–chitosan–g-PLA 2, is depicted.
CNT–chitosan–PLA, 2, and CNT–chitosan, 1 show a paramagnetic behavior and therefore relaxation times of 13C and 1H nuclei are very short. This behavior produces in the solid-state NMR spectra the intense sideband patterns. Due to the paramagnetic nature, the cross-polarization 1H-13C NMR experiments CNT–chitosan–PLA and CNT–chitosan were not successful. Their 13C MAS NMR spectra were recorded with a single pulse sequence at short relaxation delays of 0.5 s. Figure 2b. shows the 13C MAS NMR spectra. Comparison of the peaks for 1 and 2 showed distinctly the peaks associated with the PLA as 65 ppm, 20–22 ppm. Noticeable shift in the chitosan peak in 1 at 50 ppm shifted to 55 pm in 2 and a slight shift in the peak around 102–103 ppm (C-1 from chitosan).
3.1.2. Electron imaging analysis
The SEM of CNT–COCl (Fig. 3a) shows the presence of carbon nanotubes in micron length sizes, while Fig. 3b below shows that functionalized CNTs are bonded to chitosan, and that the surface texture of the CNT–Chitosan shows distinct agglomeration of CNTs, which appears to be sequestered within regions of chitosan, probably the more hydrophobic phase.
Fig. 3.

SEM results for (a) CNT–COCl, (b) CNT–g-chitosan.
The TEM (Fig. 4b) shows that coatings of chitosan on the surface of CNTs are quite visible when compared to TEM of pure CNTs (not shown). A clear indication that functionalized CNTs have attached to the surface of chitosan. TEMs for the CNT–g-chitosan–g-PLA showed the attachment of CNT to chitosan but the presence of PLA was not visible. It is believed that PLA formed a thin layer on the surface which was removed due to the sample drying in the microscope and the high pressure necessary for performing TEM.
Fig. 4.

TEM scans for (a) CNT–COCl, (b) CNT–g-chitosan.
3.1.3. TGA
The thermal behavior of CNT and CNT–chitosan derivatives is presented in Fig. 5. The as received carbon nanotubes, CNT, show less than 1% weight loss at 800 °C, and an indication of nanotubes purity and absence of little or no defect sites in the received SWCNT. The acid oxidation of carbon nano-tubes which introduces –COOH functionality created more defects. Thermogravimetric analysis showed a total weight loss of about 50% at 800 °C. It is most likely that acid oxidation using concentrated HNO3/H2SO4 mixtures also led to cutting of carbon nanotubes and creation of more defect sites, it is therefore not too surprising that the total weight loss of 50% at 800 °C is observed. The thermal analysis of pure chitosan shows two distinct weight losses, below 450 °C, a 20% weight loss observed can be attributed to amine side or N-acetyl side groups’ presence in chitosan. This degradation appears to be delayed in SWCNT–g-chitosan and in CNT–g-chitosan–g-PLA. An indication that the presence of carbon nanotubes in the chitosan enhanced the thermal stability in chitosan. The second weight loss occurred between 600 and 800 °C and may be attributed to oxidative removal of the glycosidic linkage.
Fig. 5.

TGA analysis.
4. Conclusion
Nanocomposites derived by covalent integration of functionalized carbon nanotubes and chitosan have been accomplished. The incorporation of CNTs in chitosan has been shown to improve the thermal properties of the latter. FTIR, SEM, TEM and solid state C-13 NMR confirm the bonding of the carbon nanotubes and the chitosan. Future work will focus on the bioactivity studies both in simulated body fluid to establish apatite forming ability especially if envision for application in bone tissue engineering.
Acknowledgement
Dr. Oki acknowledges the support from NIH-NIAMSD grant # ARO149172, the Welch Foundation grant to PVAMU Chemistry Department (L0002). Dr. Luo acknowledges support from the NSF grant DBI-0116835, for FE-SEM acquisition at TAMU, and the offices of the by the VP for Research, and the Texas Engineering Experiment Station. Dr. Laura Carson acknowledges support from USDA-CSREES grant # 2004-38814-15074 and the Cooperative Agriculture Research Center at Prairie View A&M University.
References
- [1].Shu XZ, Liu Y, Palumbo FS, Luo Y, Prestwich GD. Biomaterials. 2004;25:1339–48. doi: 10.1016/j.biomaterials.2003.08.014. [DOI] [PubMed] [Google Scholar]
- [2].Gutowska A, Jeong B, Jasionowski M. Anat Rec. 2001;263:342–9. doi: 10.1002/ar.1115. [DOI] [PubMed] [Google Scholar]
- [3].Noble L, Gray AI, Sadiq L, Uchegbu IF. Int J Pharm. 1999;192:173–82. doi: 10.1016/s0378-5173(99)00306-3. [DOI] [PubMed] [Google Scholar]
- [4].Majeti NV, Kumar R. React Funct Polym. 2000;46:1–27. [Google Scholar]
- [5].Boucard N, Viton C, Agay D, Mari E, Roger T, Chancerelle Y, et al. Biomaterials. 2007;28:3478–88. doi: 10.1016/j.biomaterials.2007.04.021. [DOI] [PubMed] [Google Scholar]
- [6].Chang YY, Chen SJ, Liang HC, Sung HW, Lin CC, Huang RN. Biomaterials. 2004;25:3603–11. doi: 10.1016/j.biomaterials.2003.10.039. [DOI] [PubMed] [Google Scholar]
- [7].Dutkiewicz JK. J Biomed Mater Res. 2002;63:373–81. doi: 10.1002/jbm.10231. [DOI] [PubMed] [Google Scholar]
- [8].Kurita K, Kaji Y, Mori T, Nishiyama Y. Carbohydrate. 2000;42:19–21. [Google Scholar]
- [9].Felix L, Hernandez J, Arguelles-Monal WM, Goycoolea FM. Biomacromolecules. 2005;6:2408–15. doi: 10.1021/bm0501297. [DOI] [PubMed] [Google Scholar]
- [10].Welsh ER, Schauser CL, Quadri SB, Price RR. Biomacromolecules. 2002;3:1370–4. doi: 10.1021/bm025625z. [DOI] [PubMed] [Google Scholar]
- [11].Krajewska B. Enzyme Microb Technol. 2004;35:126. [Google Scholar]
- [12].Roh IJ, Kwon ICJ. Biomater Sci. 2002;13:769. doi: 10.1163/156856202760197401. [DOI] [PubMed] [Google Scholar]
- [13].Siso MIG, Lang E, Carreno Gomez B, Becerra M, Espinar FO, Mendez JB. Proc Biochem. 1997;32:211. [Google Scholar]
- [14].Lin Y, Taylor S, Li H, Fernando SKA, Qu L, Wang W, et al. J Mater Chem. 2004;14(4):527–41. [Google Scholar]
- [15].Koerner H. Nat Mater. 2004;3(2):115. doi: 10.1038/nmat1059. [DOI] [PubMed] [Google Scholar]
- [16].Lau C, Cooney MJ, Atanssov P. Langmuir. 2008;24:7004–10. doi: 10.1021/la8005597. [DOI] [PubMed] [Google Scholar]
- [17].Wei F, Zigang W. Faming Zhuanli Shenging Gongkai Shuominhshu. 2006 patent written in Chinese Application: CN 1001-4158 20060608.
- [18].A Yao F, Chen W, Wang H, Liu H, Yao K, Sun P, et al. Polymer. 2003;44:6435–41. [Google Scholar]; B Ke G, Guan W, Tang C, Guan W, Zeng D, Deng F. Biomacromolecules. 2003;8(8):322–6. doi: 10.1021/bm0604146. [DOI] [PubMed] [Google Scholar]; C Wu Z, Feng W, Feng Y, Liu Q, Xu X, Sekino T, et al. Carbon. 2003;45:1212–8. [Google Scholar]