

Abstract
Background
Corynebacterium aurimucosum is a slightly yellowish, non-lipophilic, facultative anaerobic member of the genus Corynebacterium and predominantly isolated from human clinical specimens. Unusual black-pigmented variants of C. aurimucosum (originally named as C. nigricans) continue to be recovered from the female urogenital tract and they are associated with complications during pregnancy. C. aurimucosum ATCC 700975 (C. nigricans CN-1) was originally isolated from a vaginal swab of a 34-year-old woman who experienced a spontaneous abortion during month six of pregnancy. For a better understanding of the physiology and lifestyle of this potential urogenital pathogen, the complete genome sequence of C. aurimucosum ATCC 700975 was determined.
Results
Sequencing and assembly of the C. aurimucosum ATCC 700975 genome yielded a circular chromosome of 2,790,189 bp in size and the 29,037-bp plasmid pET44827. Specific gene sets associated with the central metabolism of C. aurimucosum apparently provide enhanced metabolic flexibility and adaptability in aerobic, anaerobic and low-pH environments, including gene clusters for the uptake and degradation of aromatic amines, L-histidine and L-tartrate as well as a gene region for the formation of selenocysteine and its incorporation into formate dehydrogenase. Plasmid pET44827 codes for a non-ribosomal peptide synthetase that plays the pivotal role in the synthesis of the characteristic black pigment of C. aurimucosum ATCC 700975.
Conclusions
The data obtained by the genome project suggest that C. aurimucosum could be both a resident of the human gut and possibly a pathogen in the female genital tract causing complications during pregnancy. Since hitherto all black-pigmented C. aurimucosum strains have been recovered from female genital source, biosynthesis of the pigment is apparently required for colonization by protecting the bacterial cells against the high hydrogen peroxide concentration in the vaginal environment. The location of the corresponding genes on plasmid pET44827 explains why black-pigmented (formerly C. nigricans) and non-pigmented C. aurimucosum strains were isolated from clinical specimens.
Background
In 2002, Corynebacterium aurimucosum was taxonomically described as new species within the genus Corynebacterium, with C. minutissimum as the nearest phylogenetic neighbour [1,2]. The phylogenetic and molecular taxonomic description was based on two isolates from human clinical specimens, one from an unknown source and one from blood cultures of a patient with bronchitis. C. aurimucosum is a non-lipophilic species, and growth is facultatively anaerobic. On Columbia blood agar colonies are generally slightly yellow in colour [1]. The isolation of C. aurimucosum from other human clinical sources has been reported, although very rarely. C. aurimucosum was detected in samples from patients with acute or chronic joint or bone infections [3,4], in infected diabetic foot wounds [5] and in a biopsy sample from a patient with rheumatoid arthritis [6]. Furthermore, C. aurimucosum 16S rDNA was detected in a bacterial population collected in the entrance area of a clean room environment in the Johnson Space Center [7,8] and in dust samples taken from office rooms in buildings located in central Finland [9].
Moreover, unusual charcoal-black-pigmented variants of C. aurimucosum were isolated in the U.S.A. and Canada from female urogenital sources, mostly from vaginal and cervical swabs [10-12]. In 2001, Shukla and co-workers reported the characterization of strain CN-1 (ATCC 700975) that was isolated from a vaginal swab of a 34-year-old female who experienced a spontaneous abortion during month six of pregnancy [10]. Subsequently, additional isolates with similar phenotypic and genotypic characteristics were recovered from the genital tract of women who had complications during pregnancy [12,13], suggesting that this newly recognized corynebacterium might be an opportunistic pathogen in pregnant women [13]. According to their black pigmentation, the name Corynebacterium nigricans was proposed for these isolates, and CN-1 was selected as type strain of this species [14]. However, morphological and biochemical analyses and sequencing of 16S rRNA genes in a polyphasic study of black-pigmented coryneform bacteria from the Centers for Disease Control and Prevention (CDC) group FCG4 suggested that C. nigricans is a later synonym for C. aurimucosum [12].
In this study, we present the complete genome sequence and bioinformatics analysis of C. aurimucosum ATCC 700975 (C. nigricans CN-1) to obtain insights into the physiology and lifestyle of this potential urogenital pathogen. Strain CN-1 was originally isolated from a female who presented with the sudden onset of premature labor during month six of gestation. The medical history of the patient was unremarkable, but the fetus died during vaginal delivery. A vaginal sample was taken at the time of delivery and revealed moderate growth of a black-pigmented corynebacterium in conjunction with other normal vaginal flora [10].
Results
Pyrosequencing and annotation of the C. aurimucosum ATCC 700975 genome
The complete genome sequence of the clinical isolate C. aurimucosum ATCC 700975 was determined by combining a whole-genome shotgun approach performed by pyrosequencing with a whole-genome scaffold generated by fosmid technology (Fig. 1A). A single run with the Genome Sequencer FLX System yielded 594,627 reads and a total number of 136,924,144 bases that were assembled into 73 large (≥ 500 bases) contigs. These assembled DNA sequences (2,736,233 bases) were combined with 215 Sanger reads performed on selected fosmid clones that covered the gaps between 70 chromosomal contigs. The resulting assembly of the DNA sequence data into a circular chromosome of 2,790,189 bp was consistent with the whole-genome scaffold deduced from 432 terminal insert sequences of randomly selected fosmid clones (Fig. 1B), corroborating the accuracy of the assembly process that was guided by the Consed program [15]. The gaps between the three remaining contigs (28,223 bases) were bridged by PCR, resulting in a circular contig of 29,037 bp in length. This assembled DNA sequence represents an endogenous plasmid of C. aurimucosum ATCC 700975, named pET44827 (Fig. 1C). The average gap size between the 70 chromosomal contigs was 1174 bp and 271 bp between the three plasmid contigs. Considering the final size of the C. aurimucosum ATCC 700975 genome, a 49-fold coverage was initially obtained by pyrosequencing. Relevant data of the C. aurimucosum ATCC 700975 genome project are summarized in Table 1.
Figure 1.

The complete genome of the clinical isolate C. aurimucosum ATCC 700975. (A), Plot of the C. aurimucosum ATCC 700975 chromosome. The circles represent from the outside: circle 1, DNA base position; circles 2 and 3, predicted protein-coding sequences transcribed clockwise and anticlockwise, respectively; circles 4 to 7, genes encoding orthologous proteins in C. diphtheriae NCTC 13129, C. jeikeium K411, C. urealyticum DSM7109, and C. kroppenstedtii DSM44385; circle 8, G+C content plotted using a 10-kb window; circle 9, G/C skew plotted using a 10-kb window. The predicted protein-coding sequences are coloured according to their functional classification into the Clusters of Orthologous Groups of proteins [93]. The genomic position of the putative prophages ϕCauri I and ϕCauri II is marked. (B), Fosmid scaffold of the C. aurimucosum ATCC 700975 chromosome. Individual fosmid clones used to build the genomic scaffold by terminal insert sequencing are represented as green segments. (C), Genetic map of plasmid pET44827 detected in C. aurimucosum ATCC 700975. The predicted protein-coding regions are shown by arrows indicating the direction of transcription. A direct repeat region is indicated as green box. Colour code: black, non-ribosomal peptide synthetase (NRPS) gene region; yellow, transposase genes and repetitive sequences; red, genes involved in plasmid replication and maintenance; blue, genes encoding hypothetical proteins.
Table 1.
Features of the C. aurimucosum ATCC 700975 genome
| Feature | Chromosome | Plasmid pET44827 |
|---|---|---|
| Total size (bp) | 2,790,189 | 29,037 |
| G+C content (%) | 60.6 | 53.3 |
| No. of protein-coding sequences | 2,531 | 20 |
| No. of pseudogenes | 4 | 8 |
| Coding density (%) | 88 | 64 |
| Average gene length (bp) | 973 | 928 |
| No. of rRNAs | 4 × 16S-23S-5S | 0 |
| No. of tRNAs | 55 | 0 |
| No. of transposase genes | 85 | 5 (3 pseudogenes) |
| No. of CRISPR 1 | 98 | 0 |
1Abbreviation: Clustered Regularly Interspaced Short Palindromic Repeats
The circular chromosome of C. aurimucosum ATCC 700975 has a size of 2,790,189 bp and a mean G+C content of 60.6% (Fig. 1A), which is slightly below the value of 63.7% that was previously determined by high-performance liquid chromatography [1]. Gene prediction and annotation of the genome sequence were performed with the GenDB system [16] and resulted in the detection of 2,531 protein-coding regions on the C. aurimucosum chromosome. On plasmid pET44827, twenty protein-coding sequences and eight pseudogenes were detected (Fig. 1C). Furthermore, 55 tRNA genes were found by the tRNAscan-SE program [17], and four rrn operons were identified on the leading strands of the C. aurimucosum chromosome, one on the right and three on the left replichore.
General architecture of the C. aurimucosum ATCC 700975 genome: moderate chromosomal reorganization in the main lineage of the genus Corynebacterium
The calculation of the G/C skew [(G-C)/(G+C)] [18] indicated a bi-directional replication mechanism of the C. aurimucosum ATCC 700975 chromosome (Fig. 1A). The origin of replication (oriC) was detected with Ori-Finder, a web-based tool for oriC prediction in bacterial genomes [19]. According to the presence and distribution of seven conserved DnaA boxes [TTATC(C/A)A(C/A)A], the oriC is located downstream of the dnaA (cauri_0001) coding region and has a computed length of 423 bp. The calculated G/C skew additionally indicated the presence of a putative dif region involved in replication termination [20] at the expected position of 180° from oriC, dividing the chromosome of C. aurimucosum ATCC 700975 in two replichores of similar size (Fig. 1A). For a more precise detection of a potential dif region, the distribution of the architecture imparting sequences G(A/T/C)GGGGGA and (T/C)GGGGGAG [21] was plotted on the leading and lagging strands of the C. aurimucosum ATCC 700975 chromosome (Fig. 2A). These octamers are present in 856 copies on the leading strands and in only 61 copies on the lagging strands of the C. aurimucosum ATCC 700975 chromosome. This characteristic distribution pattern of architecture imparting sequences revealed a putative dif region at around 1,425 kb of the chromosomal map (Fig. 2A). In accordance with this result, the respective DNA region of the C. aurimucosum ATCC 700975 chromosome contains a 28-bp sequence that showed striking similarity to the consensus sequence of actinobacterial dif sites [20] (Fig. 2B).
Figure 2.
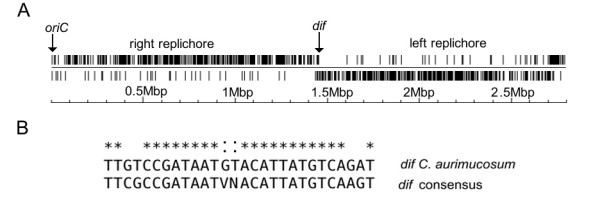
Distribution of architecture imparting sequences in the chromosome of C. aurimucosum ATCC 700975 and nucleotide sequence of the deduced dif site. (A), Distribution of the architecture imparting sequences G(A/T/C)GGGGGA and (T/C)GGGGGAG on the leading and lagging strands of the C. aurimucosum ATCC 700975 chromosome. The deduced position of the dif locus at around 1.425 Mbp is indicated in the linear representation of the chromosome. The position of the origin of replication (oriC) is marked. (B), Nucleotide sequence of the dif region in the C. aurimucosum ATCC 700975 chromosome. The 28-bp sequence is aligned with the consensus sequence of actinobacterial dif sites [20]. Identical nucleotides of the alignment are marked with asterisks. Matches to non-conserved nucleotides in the dif consensus sequence are specifically marked (:). Abbreviations: V = {A, C, G}; N = {A, C, G, T}.
Synteny analysis by reciprocal best matches with BLASTP [22] revealed a conserved order of orthologous genes between the C. aurimucosum ATCC 700975 chromosome and other pathogenic corynebacteria, in particular with C. diphtheriae NCTC 13129 displaying two breakpoints of synteny at 1,298 kb and 1,537 kb of the C. aurimucosum chromosomal map (Fig. 3). The comparison between the gene order of C. aurimucosum ATCC 700975 and C. jeikeium K411, C. urealyticum DSM7109 and C. kroppenstedtii DSM44385 revealed additional breakpoints in the chromosomal organization (Fig. 3), which is consistent with the taxonomic position of the latter three species that are part of independent branches in the phylogenetic tree of the genus Corynebacterium [2]. The two breakpoints of synteny between the chromosomes of C. aurimucosum ATCC 700975 and C. diphtheriae NCTC 13129 are indicative of a symmetric inversion of a 239-kb chromosomal DNA segment (carrying the genes cauri_1172 to cauri_1381) around the terminus of replication. The detected inversion in the C. aurimucosum ATCC 700975 chromosome provides the first example of a moderate genetic reorganization in genomes of members from the main lineage of the genus Corynebacterium, since all hitherto completely sequenced genomes (C. diphtheriae NCTC 13129, C. glutamicum ATCC 13032, C. efficiens YS-314) displayed an overall synteny of their gene order [23].
Figure 3.

Synteny between the chromosome of C. aurimucosum ATCC 700975 and those from C. diphtheriae NCTC 13129, C. jeikeium K411, C. urealyticum DSM7109, and C. kroppenstedtii DSM44385. The graphs represent X-Y plots of dots forming syntenic regions between the chromosomes of pathogenic corynebacteria. Each dot represents a predicted C. aurimucosum ATCC 700975 protein having an ortholog in another corynebacterial genome with co-ordinates corresponding to the position of the respective coding region in each genome. Orthologous proteins were detected by reciprocal best BLASTP matches. The genomic position of the putative prophages ϕCauriI and ϕCauriII is marked in the synteny plot with C. diphtheriae NCTC 13129.
Due to the symmetric inversion around the terminus of the C. aurimucosum ATCC 700975 chromosome, the preferential location of the respective genes on either the leading or lagging strands and their distance from the terminus region remained conserved after the chromosomal reorganization [24]. In general, 61% of the predicted C. aurimucosum ATCC 700975 genes are located on the leading strands of both replichores, while 39% are encoded on the lagging strands. It has been suggested that essentiality is the driving force for such type of gene-strand bias in bacterial genomes [25]. To detect candidate essential genes in the C. aurimucosum ATCC 700975 genome, the predicted gene repertoire was compared with that of C. glutamicum strain R [26], as genes shared by multiple genomes are more likely to be essential [27]. In addition, a high-density transposon mutagenesis approach in C. glutamicum R had already revealed 658 candidate essential genes in this strain, using complex medium and standard laboratory conditions for the selection of transposon mutants [28]. This number of candidate essential genes obviously represents an overestimate, at least due to polar effects caused by some transposon insertions [27]. The comparative genomics approach detected 352 coding regions of C. aurimucosum ATCC 700975 with high similarity to candidate essential genes from C. glutamicum R. Among this set of candidate essential genes, 78.8% are located on the leading strands of the C. aurimucosum ATCC 700975 chromosome, while 21.2% are encoded on the lagging strands, which is consistent with the aforementioned interpretation of gene-strand bias in bacterial genomes [25].
The annotation of the C. aurimucosum ATCC 700975 chromosome revealed the presence of two putative prophages. This observation is consistent with the lack of homology of the respective chromosomal region to the genomes of other pathogenic corynebacteria (Fig. 1A) and with a lack of synteny when compared with the genome sequence of C. diphtheriae NCTC 13129 (Fig. 3). The first prophage-like region (ϕCauriI) was detected adjacent to tRNALeu and tRNAGln genes of C. aurimucosum ATCC 700975 that may represent the integration site of ϕCauriI. This genome region has a size of about 20.7 kb and encodes 35 genes (cauri_1932 to cauri_1966). The second prophage-like region (ϕCauriII) is located directly adjacent to ϕCauriI and comprises 62 genes (cauri_1967 to cauri_2028) with a size of about 51.5 kb. Both prophage regions contain at their 3' ends genes encoding λ repressor-like transcription regulators (cauri_1965 and cauri_2027) and phage-related integrases (cauri_1966 and cauri_2028). Almost all genes of the prophage regions, with the exception of the regulatory genes, are encoded on the leading strand of the left replichore of the C. aurimucosum ATCC 700975 chromosome (Fig. 1A). The size difference between both prophage regions suggests that at least ϕCauriI is incomplete and a defective remnant of a formerly active corynephage.
Beside that, we detected with the CRISPRFinder program [29] an array of 98 clustered regularly interspaced short palindromic repeats (CRISPRs) and seven CRISPR-associated (cas) genes (cauri_0899 to cauri_0905) in the C. aurimucosum ATCC 700975 chromosome. The DNA repeats are 28 bp in length, separated by 33-bp spacers with variable nucleotide sequences and similar to repeat units of CRISPRs from C. jeikeium K411 [30], C. urealyticum DSM7109 [31] and Nocardia farcinica IFM 10152 [32]. CRISPR arrays and associated cas genes may provide acquired immunity against bacteriophages and other foreign genetic elements, with a specificity that is determined by sequence similarities between the spacers and foreign DNA sequences [33,34].
Specific features of the carbohydrate metabolism of C. aurimucosum ATCC 700975: catabolism of aromatic amines and utilization of L-tartrate
The metabolic analysis of the C. aurimucosum ATCC 700975 genome was performed with the computer program CARMEN that provided an automatic mapping of genome annotations on manually curated metabolic pathway maps [35]. These data were combined with results from a comparative genomics approach performed with the bioinformatics tool EDGAR, resulting in the detection of orthologous genes in different corynebacterial genomes and their classification as core genes or species-specific genes, so-called singletons [36]. A comparison of the gene content of pathogenic corynebacteria revealed a putative core genome consisting of 1,048 genes that are common in C. aurimucosum, C. diphtheriae, C. jeikeium, C. urealyticum, and C. kroppenstedtii. This value is very similar to data deduced previously from the comparison of gene contents in pathogenic and non-pathogenic corynebacteria [26,30]. The comparative genome analysis with EDGAR revealed also that C. aurimucosum ATCC 700975 contains 443 genes that are specific for this chromosome. The functional evaluation of the detected singletons provided characteristic features of the metabolism and lifestyle of C. aurimucosum ATCC 700975 that are described in more detail below. As the specific gene repertoire of a bacterium should in principle be shaped by the environmental conditions in the natural habitat and as we observed similarities to the gene repertoire of gut microbes, the annotation data point towards the human gut as a natural source for C. aurimucosum strains, which is consistent with the recent single report of a detection of this organism in a human fecal sample [37].
Analysis of the central carbohydrate metabolism of C. aurimucosum ATCC 700975 revealed the complete gene set for glycolysis and the pentose phosphate pathway (Fig. 4). The chromosome of C. aurimucosum ATCC 700975 provides a phosphoenolpyruvate carboxykinase (pck) and a fructose-1,6-bisphosphatase (pfkB) function for gluconeogenesis. The anaplerotic reaction of the tricarboxylic acid (TCA) cycle is mediated by pyruvate carboxylase (pyc). In contrast to other sequenced pathogenic corynebacteria that all lack the sucCD genes coding for succinyl-CoA synthetase, the TCA cycle of C. aurimucosum ATCC 700975 is complete (Fig. 4). It has been suggested that a putative succinyl-CoA:CoA transferase encoded by the cat1 gene can replace the missing succinyl-CoA synthetase activity in pathogenic corynebacteria [30,38]. Besides sucCD (cauri_2063 and cauri_2062), the C. aurimucosum ATCC 700975 chromosome also carries the respective cat1 gene (cauri_2148). On the other hand, the C. aurimucosum ATCC 700975 genome lacks the aceAB genes for the glyoxylate bypass within the TCA cycle. The lack of aceAB was previously also observed in the genome sequence of C. diphtheriae NCTC 13129 [38].
Figure 4.

Reconstruction of pathways involved in the central metabolism of C. aurimucosum ATCC 700975. The metabolic reconstruction of the carbohydrate metabolism was facilitated by using manually curated pathway maps in conjunction with the bioinformatics tool CARMEN. The resulting reconstruction was visualized with the CellDesigner software (version 3.2). Genes encoding transporters or enzymes involved in carbohydrate uptake and metabolism are shown in yellow boxes. Key metabolites are indicated by purple circles. Abbreviations for metabolites are as follows: α-Keto-G, α-ketoglutarate; β-Glcs, β-glycoside; 1,3PP-Gly, glycerate-1,3-bisphosphate; 2P-Gly, 2-phosphoglycerate; 3P-Gly, 3-phosphoglycerate; 5P-Ri-1PP, 5-phospho-ribose-1-diphosphate; 6P-β-Glcs, 6-phospho-β-glycoside; 6P-Gct, 6-phosphogluconate; 6P-Gll, 6-phosphogluconolactone; Aacetyl-CoA, acetoacetyl-CoA; Acetyl-P, acetyl-phosphate; CDP-ME, 4-(cytidine 5'-diphospho)-2-C-methyl-D-erythritol; CDP-ME2P, 2-phospho-4-(cytidine 5'-diphospho)-2-C-methyl-D-erythritol; CHM, 5-carboxymethyl-2-hydroxy-muconic acid; CHMS, 5-carboxymethyl-2-hydroxy-muconic semialdehyde; DHA, dihydroxyacetone; DHAP, dihydroxyacetone phosphate; DMAPP, dimethylallyl diphosphate; DXP, 1-deoxy-D-xylulose 5-phosphate; Ery-4P, erythrose-4-phosphate; FattyA-CoA, fatty acyl-CoA; Fru-1,6PP, fructose-1,6-bisphosphate; Fru-1P, fructose-1-phosphate; Fru-6P, fructose-6-phosphate; GA-3P, glyceraldehyde-3-phosphate; Gal-1P, galactose-1-phosphate; Glc-1P, glucose-1-phosphate; Glc-6P, glucose-6-phosphate; GlcNAc, N-acetylglucosamine; GlcNAc-6P, N-acetylglucosamine-6-phosphate; Glu-6P, glucosamine-6-phosphate; Glycerol-3P, glycerol-3-phosphate; GPDE, glycerophosphodiester; HHDD, 2-hydroxy-hept-2,4-diene-1,7-dioic acid; HHED, 2,4-dihydroxy-hept-2-ene-1,7-dioic acid; HMBDP, 1-hydroxy-2-methyl-2-butenyl 4-diphosphate; HPC, homoprotocatechuate; IPP, isopentenyl diphosphate; Man-1P, mannose-1-phosphate; Man-6P, mannose-6-phosphate; ManNAc-6P, N-acetyl-mannosamine-6-phosphate; MECDP, 2-C-methyl-D-erythritol 2,4-cyclodiphosphate; MEP, 2-C-methyl-D-erythritol 4-phosphate; OHED, 2-oxo-hept-3-ene-1,7-dioic acid; OPET, 5-oxo-pent-3-ene-1,2,5-tricarboxylic acid; PEP, phosphoenolpyruvate; Ribose-5P, ribose-5-phosphate; Sed-7P, sedoheptulose-7-phosphate; Ribu-5P, ribulose-5-phosphate; Suc-CoA, succinyl-CoA; Sucrose-6P, sucrose-6-phosphate; Sucsemiald, succinate semialdehyde; Tre-6P, trehalose-6-phosphate; UDP-Gal, UDP-galactose; UDP-Glc, UDP-glucose; Xyu-5P, xylulose-5-phosphate.
The functional analysis of singletons in the C. aurimucosum ATCC 700975 genome sequence resulted in the detection of a distinct gene cluster for the uptake and degradation of aromatic amines (cauri_2491 to cauri_2501) (Fig. 4; Fig. 5A). C. aurimucosum ATCC 700975 can apparently take up aromatic amines by a specific permease encoded by hpaX. The subsequent transformation into ammonia and the corresponding aromatic acid is catalyzed by amine oxidase (hpaY) and phenylacetaldehyde dehydrogenase (padA). The resulting metabolite is converted into pyruvate and succinic semialdehyde via the homoprotocatechuate (HPC) meta-cleavage degradation route (Fig. 5A). The hpa gene cluster encoding the respective enzymes of this pathway consists of seven genes (hpaGLEDHI and gabD2). The gabD2 gene is required for the enzymatic dehydrogenation of succinic semialdehyde to succinate that can enter the TCA cycle (Fig. 4). The hpaF function necessary to convert 5-carboxymethyl-2-hydroxy-muconic acid (CHM) into 5-oxo-pent-3-ene-1,2,5-tricarboxylic acid (OPET) is encoded elsewhere in the C. aurimucosum ATCC 700975 chromosome (cauri_0890). Two regulatory genes (hpaL and cauri_2492) are apparently involved in controlling the expression of the HPC meta-cleavage pathway and the uptake and conversion of aromatic amines. The hpa gene cluster of C. aurimucosum ATCC 700975 is similar to a previously reported cluster for the uptake and degradation of aromatic acids in E. coli [39] (Fig. 5A). Aromatic compounds may constitute a carbon source for gut microbes, as they are not only abundant in soil and water, but are also present in the gastrointestinal tract, where they mostly derive from plant constituents. As the HPC 2,3-dioxygenase (HpaD) requires molecular oxygen, utilization of the HPC meta-cleavage pathway is restricted to distinct intestinal habitats, for instance ones in close proximity to epithelial cells, where oxygen molecules might pass from the blood to the microbes attached to the epithelium [40]. Accordingly, C. aurimucosum ATCC 700975 is probably able to utilize aromatic amines as carbon and energy sources when living in an aerobic environment, such as the peripheral areas of the human gut system.
Figure 5.

Specific features of the C. aurimucosum ATCC 700975 metabolism detected by the analysis of singletons. (A), Genes and pathway for the catabolism of aromatic amines. Genetic maps of the hpa gene clusters encoding the HPC meta-cleavage pathway in C. aurimucosum and E. coli are presented. Genes with similar colour participate in the same enzymatic step of the pathway. The arrows show the direction of gene transcription. Abbreviations in the biochemistry of the pathway: DHPAL, 3,4-dihydroxyphenylacetaldehyde; HPC, homoprotocatechuate; CHMS, 5-carboxymethyl-2-hydroxy-muconic semialdehyde; CHM, 5-carboxymethyl-2-hydroxy-muconic acid; OPET, 5-oxo-pent-3-ene-1,2,5-tricarboxylic acid; HHDD, 2-hydroxy-hept-2,4-diene-1,7-dioic acid; OHED, 2-oxo-hept-3-ene-1,7-dioic acid; HHED, 2,4-dihydroxy-hept-2-ene-1,7-dioic acid. The enzymes are: HpaX, transport protein; HpaY, monoamine oxidase; PadA, phenylacetaldehyde dehydrogenase; HpaD, HPC 2,3-dioxygenase; HpaE, CHMS dehydrogenase; HpaF, CHM isomerase; HpaG, OPET decarboxylase; HpaH, hydratase; HpaI, HHED aldolase; GabD2, succinic semialdehyde dehydrogenase. (B), Genes and pathway for the catabolism of L-histidine. Genetics maps of the hut gene clusters coding for histidine catabolism in C. aurimucosum and Pseudomonas species. Genes with similar colour participate in the same enzymatic step of the pathway. The enzymes of the hut pathway are: HutH, histidine ammonia-lyase; HutU, urocanate hydratase; HutI, imidazolonepropionase; HutG, formimidoylglutamase. (C), Chromosomal gene region for selenol formate dehydrogenase and selenocysteine synthesis and incorporation in C. aurimucosum ATCC 700975. The UGA (opal) stop used for recoding the fdnG gene is indicated. The selC gene encodes the specific tRNASec.
Furthermore, the C. aurimucosum ATCC 700975 chromosome contains three singletons (cauri_2300 to cauri_2302) that are involved in the uptake and degradation of L-tartrate (Fig. 4). Uptake of L-tartrate is mediated by a tartrate/succinate antiporter encoded by yflS. The subsequent degradation to oxaloacetate is catalyzed by an (oxygen-labile) L-tartrate dehydratase (ttdAB). The respective gene cluster is homologous to previously described genes from E. coli [41,42]. Tartaric acid is found in many fruits and vegetables and is also a product of microbial metabolism in the gut ecosystem that enhances the ability of the normal gut microflora to reduce the intestinal colonization by Salmonella species and other enteropathogens [43]. L-tartrate is the most widely distributed isomer and can be degraded under aerobic and anaerobic conditions by a variety of bacteria [41]. However, E. coli can utilize L-tartrate only as reducible substrate for supporting anaerobic growth on the oxidizable co-substrate glycerol [41]. The C. aurimucosum ATCC 700975 chromosome also encodes a transporter for the uptake of glycerol (glpF) as well as glycerol-3-phosphate (glpT), gluconate (gntP) and lactate (lldT) transporters (Fig. 4). Furthermore, five genes coding for components of the bacterial phosphotransferase system (PTS) and two additional enzyme II genes (manP, nagE) encoding proteins of the fructose, glucose and glucoside (sub)families of PTS permeases [44] were identified in the chromosome of C. aurimucosum ATCC 700975 (Fig. 4). This gene repertoire, associated with the central metabolism of C. aurimucosum ATCC 700975, suggested that this bacterium is very flexible in the utilization of different carbon and energy sources, under both aerobic and anaerobic growth conditions.
Specific features of the amino acid metabolism of C. aurimucosum ATCC 700975: L-histidine catabolism and utilization of selenocysteine in formate dehydrogenase
According to the genome annotation, all known pathways for the biosynthesis of standard proteinogenic amino acids are present in the C. aurimucosum ATCC 700975 chromosome. The detection of singletons with EDGAR revealed that C. aurimucosum ATCC 700975 encodes a complete histidine utilization (hut) pathway for the conversion of histidine to glutamate (Fig. 5B). Four gene products are directly involved in histidine catabolism: The first reaction is catalyzed by histidine ammonia-lyase (hutH; cauri_1023), followed by the conversion of the resulting urocanate to 4-imidazolone-5-propanoate via urocanate hydratase (hutU; cauri_1025). The next catabolic step generates N-formimidoyl-L-glutamate by imidazolonepropionase (hutI; cauri_1026) that is finally hydrolyzed into L-glutamate and formamide by formimidoylglutamase (hutG; cauri_1019). Similar gene regions are known from saprophytic bacteria, such as Pseudomonas species, that take up and utilize L-histidine when growing in the plant environment [45,46] (Fig. 5B).
Regarding the proposal of C. aurimucosum ATCC 700975 as urogenital pathogen in pregnant women, it is interesting to note that the female histidine metabolism is affected during pregnancy, resulting in the excretion of large amounts of histidine in the urine [47,48]. Histidine excretion is largely dependent on the composition of the diet and is generally higher during the second half of gravidity [47]. Although little information regarding the utilization by bacteria of L-histidine from human sources is available, the human gut microbe Fusobacterium varium was shown to use L-histidine as carbon source for growth [49]. According to the presence of a complete hut pathway, C. aurimucosum ATCC 700975 is apparently able to utilize L-histidine as carbon and/or nitrogen source, which might be beneficial for bacterial growth when colonizing the urogenital tract of pregnant women.
Moreover, the C. aurimucosum ATCC 700975 chromosome contains the selABD genes and the tRNASec gene selC for the synthesis of the twenty-first proteinogenic amino acid selenocysteine (Sec) and its incorporation into selenoproteins (Fig. 5C). Selenocysteine is specifically incorporated into proteins by a cotranslational process that is directed by the UGA stop codon and termed recoding [50]. The first step during selenocysteine synthesis requires esterification of L-serine to the unique tRNASec by L-seryl-tRNA synthetase (serS, cauri_2392). The seryl-tRNASec is then converted to selenocysteyl-tRNASec by selenocysteine synthetase (selA, cauri_0967), using selenophosphate as selenium donor, which in turn is synthesized by selenophosphate synthetase (selD, cauri_0966). The anticodon of the specific tRNASec matches with an internal UGA stop codon in mRNAs from selenoprotein genes [51]. Furthermore, a specific elongation factor encoded by selB (cauri_0968) is required to recognize SECIS (selenocysteine insertion sequence) elements in the mRNA, such that the ribosome identifies the internal UGA stop codon as codon for selenocysteine. Translation of selenoprotein mRNAs then ends at the next in-frame stop codon to give a full-size selenoprotein [50,51].
Interestingly, the fdnG and fdxH genes (cauri_0962 and cauri_0964) encoding subunits of a formate dehydrogenase are arranged in a cluster with coding regions for the synthesis and incorporation of selenocysteine (Fig. 5C). This gene order is highly conserved in bacterial genomes that code for the incorporation of selenocysteine into proteins. Formate dehydrogenase is a heterogeneous group of enzymes that catalyze the decomposition of formate to carbon dioxide and contribute to mixed acid fermentation. Some formate dehydrogenase enzymes can contain an intrinsic selenocysteine residue [52]. The fdnG coding region of C. aurimucosum ATCC 700975 includes an internal UGA stop codon, suggesting that a formate dehydrogenase of the selenoenzyme type is encoded by this DNA region. The C. aurimucosum ATCC 700975 chromosome carries an additional gene for a standard (thiol) formate dehydrogenase (fdhD, cauri_0426) that is located adjacent to a putative formate transporter gene (cauri_0425). The utilization of the selenoenzyme form of formate dehydrogenase may enhance the adaptability of C. aurimucosum ATCC 700975 to changing environments, due to the strikingly different pH-dependence when compared with thiol forms, with selenols being active at much lower pH [53]. This biochemical feature of selenoenzymes is remarkable when considering the lifestyle of C. aurimucosum ATCC 700975, since acidophilic lactobacilli create a low-pH environment in the human vagina by producing lactic acid [54].
Cell surface structures of C. aurimucosum ATCC 700975: synthesis of a SpaH-type pilus and potential in biofilm formation
The C. aurimucosum ATCC 700975 genome codes for 438 proteins containing predicted signal peptides for either the general secretory (sec) or the twin-arginine translocation (tat) pathway for protein secretion. Three secreted proteins of C. aurimucosum ATCC 700975 showed homology to subunits of the adhesive SpaH pilus from C. diphtheriae NCTC 13129 [55]. Adhesive pili are covalently anchored to the corynebacterial cell wall by a transpeptidylation mechanism, requiring a C-terminal sorting signal with a conserved LPXTG motif [56], and can mediate the adherence to host tissues and other bacterial cells [57]. The chromosome of C. aurimucosum ATCC 700975 contains two genes, srtD and srtE (cauri_0189 and cauri_0190), encoding putative sortases involved in the assembly of a SpaH-like pilus. The housekeeping sortase of C. aurimucosum ATCC 770975 is encoded elsewhere in the chromosome (srtB, cauri_2420). The srtDE genes of C. aurimucosum ATCC 700975 are part of a DNA region also comprising the spaHIG genes (cauri_0187, cauri_0188, cauri_0191) that encode surface-anchored proteins with C-terminal sorting motifs (Fig. 6A). These proteins showed similarity to fimbrial subunits from C. diphtheriae NCTC 13129 involved in the sortase-mediated formation of the SpaH pilus [55]. The SpaH protein represents the major pilin of this pilus type and, along with the minor pilin SpaI, forms the pilus shaft, whereas the SpaG protein is located at the tip of the pilus. According to the detected homology between the corynebacterial SpaHIG proteins, it is likely that C. aurimucosum ATCC 700975 expresses an adhesive pilus with structural similarity to the SpaH pilus of C. diphtheriae NCTC 13129 (Fig. 6B). This pilus structure can mediate the initial adhesion of C. aurimucosum ATCC 700975 to host tissues particularly during vaginal infection and/or colonization [58].
Figure 6.

The putative SpaH-like pilus of C. aurimucosum ATCC 700975. (A), Genes clusters involved in the synthesis of adhesive pili in C. aurimucosum ATCC 700975 and C. diphtheriae NCTC 13129. Colour code: green, sortases required for the assembly of the pilus; red, major pilin; dark blue, minor pilin; light blue, tip protein; grey, protein of unknown function. Specifically marked are: pilin boxes (WPK), E-boxes (Ebox), sorting signals (LPXTG), hydrophobic domains in major pilins (hatched boxes), and charged tails in minor pilins (hatched boxes). (B), Model representation of the corynebacterial SpaH-like pilus. The pilus is covalently anchored to the corynebacterial cell wall [55]. Sorting motifs (S) and pilin motifs (P) used for the pilus assembly, as well as E-boxes (E) and sorting signals used for anchoring of the minor pilins (S') are marked.
Further analysis of the genome sequence revealed that C. aurimucosum ATCC 700975 codes for a putative accumulation-associated protein (aap, cauri_1006). Bioinformatics analysis displayed five G5 domains within the amino acid sequence of Aap that can mediate the formation of biofilms by recognizing and binding to N-acetylglucosamines present in the peptidoglycan of the bacterial cell wall [59]. Moreover, the C-terminal segment of Aap contains a sorting motif (LANTG) for anchoring of the protein to the corynebacterial cell wall. The secretion mechanism of Aap remains unknown, as the protein is devoid of a typical signal peptide. The Aap protein of C. aurimucosum ATCC 700975 is also characterized by a C-terminal multiple repeat unit of six amino acids (PDEPGK), the number of which may vary among different isolates and during the course of an infection, as described for members of the biofilm-associated protein (Bap) family [60,61]. Variations in the Aap protein sequence may help the bacterium to evade the immune response of the host, as observed in the case of the alpha C protein of group B streptococci [62]. The Aap protein of C. aurimucosum ATCC 700975 shares structural similarity with the biofilm-associated protein Bap of Staphylococcus aureus [63] and the accumulation-associated protein Aap of Staphylococcus epidermidis [64]. Both staphylococcal proteins are located on the cell surface and implicated in the formation of biofilms. Cell aggregation and biofilm formation by Aap requires proteolytic processing of the protein by endogenous or host-derived proteases [64]. In the latter case, host proteins can directly induce biofilm formation upon infection, thereby enabling the pathogen to evade the clearance by phagocytes. Both the Aap protein and the SpaH-type pilus apparently represent prominent protein factors on the surface of C. aurimucosum ATCC 700975 cells that are most likely involved in adhesion and colonization processes and in the formation of biofilms.
The gene repertoire of plasmid pET44827: synthesis of the characteristic black pigment of C. aurimucosum ATCC 700975
Assembly of the C. aurimucosum ATCC 700975 genome sequence revealed the presence of the endogenous plasmid pET44827 (Fig. 1C). The average G+C content of 53.3% is below the value of 60.6% determined for the C. aurimucosum ATCC 700975 chromosome, indicating the acquisition of pET44827 by horizontal gene transfer. Coding regions displaying amino acid sequence similarities with proteins of known functions and providing information about structural features of pET44827 and the physiological role of the deduced proteins are presented below.
Replication of pET44827 is presumably mediated by a replication initiator protein of the IncW type (repW) that showed 89% amino acid sequence identity to a homologous counterpart on the multiresistance plasmid pTP10 from C. striatum M82B [65]. This indicates that pET44827 replicates by the theta mechanism, in contrast to other corynebacterial plasmids belonging to the pNG2 family that replicate via the rolling-circle mode [66]. Additionally, pET44827 encodes a putative resolvase (gcrR) ensuring the stability of the plasmid during segregation. The putative resolvase of pET44827 contains a conserved N-terminal catalytic domain of the serine recombinase family and a C-terminal DNA binding domain [67]. However, the gcrR gene region lacks the typical DNA sequence repeats described for the homologous resolvase gene of pTP10 from C. striatum M82B [65]. Furthermore, pET44827 encodes a putative relaxase encoded by traA with 52% similarity to the relaxase of plasmid pREA400 from Rhodococcus erythropolis AN12 [68]. Two genes located upstream of traA were identified as parA and parB, showing similarity to class Ib plasmid partitioning genes present on the pNG2-family plasmid pTET3 from C. glutamicum LP-6 [69]. Therefore, pET44827 seems to be an unusual corynebacterial plasmid that contains characteristic features of the pNG2 family that are combined with a replication initiator protein of the IncW type.
The most prominent feature of pET44827 is a region composed of five genes, including orf18 coding for a putative non-ribosomal peptide synthetase (NRPS) (Fig. 1C). The G+C content of the NRPS gene region is about 47%, which is below the mean value detected for pET44827 (Table 1). As the NRPS gene region is flanked by a remnant of a transposase gene (tnpET23), it might have been integrated into the backbone of pET44827 by a transpositional recombination event. The deduced gene product of orf18 shares 52% similarity with the single module type NRPS BpsA from Streptomyces lavendulae [70] and 51% similarity with the NRPS IndC from the phytopathogen Erwinia chrysanthemi [71]. Both NRPSs catalyze the modular synthesis of the blue 3,3'-bipyridyl pigment indigoidine. The predicted NRPS of pET44827 comprises ten of the eleven highly conserved amino acid sequence motifs of NRPS proteins, only lacking a typical N-terminal condensation domain. Likewise, the IndC protein from E. chrysanthemi does not contain a condensation domain, suggesting that the final condensation step in pigment synthesis is catalyzed by other proteins [71]. NRPSs are key players in the synthesis of natural products and provide a modular assembly line by maintaining reaction intermediates covalently bound on the same phosphopantetheine prosthetic group [72]. NRPSs are synthesized as apoforms, followed by activation through phophospantetheinlytransfer to generate the holoenzyme. This activation might be mediated by the gene products of orf19 and orf20, encoding a protein with a phosphoribosyltransferase domain and a protein with homology to phosphoribosylanthranilate isomerase from Syntrophus aciditrophicus [73], respectively. Moreover, the NRPS gene region of plasmid pET44827 includes the coding region orf17 with deduced amino acid sequence similarity to IndA from Thermoanaerobacter tengcongensis, representing an uncharacterized protein involved in the pigment biosynthesis pathway of this species [74]. Upstream of the NRPS gene region, plasmid pET44827 encodes a putative transcriptional regulator of the MarR protein family (orf21), showing 53% similarity to the PecS regulatory protein from E. chrysanthemi [75]. Interestingly, the PecS protein is a global regulator of various virulence factors and controls the production of the extracellular blue pigment of E. chrysanthemi [75,76]. According to this annotation of pET44827 genes, it is most likely that the characteristic black pigment of C. aurimucosum ATCC 700975 is synthesized by the concerted action of the NRPS encoded by orf18 and an unknown condensase function and that the black pigment molecule is relevant for the proposed pathogenicity of C. aurimucosum.
Discussion
The annotation and bioinformatics analysis of the complete genome sequence of the clinical isolate C. aurimucosum ATCC 700975 (C. nigricans CN-1) provides detailed insights into the metabolism and lifestyle of this bacterium. The most prominent features of C. aurimucosum strains from female urogenital sources are the unusual black pigmentation and their association with complications during pregnancy [12,13]. Due to the black pigmentation and other phenotypic characteristics, these bacteria were initially named C. nigricans [14], but were later shown to represent black-pigmented variants of C. aurimucosum [12]. Annotation of the gene content of C. aurimucosum ATCC 700975 revealed a cluster of coding regions on plasmid pET44827 that is probably involved in the synthesis of the black pigment. The protein encoded by orf18 showed similarity to the gene product of indC from the phytopathogenic bacterium E. chrysanthemi [71]. The indC gene of E. chrysanthemi codes for a non-ribosomal peptide synthetase that carries out the stepwise synthesis of the blue pigment indigoidine. Reverchon et al. demonstrated that the production of indigoidine conferred an increased resistance to oxidative stress and enhanced the tolerance of E. chrysanthemi cells to external hydrogen peroxide [71]. The protection of the bacterial cells by indigoidine is probably based on common chemical properties of many pigment molecules that comprise several carbonate double bonds and can therefore act as efficient radical scavengers [71,77]. This protective function of a bacterial pigment might be relevant also for C. aurimucosum when colonizing the human vaginal tract.
The healthy human vaginal microflora is mostly dominated by acidophilic lactobacilli that play important roles in preventing disease, such as bacterial vaginosis, by creating a low-pH environment with a product of their metabolism, lactic acid, and by producing hydrogen peroxide [54]. The synthesis of hydrogen peroxide by lactobacilli provides a major protection mechanism against opportunistic pathogens in the female genital tract [78]. Hydrogen peroxide is toxic in two ways, firstly by producing toxic hydroxyl radicals and secondly by combining with the pool of chlorine ions in the vagina to generate chloridium ions [79]. Hence, the colonization of the female genital tract by C. aurimucosum may depend on the efficient protection against cellular damage induced by external hydrogen peroxide. Although the genome of C. aurimucosum ATCC 700975 contains a standard gene repertoire for defense against oxidative stress (including catalase, superoxide dismutase and alkyl hydroperoxide reductase), efficient protection of C. aurimucosum ATCC 700975 cells against reactive oxygen species in the human vagina is additionally achieved by the synthesis of the typical black pigment that might act as radical scavenger. The location of the corresponding genes on plasmid pET44827 explains why black-pigmented and non-pigmented C. aurimucosum strains were isolated from clinical specimens. Remarkably, only black-pigmented strains were recovered from female genital sources, pointing towards the pivotal role of pigmentation for the colonization of the vaginal tract. However, when considering complications during pregnancy, such as bleeding, lactobacilli are less efficient in producing hydrogen peroxide, causing increased alkaline conditions in the vagina and thereby permitting the overgrowth of pathogenic microbes [80]. It would be interesting to screen non-pregnant women and sexually active men to see if some of them are colonized with this bacterium.
Although Corynebacterium species appeared to be present in the healthy human vaginal microflora [78,81-83] and on the vaginal epithelium [84], C. aurimucosum was hitherto not detected in specimens from the genital tract of healthy women. Which pathway of vaginal, cervical and intrauterine infection by C. aurimucosum is therefore likely? Information regarding the non-urogenital habitat of C. aurimucosum is currently based on only three reports: (i) Three 16S rDNA sequences were detected in specimens from the forearm skin of a healthy male, indicating that C. aurimucosum is part of the human skin biota [19]. (ii) A single 16S rDNA sequence was reported in the prostate fluid from a prostatitis patient [85]. (iii) An apparently pigmented C. aurimucosum strain, originally named C. nigricans AE1-3, was recovered from a fecal sample of a 35-year-old male Japanese volunteer [37]. This isolation of C. aurimucosum from a fecal sample suggested that this species (and its pigmented variants) may reside in the human intestine. The specific metabolic features of C. aurimucosum ATCC 700975 deduced from the annotation of detected singletons corroborate this microbiological observation. C. aurimucosum may therefore gain access to the lower genital tract by contamination, followed by the so-called ascending pathway from the vagina and cervix, which is the most common route of intrauterine infections [80]. However, almost all of the black-pigmented variants of C. aurimucosum have been reported from urogenital sources suggesting that it may be the primary niche for this potential pathogen. Indeed, its capability to (i) attach to mucosal surfaces through pili, (ii) make biofilms, and (iii) utilize aromatic compounds and L-histidine suggests that it is fully equipped to invade vaginal mucosa. When viewed in the context of its consistent isolation from pregnant women during complications of pregnancy, it is reasonable to assume that it plays an important role there.
In the gut ecosystem, aromatic amines and aromatic acids can be a frequent carbon and energy source for microbial growth when oxygen is available for the enzymatic ring cleavage reaction [39]. As oxygen molecules are present in close proximity to epithelial cells, where they pass from the blood through the epithelium, bacterial cells attached to the epithelium can assimilate such molecules and can thus benefit from the presence of oxygen [40]. The adherence of C. aurimucosum to epithelial cells is apparently mediated by a pilus structure of the SpaH type, consisting of the major pilin SpaH and the minor pilins SpaI and SpaG. Adherence of C. diphtheriae to pharyngeal epithelial cells can be mediated also by the minor pilin SpaB of the structurally related SpaABC pilus [57]. The SpaB protein is covalently anchored to the cell wall to provide tight contact between the bacterial cell and the host tissue in the absence of a pilus shaft. Taking into consideration a similar functioning of the SpaI subunit of the SpaH pilus, adherence of C. aurimucosum cells to host tissue may also occur in the absence of a pilus shaft. Adhesive pili can mediate the initial contact to host cell receptors, with cell-wall-linked pilins supporting the formation of an intimate zone of adhesion. This close contact between the bacterium and host cells enables additional ligand-receptor interactions and, in the case of pathogenic interactions, the efficient delivery of virulence factors [58]. Furthermore, the adherence of C. aurimucosum to epithelial cells may result in the formation of biofilms. The cross-linking of bacterial cells can be mediated by the G5 domains of the accumulation-associated protein Aap that is encoded on the C. aurimucosum ATCC 700975 chromosome and can probably interact with N-acetylglycosamine residues present in the peptidoglycan of the bacterial cell wall [59].
Conclusions
The annotation of the gene repertoire provides the attractive hypothesis to consider C. aurimucosum as resident of the human gut system that manages to reach the lower female genital tract and to establish in the vaginal environment as a contaminant. The close proximity of the vagina to the rectum raises the possibility of microbial colonization of the female genital tract in a continuous process [86]. Another possibility is that black-pigmented strains are sexually transmitted from infected men to some women who engage in sex during pregnancy. Indeed, a C. aurimucosum-like 16S rDNA sequence has been reported from a prostatitis case [85]. Further studies are necessary to confirm if C. aurimucosum is frequently found in sexually active men. When C. aurimucosum is equipped with appropriate protection mechanisms against the vaginal hydrogen peroxide produced by acidophilic lactobacilli, it can apparently establish in the vagina and cervix [11-13] and probably ascend to the amniotic cavity and the fetus. Bacterial infection by contamination is thereby followed by colonization that might cause distinct maternal or fetal responses [79,87]. The results of the C. aurimucosum genome project may help to clarify the proposed role of this bacterium as urogenital pathogen in pregnant women and to prevent preterm delivery that is a leading cause of infant morbidity and mortality in the U.S.A. and Europe [79]. If the clinical diagnosis of C. aurimucosum infection and medical intervention is timely, the prognosis for pregnant women with urogenital infections caused by C. aurimucosum appears to be reasonably good, since several antibiotics turned out recently to be useful for the treatment of infections caused by this organism [88].
Methods
Bacterial strain and growth conditions
C. aurimucosum ATCC 700975 (DSM 44827, CCUG 48176, CIP 107436), formerly designated C. nigricans CN-1 [10], was obtained as a lyophilized culture from DSMZ (Braunschweig, Germany). The strain was originally isolated from the urogenital tract of a 34-year-old woman who experienced a spontaneous abortion during month six of pregnancy [10]. C. aurimucosum was routinely cultured on solid BYT complex medium [89] at 37°C. For the preparation of genomic DNA samples, C. aurimucosum was grown for 24 h in liquid BYT medium supplemented with 1.25% (w/v) glycine.
Sequencing of the C. aurimucosum ATCC 700975 genome
Genomic DNA of C. aurimucosum ATCC 700975 was purified from 20-ml aliquots of an overnight culture by an alkaline lysis procedure [90]. The published protocol was sligthly modified as follows: (i) 20-ml aliquots of C. aurimucosum cells were incubated in a 30 mg ml-1 lysozyme solution at 37°C for 1 h. (ii) The harvested cells were lysed in 0.7 ml 10% (w/v) SDS solution at 37°C for 15 min. A total of 5 μg of purified genomic DNA was used for constructing a single-stranded template DNA (sstDNA) library. The concentration of the sstDNA library was quantified by using the Agilent RNA 6000 Nano Kit. Preparation of the sstDNA library and subsequent DNA sequencing were performed according to manufacturer's protocols (Roche Applied Science). Pyrosequencing of C. aurimucosum DNA was carried out with the Genome Sequencer FLX System (Roche Applied Science). The sequence reads were assembled with the GS Assembler Software (Version 1.1.02). Using the default cutoff of 500 bases for the classification of contigs, 73 large contigs (≥ 500 bases) and 40 small contigs were assembled to give a total size of 2,744,972 bases.
A fosmid library with a 7-fold coverage of the C. aurimucosum ATCC 700975 genome was constructed in E. coli EPI300 cells with the Copy Control Fosmid Library Production Kit and the pCC1FOS fosmid vector (Biozym Scientific). Terminal insert sequences from fosmid library clones were used to build a scaffold for the genome sequence assembly. The terminal insert sequences and the assembled genomic contigs obtained by pyrosequencing were combined by means of the Consed program [15]. To close the remaining gaps in the C. aurimucosum chromosome, additional Sanger reads were generated on purified DNA of selected "linking fosmids". The assembled contigs of plasmid pET44827 were bridged by PCR products obtained with Phusion DNA polymerase (Finnzymes) on genomic template DNA. All primers used in this study were synthesized by Eurofins MWG Operon.
Bioinformatics analysis and annotation of the genome sequence
To verify the assembly of the C. aurimucosum ATCC 700975 genome sequence, the paired-end sequence information obtained from fosmid clones was used to generate a genomic map with the BACCardI program [91]. Automated annotation of the C. aurimucosum genome sequence was carried out with the bacterial genome annotation platform GenDB [16] as described earlier [92], followed by manual curation. Proteins that are orthologous in completely sequenced pathogenic corynebacteria, C. diphtheriae, C. jeikeium, C. urealyticum, and C. kroppenstedtii [30,31,38,92], were deduced by bidirectional best BLASTP hits [22]. Two proteins were considered as orthologs if BLASTP matches with at least 30% sequence identity and a minimum coverage of 50% of the query sequence length were detected in both directions. The orthologous pair of proteins have to represent additionally the best BLASTP hits for the respective query sequence in the target genome. Genes were added to the core genome of completely sequenced pathogenic corynebacteria if orthologs of the encoded proteins were detected in the complete set of selected genomes. If none of the BLAST hits met the criteria defined above in any other genome, the genes were classified as singletons (specific) for C. aurimucosum ATCC 700975. Gene content and comparative metabolic analyses were accomplished by the computer programs EDGAR and CARMEN [35,36]. The annotated genome sequence of C. aurimucosum ATCC 700975 was submitted to the GenBank/EMBL database with accession numbers CP001601 (chromosome) and FM164414 (plasmid pET44827).
Authors' contributions
ET coordinated the project, sequenced the C. aurimucosum genome, carried out the manual annotation and prepared the manuscript. KHG purified the genomic DNA. RS, AT, PV, and WA participated in genome sequencing. SG and SSB supported the gap closure process. JS, TB, JB, BL, and AG provided bioinformatics support for the genome annotation. AP, SKS, and AT supervised the work and participated in data evaluation. All authors read and approved the final version of the manuscript.
Contributor Information
Eva Trost, Email: etrost@cebitec.uni-bielefeld.de.
Susanne Götker, Email: sgoetker@cebitec.uni-bielefeld.de.
Jessica Schneider, Email: jschneid@cebitec.uni-bielefeld.de.
Susanne Schneiker-Bekel, Email: Susanne.Schneiker@cebitec.uni-bielefeld.de.
Rafael Szczepanowski, Email: Rafael.Szczepanowski@cebitec.uni-bielefeld.de.
Alexandra Tilker, Email: atilker@iit-biotech.de.
Prisca Viehoever, Email: viehoeve@cebitec.uni-bielefeld.de.
Walter Arnold, Email: warnold@cebitec.uni-bielefeld.de.
Thomas Bekel, Email: tbekel@cebitec.uni-bielefeld.de.
Jochen Blom, Email: jblom@cebitec.uni-bielefeld.de.
Karl-Heinz Gartemann, Email: Karl.Gartemann@uni-bielefeld.de.
Burkhard Linke, Email: blinke@cebitec.uni-bielefeld.de.
Alexander Goesmann, Email: agoesman@cebitec.uni-bielefeld.de.
Alfred Pühler, Email: puehler@cebitec.uni-bielefeld.de.
Sanjay K Shukla, Email: Shukla.Sanjay@mcrf.mfldclin.edu.
Andreas Tauch, Email: tauch@cebitec.uni-bielefeld.de.
Acknowledgements
ET and JS acknowledge the receipt of a scholarship from the CLIB Graduate Cluster Industrial Biotechnology. The authors thank the sequencing team from IIT GmbH for the help during the gap closure process.
References
- Yassin AF, Steiner U, Ludwig W. Corynebacterium aurimucosum sp. nov. and emended description of Corynebacterium minutissimum Collins and Jones (1983) Int J Syst Evol Microbiol. 2002;52(Pt 3):1001–1005. doi: 10.1099/ijs.0.02045-0. [DOI] [PubMed] [Google Scholar]
- Khamis A, Raoult D, La Scola B. rpoB gene sequencing for identification of Corynebacterium species. J Clin Microbiol. 2004;42(9):3925–3931. doi: 10.1128/JCM.42.9.3925-3931.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux V, Drancourt M, Stein A, Riegel P, Raoult D, La Scola B. Corynebacterium species isolated from bone and joint infections identified by 16S rRNA gene sequence analysis. J Clin Microbiol. 2004;42(5):2231–2233. doi: 10.1128/JCM.42.5.2231-2233.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drancourt M, Berger P, Raoult D. Systematic 16S rRNA gene sequencing of atypical clinical isolates identified 27 new bacterial species associated with humans. J Clin Microbiol. 2004;42(5):2197–2202. doi: 10.1128/JCM.42.5.2197-2202.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein EJ, Citron DM, Merriam CV, Warren YA, Tyrrell KL, Fernandez HT. In vitro activities of doripenem and six comparator drugs against 423 aerobic and anaerobic bacterial isolates from infected diabetic foot wounds. Antimicrob Agents Chemother. 2008;52(2):761–766. doi: 10.1128/AAC.01128-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siala M, Jaulhac B, Gdoura R, Sibilia J, Fourati H, Younes M, Baklouti S, Bargaoui N, Sellami S, Znazen A, Barthel C, Collin E, Hammami A, Sghir A. Analysis of bacterial DNA in synovial tissue of Tunisian patients with reactive and undifferentiated arthritis by broad-range PCR, cloning and sequencing. Arthritis Res Ther. 2008;10(2):R40. doi: 10.1186/ar2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Duc MT, Dekas A, Osman S, Moissl C, Newcombe D, Venkateswaran K. Isolation and characterization of bacteria capable of tolerating the extreme conditions of clean room environments. Appl Environ Microbiol. 2007;73(8):2600–2611. doi: 10.1128/AEM.03007-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moissl C, Osman S, La Duc MT, Dekas A, Brodie E, DeSantis T, Venkateswaran K. Molecular bacterial community analysis of clean rooms where spacecraft are assembled. FEMS Microbiol Ecol. 2007;61(3):509–521. doi: 10.1111/j.1574-6941.2007.00360.x. [DOI] [PubMed] [Google Scholar]
- Rintala H, Pitkaranta M, Toivola M, Paulin L, Nevalainen A. Diversity and seasonal dynamics of bacterial community in indoor environment. BMC Microbiol. 2008;8:56. doi: 10.1186/1471-2180-8-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla SK, Vevea DN, Frank DN, Pace NR, Reed KD. Isolation and characterization of a black-pigmented Corynebacterium sp. from a woman with spontaneous abortion. J Clin Microbiol. 2001;39(3):1109–1113. doi: 10.1128/JCM.39.3.1109-1113.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard KA, Munro C, Wiebe D, Ongsansoy E. Characteristics of rare or recently described Corynebacterium species recovered from human clinical material in Canada. J Clin Microbiol. 2002;40(11):4375–4381. doi: 10.1128/JCM.40.11.4375-4381.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daneshvar MI, Hollis DG, Weyant RS, Jordan JG, MacGregor JP, Morey RE, Whitney AM, Brenner DJ, Steigerwalt AG, Helsel LO, Raney PM, Patel JB, Levett PN, Brown JM. Identification of some charcoal-black-pigmented CDC fermentative coryneform group 4 isolates as Rothia dentocariosa and some as Corynebacterium aurimucosum: proposal of Rothia dentocariosa emend. Georg and Brown 1967, Corynebacterium aurimucosum emend. Yassin et al. 2002, and Corynebacterium nigricans Shukla et al. 2003 pro synon. Corynebacterium aurimucosum. J Clin Microbiol. 2004;42(9):4189–4198. doi: 10.1128/JCM.42.9.4189-4198.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla SK, Harney M, Jhaveri B, Andrews K, Reed KD. Is a black-pigmented Corynebacterium species an opportunistic pathogen during pregnancy? Literature review and report of 3 new cases. Clin Infect Dis. 2003;37(6):834–837. doi: 10.1086/377233. [DOI] [PubMed] [Google Scholar]
- Shukla SK, Bernard KA, Harney M, Frank DN, Reed KD. Corynebacterium nigricans sp. nov.: proposed name for a black-pigmented Corynebacterium species recovered from the human female urogenital tract. J Clin Microbiol. 2003;41(9):4353–4358. doi: 10.1128/JCM.41.9.4353-4358.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D, Abajian C, Green P. Consed: a graphical tool for sequence finishing. Genome Res. 1998;8(3):195–202. doi: 10.1101/gr.8.3.195. [DOI] [PubMed] [Google Scholar]
- Meyer F, Goesmann A, McHardy AC, Bartels D, Bekel T, Clausen J, Kalinowski J, Linke B, Rupp O, Giegerich R, Pühler A. GenDB--an open source genome annotation system for prokaryote genomes. Nucleic Acids Res. 2003;31(8):2187–2195. doi: 10.1093/nar/gkg312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25(5):955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoriev A. Analyzing genomes with cumulative skew diagrams. Nucleic Acids Res. 1998;26(10):2286–2290. doi: 10.1093/nar/26.10.2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Tseng CH, Pei Z, Blaser MJ. Molecular analysis of human forearm superficial skin bacterial biota. Proc Natl Acad Sci USA. 2007;104(8):2927–2932. doi: 10.1073/pnas.0607077104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson H, Lawrence JG. Mutational bias suggests that replication termination occurs near the dif site, not at Ter sites. Mol Microbiol. 2007;64(1):42–56. doi: 10.1111/j.1365-2958.2007.05596.x. [DOI] [PubMed] [Google Scholar]
- Hendrickson H, Lawrence JG. Selection for chromosome architecture in bacteria. J Mol Evol. 2006;62(5):615–629. doi: 10.1007/s00239-005-0192-2. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura M, Canchaya C, Tauch A, Chandra G, Fitzgerald GF, Chater KF, van Sinderen D. Genomics of Actinobacteria: tracing the evolutionary history of an ancient phylum. Microbiol Mol Biol Rev. 2007;71(3):495–548. doi: 10.1128/MMBR.00005-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackiewicz P, Mackiewicz D, Kowalczuk M, Cebrat S. Flip-flop around the origin and terminus of replication in prokaryotic genomes. Genome Biol. 2001;2(12) doi: 10.1186/gb-2001-2-12-interactions1004. INTERACTIONS1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha EP, Danchin A. Essentiality, not expressiveness, drives gene-strand bias in bacteria. Nat Genet. 2003;34(4):377–378. doi: 10.1038/ng1209. [DOI] [PubMed] [Google Scholar]
- Yukawa H, Omumasaba CA, Nonaka H, Kos P, Okai N, Suzuki N, Suda M, Tsuge Y, Watanabe J, Ikeda Y, Vertès AA, Inui M. Comparative analysis of the Corynebacterium glutamicum group and complete genome sequence of strain R. Microbiology. 2007;153(Pt 4):1042–1058. doi: 10.1099/mic.0.2006/003657-0. [DOI] [PubMed] [Google Scholar]
- Gil R, Silva FJ, Pereto J, Moya A. Determination of the core of a minimal bacterial gene set. Microbiol Mol Biol Rev. 2004;68(3):518–537. doi: 10.1128/MMBR.68.3.518-537.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki N, Okai N, Nonaka H, Tsuge Y, Inui M, Yukawa H. High-throughput transposon mutagenesis of Corynebacterium glutamicum and construction of a single-gene disruptant mutant library. Appl Environ Microbiol. 2006;72(5):3750–3755. doi: 10.1128/AEM.72.5.3750-3755.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grissa I, Vergnaud G, Pourcel C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007. pp. W52–57. [DOI] [PMC free article] [PubMed]
- Tauch A, Kaiser O, Hain T, Goesmann A, Weisshaar B, Albersmeier A, Bekel T, Bischoff N, Brune I, Chakraborty T, Chakraborty T, Kalinowski J, Meyer F, Rupp O, Schneiker S, Viehoever P, Pühler A. Complete genome sequence and analysis of the multiresistant nosocomial pathogen Corynebacterium jeikeium K411, a lipid-requiring bacterium of the human skin flora. J Bacteriol. 2005;187(13):4671–4682. doi: 10.1128/JB.187.13.4671-4682.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauch A, Schneider J, Szczepanowski R, Tilker A, Viehoever P, Gartemann KH, Arnold W, Blom J, Brinkrolf K, Brune I, Götker S, Weisshaar B, Goesmann A, Dröge M, Pühler A. Ultrafast pyrosequencing of Corynebacterium kroppenstedtii DSM44385 revealed insights into the physiology of a lipophilic corynebacterium that lacks mycolic acids. J Biotechnol. 2008;136(1-2):22–30. doi: 10.1016/j.jbiotec.2008.03.004. [DOI] [PubMed] [Google Scholar]
- Ishikawa J, Yamashita A, Mikami Y, Hoshino Y, Kurita H, Hotta K, Shiba T, Hattori M. The complete genomic sequence of Nocardia farcinica IFM 10152. Proc Natl Acad Sci USA. 2004;101(41):14925–14930. doi: 10.1073/pnas.0406410101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315(5819):1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- Haft DH, Selengut J, Mongodin EF, Nelson KE. A guild of 45 CRISPR-associated (Cas) protein families and multiple CRISPR/Cas subtypes exist in prokaryotic genomes. PLoS Comput Biol. 2005;1(6):e60. doi: 10.1371/journal.pcbi.0010060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goesmann A, Schneider J, Trost E, Tauch A. Entschlüsselung mikrobieller Genomsequenzen im Schnellverfahren. BIOspektrum. 2009;15(3):279–281. [Google Scholar]
- Blom J, Albaum SP, Doppmeier D, Pühler A, Vorhölter FJ, Zakrzewski M, Goesmann A. EDGAR: a software framework for the comparative analysis of prokaryotic genomes. BMC Bioinformatics. 2009;10:154. doi: 10.1186/1471-2105-10-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita H, Kuwahara T, Ohshima K, Sasamoto H, KItoh K, Hattori M, Hayashi T, Takami H. An improved DNA isolation method for metagenomic analysis of the microbial flora of the human intestine. Microbes Environ. 2007;22(3):214–222. doi: 10.1264/jsme2.22.214. [DOI] [Google Scholar]
- Cerdeno-Tarraga AM, Efstratiou A, Dover LG, Holden MT, Pallen M, Bentley SD, Besra GS, Churcher C, James KD, De Zoysa A, Chillingworth T, Cronin A, Dowd L, Feltwell T, Hamlin N, Holroyd S, Jagels K, Moule S, Quail MA, Rabbinowitsch E, Rutherford KM, Thomson NR, Unwin L, Whitehead S, Barrell BG, Parkhill J. The complete genome sequence and analysis of Corynebacterium diphtheriae NCTC13129. Nucleic Acids Res. 2003;31(22):6516–6523. doi: 10.1093/nar/gkg874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz E, Ferrandez A, Prieto MA, Garcia JL. Biodegradation of aromatic compounds by Escherichia coli. Microbiol Mol Biol Rev. 2001;65(4):523–569. doi: 10.1128/MMBR.65.4.523-569.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage DC. Microbial ecology of the gastrointestinal tract. Annu Rev Microbiol. 1977;31:107–133. doi: 10.1146/annurev.mi.31.100177.000543. [DOI] [PubMed] [Google Scholar]
- Reaney SK, Begg C, Bungard SJ, Guest JR. Identification of the L-tartrate dehydratase genes (ttdA and ttdB) of Escherichia coli and evolutionary relationship with the class I fumarase genes. J Gen Microbiol. 1993;139(7):1523–1530. doi: 10.1099/00221287-139-7-1523. [DOI] [PubMed] [Google Scholar]
- Kim OB, Unden G. The L-tartrate/succinate antiporter TtdT (YgjE) of L-tartrate fermentation in Escherichia coli. J Bacteriol. 2007;189(5):1597–1603. doi: 10.1128/JB.01402-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinton A Jr, Hume ME. Inhibition of Salmonella typhimurium by the products of tartrate metabolism by a Veillonella species. J Appl Bacteriol. 1996;81(2):188–190. doi: 10.1111/j.1365-2672.1996.tb04498.x. [DOI] [PubMed] [Google Scholar]
- Barabote RD, Saier MH Jr. Comparative genomic analyses of the bacterial phosphotransferase system. Microbiol Mol Biol Rev. 2005;69(4):608–634. doi: 10.1128/MMBR.69.4.608-634.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janiyani KL, Ray MK. Cloning, sequencing, and expression of the cold-inducible hutU gene from the antarctic psychrotrophic bacterium Pseudomonas syringae. Appl Environ Microbiol. 2002;68(1):1–10. doi: 10.1128/AEM.68.1.1-10.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XX, Rainey PB. Genetic analysis of the histidine utilization (hut) genes in Pseudomonas fluorescens SBW25. Genetics. 2007;176(4):2165–2176. doi: 10.1534/genetics.107.075713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapeller-Adler R, Schiller W. Über die Histidinausscheidung in der Schwangerschaft unter dem Einfluss verschiedenartiger Ernährung. Klin Wochenschr. 1935;14(50):1790–1792. doi: 10.1007/BF01779940. [DOI] [Google Scholar]
- Tasch H. Zur Frühdiagnose der Schwangerschaft. Arch Gynäk. 1941;171:581–602. doi: 10.1007/BF01714309. [DOI] [Google Scholar]
- Potrykus J, White RL, Bearne SL. Proteomic investigation of amino acid catabolism in the indigenous gut anaerobe Fusobacterium varium. Proteomics. 2008;8(13):2691–2703. doi: 10.1002/pmic.200700437. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Turanov AA, Hatfield DL, Gladyshev VN. In silico identification of genes involved in selenium metabolism: evidence for a third selenium utilization trait. BMC Genomics. 2008;9:251. doi: 10.1186/1471-2164-9-251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtman TC. Selenocysteine. Annu Rev Biochem. 1996;65:83–100. doi: 10.1146/annurev.bi.65.070196.000503. [DOI] [PubMed] [Google Scholar]
- Jormakka M, Byrne B, Iwata S. Formate dehydrogenase--a versatile enzyme in changing environments. Curr Opin Struct Biol. 2003;13(4):418–423. doi: 10.1016/S0959-440X(03)00098-8. [DOI] [PubMed] [Google Scholar]
- Wessjohann LA, Schneider A, Abbas M, Brandt W. Selenium in chemistry and biochemistry in comparison to sulfur. Biol Chem. 2007;388(10):997–1006. doi: 10.1515/BC.2007.138. [DOI] [PubMed] [Google Scholar]
- Zhou X, Bent SJ, Schneider MG, Davis CC, Islam MR, Forney LJ. Characterization of vaginal microbial communities in adult healthy women using cultivation-independent methods. Microbiology. 2004;150(Pt 8):2565–2573. doi: 10.1099/mic.0.26905-0. [DOI] [PubMed] [Google Scholar]
- Swierczynski A, Ton-That H. Type III pilus of corynebacteria: Pilus length is determined by the level of its major pilin subunit. J Bacteriol. 2006;188(17):6318–6325. doi: 10.1128/JB.00606-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ton-That H, Marraffini LA, Schneewind O. Protein sorting to the cell wall envelope of Gram-positive bacteria. Biochim Biophys Acta. 2004;1694(1-3):269–278. doi: 10.1016/j.bbamcr.2004.04.014. [DOI] [PubMed] [Google Scholar]
- Mandlik A, Swierczynski A, Das A, Ton-That H. Corynebacterium diphtheriae employs specific minor pilins to target human pharyngeal epithelial cells. Mol Microbiol. 2007;64(1):111–124. doi: 10.1111/j.1365-2958.2007.05630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandlik A, Swierczynski A, Das A, Ton-That H. Pili in Gram-positive bacteria: assembly, involvement in colonization and biofilm development. Trends Microbiol. 2008;16(1):33–40. doi: 10.1016/j.tim.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman A, Holden MT, Yeats C. The G5 domain: a potential N-acetylglucosamine recognition domain involved in biofilm formation. Bioinformatics. 2005;21(8):1301–1303. doi: 10.1093/bioinformatics/bti206. [DOI] [PubMed] [Google Scholar]
- Shankar V, Baghdayan AS, Huycke MM, Lindahl G, Gilmore MS. Infection-derived Enterococcus faecalis strains are enriched in esp, a gene encoding a novel surface protein. Infect Immun. 1999;67(1):193–200. doi: 10.1128/iai.67.1.193-200.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cucarella C, Tormo MA, Ubeda C, Trotonda MP, Monzon M, Peris C, Amorena B, Lasa I, Penades JR. Role of biofilm-associated protein Bap in the pathogenesis of bovine Staphylococcus aureus. Infect Immun. 2004;72(4):2177–2185. doi: 10.1128/IAI.72.4.2177-2185.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madoff LC, Michel JL, Gong EW, Kling DE, Kasper DL. Group B streptococci escape host immunity by deletion of tandem repeat elements of the alpha C protein. Proc Natl Acad Sci USA. 1996;93(9):4131–4136. doi: 10.1073/pnas.93.9.4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tormo MA, Knecht E, Gotz F, Lasa I, Penades JR. Bap-dependent biofilm formation by pathogenic species of Staphylococcus: evidence of horizontal gene transfer? Microbiology. 2005;151(Pt 7):2465–2475. doi: 10.1099/mic.0.27865-0. [DOI] [PubMed] [Google Scholar]
- Rohde H, Burdelski C, Bartscht K, Hussain M, Buck F, Horstkotte MA, Knobloch JK, Heilmann C, Herrmann M, Mack D. Induction of Staphylococcus epidermidis biofilm formation via proteolytic processing of the accumulation-associated protein by staphylococcal and host proteases. Mol Microbiol. 2005;55(6):1883–1895. doi: 10.1111/j.1365-2958.2005.04515.x. [DOI] [PubMed] [Google Scholar]
- Tauch A, Krieft S, Kalinowski J, Pühler A. The 51,409-bp R-plasmid pTP10 from the multiresistant clinical isolate Corynebacterium striatum M82B is composed of DNA segments initially identified in soil bacteria and in plant, animal, and human pathogens. Mol Gen Genet. 2000;263(1):1–11. doi: 10.1007/PL00008668. [DOI] [PubMed] [Google Scholar]
- Tauch A, Bischoff N, Brune I, Kalinowski J. Insights into the genetic organization of the Corynebacterium diphtheriae erythromycin resistance plasmid pNG2 deduced from its complete nucleotide sequence. Plasmid. 2003;49(1):63–74. doi: 10.1016/S0147-619X(02)00115-4. [DOI] [PubMed] [Google Scholar]
- Marchler-Bauer A, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, He S, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Liebert CA, Liu C, Lu F, Lu S, Marchler GH, Mullokandov M, Song JS, Tasneem A, Thanki N, Yamashita RA, Zhang D, Zhang N, Bryant SH. CDD: specific functional annotation with the Conserved Domain Database. Nucleic Acids Res. 2009. pp. D205–210. [DOI] [PMC free article] [PubMed]
- Yang JC, Lessard PA, Sengupta N, Windsor SD, O'Brien XM, Bramucci M, Tomb JF, Nagarajan V, Sinskey AJ. TraA is required for megaplasmid conjugation in Rhodococcus erythropolis AN12. Plasmid. 2007;57(1):55–70. doi: 10.1016/j.plasmid.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Tauch A, Götker S, Pühler A, Kalinowski J, Thierbach G. The 27.8-kb R-plasmid pTET3 from Corynebacterium glutamicum encodes the aminoglycoside adenyltransferase gene cassette aadA9 and the regulated tetracycline efflux system Tet 33 flanked by active copies of the widespread insertion sequence IS6100. Plasmid. 2002;48(2):117–129. doi: 10.1016/S0147-619X(02)00120-8. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Kumagai T, Kitani K, Mori M, Matoba Y, Sugiyama M. Cloning and characterization of a Streptomyces single module type non-ribosomal peptide synthetase catalyzing a blue pigment synthesis. J Biol Chem. 2007;282(12):9073–9081. doi: 10.1074/jbc.M611319200. [DOI] [PubMed] [Google Scholar]
- Reverchon S, Rouanet C, Expert D, Nasser W. Characterization of indigoidine biosynthetic genes in Erwinia chrysanthemi and role of this blue pigment in pathogenicity. J Bacteriol. 2002;184(3):654–665. doi: 10.1128/JB.184.3.654-665.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donadio S, Monciardini P, Sosio M. Polyketide synthases and nonribosomal peptide synthetases: the emerging view from bacterial genomics. Nat Prod Rep. 2007;24(5):1073–1109. doi: 10.1039/b514050c. [DOI] [PubMed] [Google Scholar]
- McInerney MJ, Rohlin L, Mouttaki H, Kim U, Krupp RS, Rios-Hernandez L, Sieber J, Struchtemeyer CG, Bhattacharyya A, Campbell JW, Gunsalus RP. The genome of Syntrophus aciditrophicus: life at the thermodynamic limit of microbial growth. Proc Natl Acad Sci USA. 2007;104(18):7600–7605. doi: 10.1073/pnas.0610456104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao Q, Tian Y, Li W, Xu Z, Xuan Z, Hu S, Dong W, Yang J, Chen Y, Xue Y, Xu Y, Lai X, Huang L, Dong X, Ma Y, Ling L, Tan H, Chen R, Wang J, Yu J, Yang H. A complete sequence of the T. tengcongensis genome. Genome Res. 2002;12(5):689–700. doi: 10.1101/gr.219302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reverchon S, Nasser W, Robert-Baudouy J. pecS: a locus controlling pectinase, cellulase and blue pigment production in Erwinia chrysanthemi. Mol Microbiol. 1994;11(6):1127–1139. doi: 10.1111/j.1365-2958.1994.tb00389.x. [DOI] [PubMed] [Google Scholar]
- Hommais F, Oger-Desfeux C, Van Gijsegem F, Castang S, Ligori S, Expert D, Nasser W, Reverchon S. PecS is a global regulator of the symptomatic phase in the phytopathogenic bacterium Erwinia chrysanthemi 3937. J Bacteriol. 2008;190(22):7508–7522. doi: 10.1128/JB.00553-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giese B. Radicals in organic synthesis: formation of carbon-carbon bonds. Oxford: Pergamon; 1986. [Google Scholar]
- Coolen MJ, Post E, Davis CC, Forney LJ. Characterization of microbial communities found in the human vagina by analysis of terminal restriction fragment length polymorphisms of 16S rRNA genes. Appl Environ Microbiol. 2005;71(12):8729–8737. doi: 10.1128/AEM.71.12.8729-8737.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pararas MV, Skevaki CL, Kafetzis DA. Preterm birth due to maternal infection: Causative pathogens and modes of prevention. Eur J Clin Microbiol Infect Dis. 2006;25(9):562–569. doi: 10.1007/s10096-006-0190-3. [DOI] [PubMed] [Google Scholar]
- Deb K, Chaturvedi MM, Jaiswal YK. Comprehending the role of LPS in Gram-negative bacterial vaginosis: ogling into the causes of unfulfilled child-wish. Arch Gynecol Obstet. 2004;270(3):133–146. doi: 10.1007/s00404-004-0623-0. [DOI] [PubMed] [Google Scholar]
- Onderdonk AB, Zamarchi GR, Walsh JA, Mellor RD, Munoz A, Kass EH. Methods for quantitative and qualitative evaluation of vaginal microflora during menstruation. Appl Environ Microbiol. 1986;51(2):333–339. doi: 10.1128/aem.51.2.333-339.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onderdonk AB, Zamarchi GR, Rodriguez ML, Hirsch ML, Munoz A, Kass EH. Qualitative assessment of vaginal microflora during use of tampons of various compositions. Appl Environ Microbiol. 1987;53(12):2779–2784. doi: 10.1128/aem.53.12.2779-2784.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funke G, Frodl R. Comprehensive study of Corynebacterium freneyi strains and extended and emended description of Corynebacterium freneyi Renaud, Aubel, Riegel, Meugnier, and Bollet 2001. J Clin Microbiol. 2008;46(2):638–643. doi: 10.1128/JCM.01491-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman RW, Fukushima M, Diamond L, Kumm J, Giudice LC, Davis RW. Microbes on the human vaginal epithelium. Proc Natl Acad Sci USA. 2005;102(22):7952–7957. doi: 10.1073/pnas.0503236102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner MA, Shoskes D, Shahed A, Pace NR. Prevalence of corynebacterial 16S rRNA sequences in patients with bacterial and "nonbacterial" prostatitis. J Clin Microbiol. 1999;37(6):1863–1870. doi: 10.1128/jcm.37.6.1863-1870.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid G, Bruce AW. Urogenital infections in women: can probiotics help? Postgrad Med J. 2003;79(934):428–432. doi: 10.1136/pmj.79.934.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casadevall A, Pirofski LA. Host-pathogen interactions: basic concepts of microbial commensalism, colonization, infection, and disease. Infect Immun. 2000;68(12):6511–6518. doi: 10.1128/IAI.68.12.6511-6518.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Roblas R, Adames H, Martin-de-Hijas NZ, Almeida DG, Gadea I, Esteban J. In vitro activity of tigecycline and 10 other antimicrobials against clinical isolates of the genus Corynebacterium. Int J Antimicrob Agents. 2009;33(5):453–455. doi: 10.1016/j.ijantimicag.2008.11.001. [DOI] [PubMed] [Google Scholar]
- Tauch A, Bischoff N, Pühler A, Kalinowski J. Comparative genomics identified two conserved DNA modules in a corynebacterial plasmid family present in clinical isolates of the opportunistic human pathogen Corynebacterium jeikeium. Plasmid. 2004;52(2):102–118. doi: 10.1016/j.plasmid.2004.05.003. [DOI] [PubMed] [Google Scholar]
- Hopwood DA. Genetic Manipulation of Streptomyces: A Laboratory Manual. New York: Cold Spring Harbor; 1985. [Google Scholar]
- Bartels D, Kespohl S, Albaum S, Druke T, Goesmann A, Herold J, Kaiser O, Pühler A, Pfeiffer F, Raddatz G, Stoye J, Meyer F, Schuster SC. BACCardI--a tool for the validation of genomic assemblies, assisting genome finishing and intergenome comparison. Bioinformatics. 2005;21(7):853–859. doi: 10.1093/bioinformatics/bti091. [DOI] [PubMed] [Google Scholar]
- Tauch A, Trost E, Tilker A, Ludewig U, Schneiker S, Goesmann A, Arnold W, Bekel T, Brinkrolf K, Brune I, Götker S, Kalinowski J, Kamp PB, Lobo FP, Viehoever P, Weisshaar B, Soriano F, Dröge M, Pühler A. The lifestyle of Corynebacterium urealyticum derived from its complete genome sequence established by pyrosequencing. J Biotechnol. 2008;136(1-2):11–21. doi: 10.1016/j.jbiotec.2008.02.009. [DOI] [PubMed] [Google Scholar]
- Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28(1):33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]