


Abstract
Acetyl-CoA Carboxylase catalyzes the first committed step in fatty acid synthesis. Escherichia coli acetyl-CoA carboxylase is composed of biotin carboxylase, carboxyltransferase and biotin carboxyl carrier protein functions. The accA and accD genes that code for the α- and β-subunits, respectively, are not in an operon, yet yield an α2β2 carboxyltransferase. Here, we report that carboxyltransferase regulates its own translation by binding the mRNA encoding its subunits. This interaction is mediated by a zinc finger on the β-subunit; mutation of the four cysteines to alanine diminished nucleic acid binding and catalytic activity. Carboxyltransferase binds the coding regions of both subunit mRNAs and inhibits translation, an inhibition that is relieved by the substrate acetyl-CoA. mRNA binding reciprocally inhibits catalytic activity. Preferential binding of carboxyltransferase to RNA in situ was shown using fluorescence resonance energy transfer. We propose an unusual regulatory mechanism by which carboxyltransferase acts as a ‘dimmer switch’ to regulate protein production and catalytic activity, while sensing the metabolic state of the cell through acetyl-CoA concentration.
INTRODUCTION
Acetyl-CoA carboxylase (ACCase, EC 6.4.1.2) catalyzes the first committed step in fatty acid biosynthesis in all organisms. The overall reaction is the biotin-dependent carboxylation of acetyl-CoA to form malonyl-CoA and it takes place in two separate half-reactions.
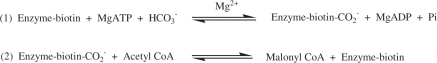
The first half-reaction is catalyzed by biotin carboxylase, where ATP is used to activate bicarbonate for carboxylation of biotin. The natural substrate is the biotin carboxyl carrier protein (designated as Enzyme-biotin in the above scheme). The second half-reaction, catalyzed by carboxyltransferase, transfers the activated carboxyl group from carboxybiotin to acetyl-CoA to form malonyl-CoA. In eukaryotes, each of the three different proteins, biotin carboxylase, biotin carboxyl carrier protein and carboxyltransferase form individual domains in a single polypeptide chain. In contrast, Escherichia coli encodes separate polypeptides generating a homodimeric biotin carboxylase (BC), biotin carboxyl carrier protein (BCCP) and an α2β2 heterotetrameric carboxyltransferase (CT) (1).
Because the reaction involves two separate half-reactions, coordinated expression of the genes coding for the various subunits of bacterial ACCase is essential in order to achieve efficient conversion of substrate to product. This is readily accomplished for the first half-reaction because the genes coding for biotin carboxylase (accC) and the biotin carboxyl carrier protein (accB) are in a single operon that is transcriptionally controlled (2,3). However, the genes encoding the CT α (accA) and CT β (accD) subunits are not in an operon, but are found on nearly opposite poles of the chromosome (4.5′ and 52.4′, respectively) in opposing directions (4,5). Yet, the expression of accA and accD should yield stoichiometric amounts of the α- and β-subunits to form the α2β2 heterotetramer of CT. Sequence analyses reveal no obvious clues to a mechanism of concerted expression of accA and accD, however, an alternative to transcriptional control for modulating expression of accA and accD has been suggested (2).
The crystal structures of both E. coli (PDB: 2f9y) and Staphylococcus aureus (PDB: 2f9i) CT reveal a Cys4-type zinc ribbon motif on the β-subunit (6) (Figure 1). The zinc domain forms part of a saddle-like structure with a net electropositive surface potential, whereas the rest of the protein has a net electronegative surface potential (6). Zinc-finger domains are commonly associated with nucleic acid-binding proteins and previous studies have shown that CT does bind DNA, albeit non-specifically (7). Notably, DNA binding inhibited catalysis while the substrates inhibited DNA binding (7). In other words, DNA binding and catalysis are reciprocally related, implying a physiological role for nucleic acid binding.
Figure 1.

The Cys4-type Zn ribbon of E. coli carboxyltransferase with the Zn atom shown in gray coordinated by four cysteine residues (PDBID: 2f9y).
In this report, we show that an unusual regulatory mechanism exists by which CT autoregulates activity in response to metabolic needs. Specifically, we show that the zinc domain of CT is not only required for catalysis, but that it also mediates binding to the mRNA coding for the α- and β-subunits of carboxyltransferase so that CT can act as a ‘dimmer switch’ to regulate translation. These properties of the zinc domain, which is unique to bacterial CT, provide a mechanism for regulating enzymatic activity and for coordinating expression of the genes coding for the α- and β-subunits of carboxyltransferase.
MATERIALS AND METHODS
Reagents
Restriction enzymes and T4 DNA ligase were from New England Biolabs. Pfu Turbo DNA polymerase was from Stratagene. The cloning and expression vector pETBlue-2 and the E. coli host strains NovaBlue and BL21(DE3) were from Novagen. Primers were synthesized by MWG Biotech. In vitro transcription kit was from Ambion. T7 in vitro transcription/translation kit was from Qiagen. [α-32P]UTP (3000 Ci/mmol) and [35S]l-Methionine (>1000 Ci/mmol) were from PerkinElmer. His-binding cartridges and all other reagents were from Sigma.
Construction of expression vectors
The genes accA and accD coding for the α- and β-subunits of carboxyltransferase were overexpressed linked in a minioperon similar to the one described by Blanchard and Waldrop (8) except that the expression vector pETBlue-2 was used and the his-tag was placed on the C-terminus of the β-subunit. To construct the minioperon, the gene encoding the α-subunit of E. coli CT was amplified by PCR using pCZB2 (8) as template and primers CTa5 and CTa3 (Table 1), digested with NcoI and HindIII, and cloned into equivalently digested pETBlue-2 to generate the plasmid pGDM8. To make possible the use of the NcoI restriction site, an amino acid mutation, S2A, was introduced near the N-terminus of accA. This mutation had no effect on the functions of the enzyme, either nucleic acid binding or catalytic activity. The CTa3 primer allowed incorporation of the modified ribosomal binding site after the accA stop codon. The gene for the wild-type β-subunit of E. coli CT was amplified by PCR using pCZB1 (8) as template and primers CTb5 and CTb3, digested with HindIII and XhoI, and cloned into equivalently digested pGDM8 to generate the plasmid pGDM9, which was used for coexpression of both genes.
Table 1.
List of primers used for mutagenesis
 |
The boxed nucleotides are the mutations from wild-type.
Site-directed mutagenesis
The cysteines in the zinc domain of the CT β-subunit were mutated to alanine using the method of overlap extension (9). Four single variants were generated, resulting in plasmids pGDM12-15. Complementary oligonucleotides containing specific alterations in the nucleotide sequence (e.g. C30A5 and C30A3) were used to generate two DNA fragments having overlapping ends. These fragments were combined and used as the template in a subsequent PCR reaction using primers CTb5 and CTb3 (Table 1) to generate the full-length accD gene with the desired mutation (e.g. C30A). Three multiple cysteine to alanine variants were generated by sequential site-directed mutagenesis of the C27A accD gene using the pairs of primers C46A5 and C46A3, C46/49A5 and C46/49A3, and C27/30A5 and C27/30A3. All constructs were sequenced to confirm the mutations and verify there were no other changes.
Expression, purification and enzymatic assay
The plasmids encoding wild-type and CT variants were transformed into E. coli BL21(DE3) and grown as described by Blanchard and Waldrop (8) with the following exceptions. The temperature was reduced to 25°C prior to induction with 2.8 mM β-lactose. Expressed protein was purified by nickel affinity chromatography using a HisSelect cartridge (Sigma) following the manufacturer’s protocol. The wash buffer contained 10 mM imidazole, 300 mM NaCl, 50 mM KH2PO4, pH 7.0 and the elution buffer contained 200 mM imidazole, 500 mM NaCl, 20 mM Tris–Cl, pH 8.0. Eluents were subjected to 24 h dialysis first against 134 mM KCl, 1 mM Na2EDTA, 10 mM KH2PO4, pH 7.0 and second against 500 mM KCl, 10 mM HEPES, pH 7.0, followed by concentration to 0.5–10 mg/ml using Amicon Ultra 4 (Millipore) centrifugal concentrators with a 10 000 molecular weight cutoff. CT activity was measured in the reverse, or non-physiological, direction with a spectrophotometric assay in which the production of acetyl-CoA was coupled to citrate synthase and malate dehydrogenase reactions (8). NADH formation was followed spectrophotometrically at 340 nm using a Uvikon 810 (Kontron Instruments) spectrophotometer interfaced to a computer equipped with a data acquisition program.
Inactivation studies
Enzyme modification by N-ethylmaleimide (NEM) was performed in 500 mM KCl, 10 mM HEPES, pH 7.0. For NEM inactivation, reactions were initiated by the addition of NEM to the CT solution at a final concentration of 4 mM. An aliquot was removed every 5 min and assayed for enzymatic activity. The aliquots were of sufficiently small volume compared with the assay volume such that the modifying agents were diluted 50-fold.
Data analysis
The kinetic parameters Km and Vmax were determined by nonlinear regression analysis. The initial velocities versus the substrate concentration were fitted to the Michaelis–Menten equation using the programs of Cleland (10). The dissociation constant (Kd) for RNA binding was determined using the program Enzfitter. The binding isotherms for CT binding to RNA were analyzed by fitting the data to Equation 1, where Kd reflects half maximal saturation, Y is the fractional complex, X is concentration of CT and Ymax is the maximal complex or horizontal asymptote.
| 1 |
.
Zinc determination and circular dichroism
The molar ratio of Zn to CT tetramer was determined using Atomic Absorption Spectroscopy using a Varian Spectra AA 220 Atomic Absorption Spectrometer equipped with a zinc lamp and absorption recorded at 213.9 nm. Protein concentrations for wild-type and variants ranged from 15 to 50 µM. CD spectra were recorded on an AVIV Model 202 CD spectropolarimeter using a 1 cm pathlength quartz cuvette. Data points were averaged from triplicate scans from 200 to 250 nm. Spectra for wild-type and each variant were recorded at 25°C at a protein concentration of 0.1 mg/ml in a buffer of 500 mM KCl, 10 mM HEPES, pH 7.0.
DNA Electrophoretic mobility shift assay
Wild-type CT and variants were incubated for 0.5 h at 25°C with an 800 bp linear dsDNA at final concentrations of 0.88 µM and 50 nM, respectively. The DNA was obtained by gel extraction following PCR using primers C46A5 and CTb3 and the accD gene as the template. After addition of glycerol to a final concentration of 2.5%, samples were resolved on 0.8% (w/v) agarose gels in 1 × TAE (40 mM Tris–Cl at pH 8.0, 20 mM acetate, 1 mM Na2EDTA) followed by staining with ethidium bromide and visualized by UV irradiation.
In vitro transcription and RNA electrophoretic mobility shift assay
A cell-free extract was used to produce mRNA using the MEGAscript Kit (Ambion) following the manufacturer’s protocol with 0.2 µg PCR product as the template. Each 20 µl reaction contained [α-32P]UTP at 167 nM. The reactions also contained 7.5 mM ATP, CTP, GTP and 3.75 mM UTP. The resultant mRNA was purified according to the manufacturer’s instructions with the MEGAclear Kit (Ambion) and eluted in 100 µl, to which was added 20 µl of 80% glycerol and 20 µl of 10 × EMSA buffer (200 mM Tris–Cl at pH 8.0, 1 mM Na2EDTA, 500 mM NaCl, 50 mM MgCl2). Seven microliter of this solution was added to each 10 µl reaction, with CT titrated from 0 to 1 µM in the remaining 3 µl. For the inhibition of RNA binding, acetyl-CoA was titrated from 0 to 4.0 mM. Reactions were incubated on ice for 0.5 h. Samples were loaded with the power on onto prerun 5.0% (w/v) native polyacrylamide gels (39: 1 acrylamide:bisacrylamide) and electrophoresed at room temperature in TBE buffer (45 mM Tris borate at pH 8.3, 1.25 mM Na2EDTA). After 3–6 h of electrophoresis, the gels were dried and complexes were visualized by phosphorimaging and quantified using Molecular Dynamics ImageQuant software. The protein–RNA complex was defined as the slowest migrating band on the gel, while free RNA was the lower band. For calculating percent complex formation, the region on the gel from the slowest migrating complex to the free nucleic acid was considered as complex. The percent complex was calculated as complex intensity divided by total intensity (complex plus free RNA). Each gel shift was done in at least duplicate.
Coupled in vitro transcription/translation
Translation repression of the mRNA encoding the α- and β-subunits via CT binding was examined using a coupled in vitro transcription/translation system. The EasyXpress Protein Synthesis System (Qiagen) was used following the manufacturer’s protocol with 1 µg (∼10 nM) plasmid or unlabeled mRNA as templates. To examine the effect of CT on the expression of both genes simultaneously, pCZB5 was used as a template. The plasmid pCZB5 is an expression vector of similar construction to pGDM9 and contains the accA and accD genes in tandem with high levels of overexpression from those genes (8). The ability of CT to attenuate translation of only the mRNA for the β-subunit utilized the vector pGLW39, which contains only the accD gene cloned into pET28. The proteins expressed from these templates were visualized by polyacrylamide gel electrophoresis, where each 50 µl reaction contained 5 µCi of [35S]l-Methionine. The reaction mixtures were run on 5.0% (w/v) denaturing polyacrylamide gels (39:1 acrylamide:bisacrylamide, 2.5% SDS) at 2 V/cm for 10–12 h and dried. Bands were visualized by phosphorimaging. Each assay was done in triplicate.
Detection of CT–RNA interaction in situ
Detection of CT binding to RNA in situ was accomplished using a fluorescence resonance energy transfer (FRET) assay with a CT β-enhanced yellow fluorescent protein (eYFP) fusion protein as the donor and the RNA binding dye Sytox Orange (Molecular Probes) as the acceptor. The gene for eYFP was amplified with primers eYFP5 and eYFP3, digested with XhoI and PacI, and cloned into equivalently digested pGDM9 and pGDM18 to generate pGDM24 and pGDM25, respectively. The use of XhoI and PacI restriction sites eliminates the vector-encoded His6-tag and stop codon, while placing the stop codon following the coding region for the CT β-eYFP fusion protein. Plasmid pGDM24 contains wild-type CT-eYFP fusion and pGDM25 contains the quadruple cysteine variant of CT-eYFP fusion. E. coli BL21(DE3) harboring the plasmids pGDM24 and pGDM25 were grown at 37°C until the A600 reached 0.7, at which point lactose was added to 28 mM. Samples of 100 µl were collected just prior to induction with lactose, and every 0.5 h afterward for 3 h and flash frozen in liquid nitrogen. Each sample was then thawed and divided equally into thirds. The divided samples were either left as is, subjected to 0.3 µg/µl RNase A (Qiagen), or 1.0 U/µl DNase I for 0.5 h at 37°C. Sytox Orange is a high-affinity nucleic acid stain that readily enters cells with compromised membranes. The dye was added to control and nuclease treated cells to a final concentration of 250 nM. There is considerable overlap of YFP emission and Sytox Orange absorption spectra; the Föerster distance (R0) is 5.6 nm. Spectra were collected using a Jasco FP-6300 spectrofluorometer equipped to hold a 3 × 3 mm quartz cuvette. A 500 nm incident light was used to achieve sufficient FRET with minimal direct excitation of the acceptor. Emission spectra were measured perpendicular to the incident light at 540–600 nm to avoid the peak due to refracted light and to sufficiently span the absorption maximum of 573 nm when the stain is bound to nucleic acid. Each assay was done in triplicate.
RESULTS
The Zn domain is required for catalysis and nucleic acid binding
The Zn finger of CT is a member of the Cys4-type family, with the divalent metal coordinated in a tetrahedral fashion by four cysteine sulfhydryl groups (Figure 1). Each of the cysteines (residues 27, 30, 46 and 49) coordinating the zinc atom were mutated to alanine individually followed by multiple mutations to remove two (double; C27A, C46A), three (triple; C27A, C46A, C49A), and all four (quadruple) cysteines.
The effect of the cysteine mutations on enzymatic activity was assessed using a coupled enzyme assay where the reaction is run in the non-physiological direction in which malonyl-CoA and biocytin [biocytin is a biotin molecule with a lysine affixed to the carboxyl group of the valeric acid side chain via an amide linkage at the ε-amino group and is preferred over biotin in this assay because it produces a maximal velocity three orders of magnitude greater than does biotin (8)] are the substrates. The kinetic results indicate that mutation of individual cysteine residues 27, 46 and 49 decreases the Vmax only 8- to 11-fold, whereas the Vmax for the C30A variant decreases by over 100-fold (Table 2). Similar results were obtained with analogous mutations to the Cys4-type zinc finger of pea plastidic ACCase CT β (11). Mutation of a single cysteine has only small effects on the Michaelis constant for both substrates, biocytin and malonyl-CoA. None of the multiple site variants have measureable activity. The results suggest that the Zn-finger cysteines are not directly involved in substrate binding, but rather play a role in the catalytic step or product release.
Table 2.
Steady state kinetic parameters for wild-type CT and variants
| Construct | Vmax (min−1) | Km (biocytin, mM) | Km (malonyl-CoA, µM) |
|---|---|---|---|
| Wild-type CT | 211 ± 7 | 11.4 ± 0.4 | 40.0 ± 5.6 |
| C27A | 22.8 ± 1.4 | 11.5 ± 0.5 | 53.2 ± 9.2 |
| C30A | 1.5 ± 0.2 | 5.0 ± 1.1 | 20.2 ± 2.4 |
| C46A | 18.8 ± 0.7 | 4.0 ± 0.7 | 72.2 ± 10.4 |
| C49A | 26.7 ± 0.9 | 9.7 ± 0.6 | 106.8 ± 9.8 |
| C27/46A ‘double’ | 0 | ||
| C27/46/49A ‘triple’ | 0 | ||
| C27/30/46/49A ‘quadruple’ | 0 |
The apparent maximal velocities and Michaelis constant for biocytin were obtained by varying biocytin while holding malonyl-CoA constant at 100 µM. The apparent Michaelis constant for malonyl-CoA was obtained by varying malonyl-CoA while holding biocytin constant at 20 mM. The standard errors on Vmax and Km were determined from the nonlinear regression analysis.
The decrease in activity as a result of mutating the cysteines in the zinc domain explains earlier observations that the thiol modifying reagent N-ethylmaleimide (NEM) inactivated carboxyltransferase and that the substrate malonyl-CoA protected against inactivation (8). Reaction of NEM with the most active single-site variants (C27A, C46A and C49A) showed less susceptibility to NEM when normalized and compared to wild-type (data not shown), which is consistent with NEM modifying these cysteine residues in the zinc domain and in turn affecting enzymatic activity. In addition, the location of the zinc domain on the β-subunit (which contains the malonyl-CoA binding site) is consistent with malonyl-CoA protecting against inactivation.
Mutation of the cysteines in the zinc domain not only affected catalytic activity but also nucleic acid binding. An electrophoretic mobility shift assay (EMSA) was used to assess the effect of the mutations on CT binding to DNA (Figure 2). CT with single substitutions at positions 27, 46 and 49 appear to exhibit diminished DNA-binding capacity as evidenced by complex dissociation during electrophoresis, while the C30A variant and the multiple cysteine variants fail to form stable complexes with the DNA. Thus, the zinc-finger cysteines are required for DNA-binding, as expected.
Figure 2.

An ethidium bromide-stained 0.8% agarose gel image of DNA binding by CT and variants. Lanes 1–9 contain an 800 bp linear dsDNA with the following proteins: 1, no protein; 2, wtCT; 3, C27A; 4, C30A; 5, C46A; 6, C49A; 7, double variant; 8, triple variant; 9, quadruple variant.
Substitution of the cysteines likely altered the electrostatic structure of the zinc finger, resulting in the loss of zinc coordination. The effect of the mutations on the molar zinc content was determined using atomic absorption spectroscopy. Wild-type CT had two zinc atoms per CT tetramer as predicted from the structure. CT with single-site substitutions at cysteines 27, 46 and 49 contained 1.2–1.3 zinc atoms per CT tetramer, while the C30A variant had only 0.76 zinc atoms per CT tetramer. CT variants with multiple substitutions show progressively lower occupancies decreasing from 0.52 to 0.29 molar ratios. Circular dichroism spectroscopy was used to determine if the mutations had an effect on the overall secondary structure. CT variants C30A, C46A and C49A show spectra similar to wild-type CT (Supplementary Figure S1). The spectra for the C27A and variants with double and triple substitutions show successive changes in the 205–215 nm range; the spectrum of CT lacking all four cysteines has much more extensive changes ranging from 205 to as far as 240 nm. The effects of the mutations on catalysis and nucleic acid binding are likely due to loss of the zinc atom and alteration in secondary structure and they clearly demonstrate that the zinc domain is the structural feature of CT that links nucleic acid binding with catalysis.
Carboxyltransferase binds preferentially to the α- and β-subunit mRNA
Earlier DNA binding studies showed that DNA binding was non-specific and cooperative with half-maximal saturation of about 1 µM for a variety of DNA sequences, indicating that DNA binding was not likely to be the physiological role of the zinc domain of carboxyltransferase (7). However, considering that the closest structural homolog of the carboxyltransferase Zn-domain is the zinc domain from the 50S ribosomal protein L37Ae (6), the ability of carboxyltransferase to bind RNA was investigated. Four different types of RNA were tested for their ability to inhibit formation of the CT–DNA complex. The RNA molecules considered included mRNA coding for the α-subunit, mRNA coding for the β-subunit, a total RNA extract from E. coli, and tRNA from Saccharomyces cerevisiae. All four types of RNA were found to inhibit formation of the CT–DNA complex (data not shown). Notably, a qualitative comparison of the data showed that mRNA coding for the α- and β-subunits inhibited complex formation more effectively. A transcript including the α-subunit mRNA starting 213 nt upstream of the AUG through the end of the structural gene was therefore used in an EMSA (Figure 3). In contrast to the sigmoidal binding isotherm obtained for carboxyltransferase binding to DNA, fractional carboxyltransferase–RNA complex formation is best fit to a hyperbolic curve, yielding a half-maximal binding constant (Kd) of 145 ± 9 nM, which is about 7-fold lower than the half-maximal saturation for DNA binding. When the data were fitted to the Hill equation a Hill coefficient of ∼1 was calculated, indicating a lack of cooperativity (not shown). Moreover, the data were also analyzed according to Hensley et al. (12), which uses various linear transformations to detect a low degree of positive cooperativity. These analyses also indicate that the degree of cooperativity for subunit mRNA binding to carboxyltransferase is either very small or nonexistent. The tighter binding and hyperbolic binding isotherm for carboxyltransferase mRNA complex formation may suggest preferred binding to specific sites.
Figure 3.

(A) Electrophoretic analysis of the transcript from CT α-subunit titrated with CT. (A) All lanes contain 50 fmol 32P-labeled mRNA, while lanes 2–15 contain 1.0, 5.0, 10, 25, 40, 55, 70, 85, 100, 200, 300, 400, 500 and 600 nM CT. (B) Binding isotherm for CT binding to the transcript from CT α-subunit. The points represent averages from triplicate measurements. The error bars shown are the standard deviation. The curve represents the best fit of the data to Equation 1. No conclusions about the stoichiometry of binding can be made from this curve.
The CT binding site is within the α- and β-subunit mRNA coding region
To determine the region(s) of mRNA to which CT is binding, a series of linear DNA templates were constructed from which mRNAs of various lengths were transcribed using runoff transcription (Figure 4). Electrophoretic mobility shift assays were used to measure the Kd for each RNA construct binding to wild-type CT. Starting with the natural transcription start site of accA and ending with the stop codon, the 1183 nt transcript yielded a Kd of 136 ± 10 nM. Systematic truncation of the 3′-end by successive ∼300 nt deletions generated a 886 nt CT α RNA to which CT bound more tightly with a Kd of 48 ± 8 nM, while further truncation resulted in a marked decrease in the ability of CT to bind the RNA. The shortest transcripts consisting only of the 5′ UTR yielded no complex on incubation with CT. To confirm that the 5′ UTR region is not involved in CT binding, similar sized templates were made without the 5′ UTR and tested for CT binding. The longest transcript lacking the 5′ UTR but containing the entire α-subunit coding region (966 nt) bound to CT with a Kd of 250 ± 20 nM. The 684 nt transcript representing a 3′-truncation yielded a CT–RNA complex, however the fractional complex could not be measured due to the existence of numerous CT–RNA complexes comigrating with free RNA. The shortest transcript (328 nt) showed no ability to bind to CT.
Figure 4.

mRNA in vitro transcription products.
A series of templates similar to the ones described above for accA were also made for the accD coding region (Figure 4). The 5′ UTR was excluded from the accD transcripts because it was found not to play a role in CT binding to accA mRNA. The transcript (912 nt) containing the entire open reading frame of CT β had a Kd of 290 ± 20 nM, while truncating 228 nt from the 3′-end (684 nt transcript) resulted in a decrease in Kd to 83 ± 9 nM. This pattern for the two longest transcripts was also observed with accA. Moreover, just as with accA, shorter transcripts of accD bound CT only poorly.
Comparison of the α- and β-subunit mRNA regions required for CT binding reveals a striking similarity. For both subunits, 684 nt fragments beginning at the start sites were optimal. Within this sequence were 356 nt in α mRNA, and 335 nt in β mRNA that translate into regions of higher amino acid sequence homology (41%) compared to the alignment of the entire protein sequence (30%). This region of the mRNA of the α- and β-subunits will be hereafter referred to as the ‘symmetry box’. All RNA constructs from either subunit that excluded this region lacked the ability to bind CT, suggesting its contribution to enzyme recognition. Two additional templates were constructed consisting of only the regions from each gene representing this symmetry box to determine whether these regions are not only necessary, but sufficient for CT binding. Neither transcript had any affinity for CT, indicating that regions upstream of the symmetry box are also necessary for CT binding. The upstream region and the symmetry box must be connected for binding to CT because simply combining the two regions individually did not result in CT binding for either mRNA (data not shown).
Carboxyltransferase inhibits translation of α- and β-subunit mRNA
If CT binds to the mRNA coding for the α- and β-subunits, then does CT binding inhibit translation of the mRNA? This question was addressed using an in vitro coupled transcription/translation reaction. The mRNA for both α- andβ-subunits was generated by transcription from a plasmid containing the genes for both subunits. The reaction was carried out in the presence of 35S-labeled methionine, and products separated on a polyacrylamide gel and visualized by phosphorimaging (Figure 5). Increasing amounts of unlabeled wild-type CT added to the reaction prior to initiation by addition of plasmid progressively decreased the in vitro synthesis of CT (Figure 5, lanes 6–10). The decrease in expression in the presence of unlabeled CT is not due to an inhibition of transcription because CT also inhibited translation when an mRNA transcript was used to initiate expression (Figure 5A, lanes 3 and 4). The inhibition of translation was partially reversed by the addition of the substrate acetyl-CoA, indicating a reversible competition between mRNA and substrate and consistent with the notion that nucleic acid binding and catalysis are mutually exclusive (Figure 5A, lanes 11–15). To ensure that CT binding to α mRNA does not affect translation of the β-subunit, the ability of CT to inhibit translation of the β-subunit mRNA was examined. The coupled transcription/translation reaction from a plasmid containing only the accD gene showed that CT does indeed inhibit translation of the β-subunit mRNA (Figure 5B, lanes 4–7). The inhibition was again relieved by the substrate acetyl-CoA (Figure 5B, lanes 8–11). The inhibition of translation was specific for the mRNA coding for the α- and β-subunits because CT did not inhibit translation of the gene coding for the 32 kDa elongation factor EF-Ts which was provided as a control in the in vitro coupled transcription/translation reaction (Figure 5C). Reinforcing the role of the zinc domain in nucleic acid binding, addition of the quadruple variant of CT had no effect on the level of translation (data not shown.).
Figure 5.

Five percent denaturing polyacrylamide gels of in vitro coupled transcription/translation reactions expressing 35S-labeled proteins. (A) Lane 1, no DNA template. Lane 2, positive control test plasmid encoding the 32 kDa elongation factor EF-Ts protein. Lane 3, polycistronic mRNA generated in vitro from pCZB5 plasmid showing CT α (37 kDa) and CT β (33 kDa). Lane 4, same as lane 3 with the addition of 166nM unlabeled wild-type CT to the transcription/translation reactions. Lanes 5–15 contain 1 µg (∼10 nM) pCZB5 plasmid. Lanes 5–10 contain 0, 63, 125, 250, 500 and 1000 nM unlabeled wild-type CT. Lanes 10–15 contain 1 µM unlabeled wild-type CT and acetyl-CoA at 0, 88, 175, 350, 700 and 1400 µM. (B) Lane 1, no DNA template. Lane 2, positive control test plasmid showing a 32kDa protein. Lane 3–11 contain 1 µg (∼10nM) pGLW39 plasmid. Lanes 3–7 contain 0, 63, 125, 250 and 1000 nM unlabeled wild-type CT. Lanes 7–11 contain 1 µM unlabeled wild-type CT and acetyl-CoA at 0, 175, 350, 700 and 1400 µM. (C) Lane 1, no DNA template. Lanes 2–8 contain positive control test plasmid as in (A), lane 2. Lanes 3–8 contain 31, 63, 125, 250, 500 and 1000 nM unlabeled wild-type CT.
To confirm that acetyl-CoA relieved inhibition of translation by reversible competition between mRNA and the substrate acetyl-CoA, inhibition of CT–RNA complex formation by acetyl-CoA was examined. The transcript (912 nt) containing the entire open reading frame of CT β was incubated with 166 nM CT and increasing amounts of acetyl-CoA (Figure 6A). With increasing [acetyl-CoA], a concomitant decrease in CT–RNA complex formation is evident. This demonstrates reversible competition between mRNA and substrate. Conversely, since the substrate acetyl-CoA inhibited the binding of CT to RNA, and given that catalysis and nucleic acid binding in CT are linked through the zinc domain, RNA should inhibit catalysis as well. An 1183 nt CT α mRNA was found to inhibit CT activity in a dose-dependent manner, with nearly a 4-fold drop in activity at 90 nM (Figure 6B). As a control, CT retained >80% activity in the presence of a 90 nM 1040 nt mRNA coding for EF-Ts (Figure 6B); that EF-Ts inhibits CT activity at higher concentrations is consistent with the nonspecific binding to nucleic acids reflected in DNA binding. These results confirm that mRNA binding to CT inhibits enzymatic activity.
Figure 6.

(A) Electrophoretic analysis of the transcript from CT β-subunit titrated with CT. All lanes contain 50 fmol 32P-labeled mRNA, lane 1 contains only mRNA, while lanes 2–14 contain 166 nM CT and acetyl-CoA at 0, 20, 50, 100, 200, 300, 400, 500, 600, 750, 1000, 2000 and 4000 µM. (B) RNA inhibition of CT activity. Circles represent averages from triplicate measurements for reactions containing the 1040 nt EF-Ts transcript at 0, 11.3, 22.6, 45.3 and 90.6 nM. Triangles represent averages from triplicate measurements for reactions containing the 1183 nt CT α transcript at 0, 11.6, 23.2, 46.4 and 92.8 nM. The error bars shown are the standard deviation. Note that enzymatic reactions contain only 1 mM KCl, conditions under which the affinity of CT for RNA is higher than that determined from EMSA assays.
CT–RNA interaction occurs in situ
CT binds to RNA in vitro, but does CT interact with RNA in the context of a cell? To address this issue, a FRET (fluorescence resonance energy transfer) assay was used. The assay is based on the work of Lorenz (13) and utilized eYFP (enhanced yellow fluorescent protein) fused to wildtype or the quadruple variant of CT β as the donor and the nucleic acid stain Sytox Orange as the acceptor. Cells containing plasmids where the genes for CT α and either wildtype CT β fused to eYFP or quadruple variant CT β fused to eYFP were induced to express the CT α and CT β-eYFP fusion proteins. After 0–3 h, samples were freeze-thawed followed by various nuclease digestions. Sytox Orange was added to the treated cells and fluorescence was measured to determine proximity of CT β-eYFP to nucleic acid-bound stain. As shown in Figure 7 a FRET signal was observed only after induction of protein expression (blue trace). The maximal signal occurred 1 h after induction with no change up to 3 h post-induction. Little to no signal was detected prior to induction (purple trace). Since Sytox Orange can also bind to DNA, cells were treated with DNase or RNase in order to confirm the signal is a result of RNA interaction. Treatment with DNase I had little effect on the fluorescence signal (red trace); however, digestion with RNase A attenuated the signal to near pre-induction levels (green trace). The quadruple variant-eYFP fusion protein showed no FRET signal consistent with its inability to bind nucleic acids (orange trace). These data indicate that in situ the primary nucleic acid binding target of CT is RNA. While these results show that CT prefers to bind to RNA over DNA in the cell, they do not address the question of RNA specificity. As a control, the CT β-eYFP fusion protein was expressed, purified, and shown in vitro to exhibit fluorescence with 500 nm incident light. The emission peak at 547 nm drops significantly with a concomitant appearance of a new emission peak at 574 nm upon addition of Sytox Orange to the sample (data not shown). This indicates a functional donor/acceptor pair for FRET with an excitation wavelength of 500 nm.
Figure 7.

FRET emission scan showing absorption peak generated by incident light at 500 nm. Blue trace, wild-type CT fused to eYFP, 1 h post-induction, not subjected to nucleic acid digestion. Purple trace, wild-type CT fused to eYFP, pre-induction, not subjected to nucleic acid digestion. Red trace, wild-type CT fused to eYFP, 1 h post-induction, treated with DNase. Green trace, wild-type CT fused to eYFP, 1 h post-induction, treated with RNase. Orange trace, quadruple variant CT fused to eYFP, 1 h post-induction, not subjected to nucleic acid digestion.
DISCUSSION
The zinc domain is required for enzymatic activity and translational repression
Site-directed mutagenesis of the zinc domain conclusively establishes that it is the structural link between nucleic acid binding and catalysis. The coupling of nucleic acid binding to catalysis implies a selective pressure to maintain the zinc domain on the β-subunit and that the ability to bind nucleic acids has a specific purpose; consistent with this inference, we find that CT binds RNA in situ. Notably, in vitro analyses demonstrate that CT preferentially binds the mRNA encoding its own α- and β-subunits, implying the existence of an uncommon regulatory mechanism.
The binding of CT to the mRNA coding for the α- and β-subunits inhibited translation in a dose-dependent manner, while the substrate acetyl-CoA inhibited RNA binding and relieved the inhibition of translation also in a dose-dependent manner. Consistent with the coupled nature of nucleic acid binding and catalysis by CT, the mRNA also inhibited catalysis. These findings indicate a physiological role for the zinc domain in regulating not only production of the α- and β-subunits of CT, but also its enzymatic activity. The basic premise of the model of regulation illustrated in Figure 8 is that in bacteria, fatty acids are only used for membrane biogenesis, hence carboxyltransferase is only needed when nutrients are abundant. In contrast, during the stationary phase of E. coli growth where nutrients are limited, carboxyltransferase would act as a ‘dimmer switch’ on the expression of the α- and β-subunits by binding to the mRNA and inhibiting translation. Reciprocally, RNA binding by CT renders it catalytically inactive, limiting unneeded and wasteful conversion of acetyl-CoA to malonyl-CoA. During the growth phase when nutrients are abundant, the level of acetyl-CoA would increase dramatically and compete with the mRNA for binding to carboxyltransferase. This alleviates both the inhibition of catalytic activity and the repression of translation characteristic of stationary phase, leading to increased levels of CT and ultimately fatty acids. The levels of acetyl-CoA in E. coli during the log phase (600 µM) and stationary phase (20 µM) (14) are in accord with the data in Figure 5A, because relief of translational repression was observed at 350 µM acetyl-CoA, whereas very little protein production was observed at 88 µM acetyl-CoA. This model of regulation is also consistent with reports that levels of the mRNA coding for the α- and β-subunits do not change appreciably between the log and stationary phases (15,16).
Figure 8.

Proposed mechanism for autoregulation of CT enzymatic activity and mRNA translation. The RNA-binding and catalytic functions of CT are sensitive to the metabolic state of the cell. During stationary phase when de novo fatty acid synthesis is unneeded, glucose and thus acetyl-CoA levels are low, allowing CT to bind the mRNA for its subunits, attenuating expression of the enzyme as well as enzymatic activity on remaining acetyl-CoA stores. During log phase growth membrane biogenesis is needed. Glucose is abundant, increasing the availability of acetyl-CoA for fatty acid synthesis. Acetyl-CoA binds to CT permitting translation of the mRNA for the α- and β-subunits. This ‘dimmer switch’ model gives the cell a means of rapid response to changes in cellular metabolic state.
The model of regulation in Figure 8 also provides a plausible explanation for how the cell maintains stoichiometric amounts of the α- and β-subunits. Instead of transcriptional control of gene expression of the genes for the α- and β-subunits, bacteria appear to utilize translational repression where CT binds to the mRNA for both of its subunits.
The mechanism of gene regulation proposed here is not without precedent. For example, thymidylate synthase from both E. coli and humans contains a Cys4-type zinc-binding motif and binds its own mRNA within the protein-coding region, repressing translation (17–19). Also, it is important to note that this type of regulation is not found in eukaryotic carboxyltransferase because the α- and β-subunits are fused to one another as well as to the BC and BCCP components to form one polypeptide chain and because the zinc domain is absent (20). The one exception to the multidomain homomeric ACCase found in eukaryotes is the heteromeric ACCase found in plastids. The chloroplast ACCase of all plants except for the grasses (Graminae) consists of the same four subunits as E. coli ACCase, but their genetic origins are different (21). The BC, BCCP and CT α peptides originate from nuclear-encoded genes and are shuttled to the chloroplast (22–24). The CT β protein, however, is expressed from a chloroplast-encoded gene, and like the E. coli accD gene, encodes a Cys4-type zinc-binding domain, which is consistent with the theory that chloroplasts evolved from bacterial symbiosis (24). Perhaps the zinc domain in chloroplast CT β binds to the ;β mRNA to regulate translation in a manner similar to the model presented here for E. coli CT.
A symmetry-based model
The fundamental tenet of the gene regulation model presented in Figure 8 rests on the ability of CT to bind the mRNA encoding both α- and β-subunits. The molecular basis of how it does so is rooted in the symmetry of the chemical mechanism and 3D structure of CT. Transfer of the carboxyl group from carboxybiotin to acetyl-CoA involves formation of enolate or enolate-like anions in both carboxybiotin and acetyl-CoA. To this end, both the α- and β-subunits have evolved tertiary folds that stabilize oxyanions. Acetyl-CoA binds to the β-subunit, while carboxybiotin binds to the α-subunit and the tertiary structure of both the α- and β-subunits contain a similar α/β spiral core of two β-sheets surrounded by α-helices suggesting a gene duplication event. This type of fold places carboxyltransferase in the crotonase superfamily of enzymes whose members all catalyze reactions that generate enolate anions (25,26). Within the spiral core of both the α- and β-subunits of CT are oxyanion holes formed by main chain amides from glycine residues (6). The regions of the mRNA for the α- and β-subunits that bind to CT also reveal a symmetrical pattern. CT binds only to the coding region of the mRNA of the α- and β-subunits, a region that included the symmetry box, which corresponds to the spiral core of the tertiary structure containing the oxyanion holes. While the symmetry box is required for CT binding, it is not sufficient. The mRNA upstream of the symmetry box is also needed for binding, probably to achieve the proper 3D fold of mRNA to interact with CT. This observation is consistent with the notion that mRNA sequences close to the start site are more likely to be involved in regulatory events. Thus, while there are numerous examples of how the chemistry of a reaction drives the evolution of enzyme structure, the results presented here for carboxyltransferase not only suggest a physiological role for the zinc domain but also extend the concept of chemistry directing enzyme evolution to also include directing the mechanism of gene regulation.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Funding for open access charge: National Science Foundation (MCB-0841134 to G.L.W.).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Dr N. Kato for the plasmid containing the eYFP gene. We thank F. Kizilkaya for assistance with the Zn atomic absorption spectroscopy measurements.
REFERENCES
- 1.Cronan JE, Jr, Waldrop GL. Multi-subunit acetyl-CoA carboxylases. Prog. Lipid Res. 2002;41:407–435. doi: 10.1016/s0163-7827(02)00007-3. [DOI] [PubMed] [Google Scholar]
- 2.Li SJ, Cronan JE., Jr Growth rate regulation of Escherichia coli acetyl coenzyme A carboxylase, which catalyzes the first committed step of lipid biosynthesis. J. Bacteriol. 1993;175:332–340. doi: 10.1128/jb.175.2.332-340.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.James ES, Cronan JE. Expression of two Escherichia coli acetyl-CoA carboxylase subunits is autoregulated. J. Biol. Chem. 2004;279:2520–2527. doi: 10.1074/jbc.M311584200. [DOI] [PubMed] [Google Scholar]
- 4.Li SJ, Cronan JE., Jr The genes encoding the two carboxyltransferase subunits of Escherichia coli acetyl-CoA carboxylase. J. Biol. Chem. 1992;267:16841–16847. [PubMed] [Google Scholar]
- 5.Blattner FR, Plunkett G., III, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1474. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 6.Bilder P, Lightle S, Bainbridge G, Ohren J, Finzel B, Sun F, Holley S, Al-Kassim L, Spessard C, Melnick M, et al. The structure of the carboxyltransferase component of acetyl-CoA carboxylase reveals a zinc-binding motif unique to the bacterial enzyme. Biochemistry. 2006;45:1712–1722. doi: 10.1021/bi0520479. [DOI] [PubMed] [Google Scholar]
- 7.Benson BK, Meades G., Jr, Grove A, Waldrop GL. DNA inhibits catalysis by the carboxyltransferase subunit of acetyl-CoA carboxylase: implications for active site communication. Protein Sci. 2008;17:34–42. doi: 10.1110/ps.073186408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blanchard CZ, Waldrop GL. Overexpression and kinetic characterization of the carboxyltransferase component of acetyl-CoA carboxylase. J. Biol. Chem. 1998;273:19140–19145. doi: 10.1074/jbc.273.30.19140. [DOI] [PubMed] [Google Scholar]
- 9.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 10.Cleland WW. Statistical analysis of enzyme kinetic data. Methods Enzymol. 1979;63:103–138. doi: 10.1016/0076-6879(79)63008-2. [DOI] [PubMed] [Google Scholar]
- 11.Kozaki A, Mayumi K, Sasaki Y. Thiol-disulfide exchange between nuclear-encoded and chloroplast-encoded subunits of pea acetyl-CoA carboxylase. J. Biol. Chem. 2001;276:39919–39925. doi: 10.1074/jbc.M103525200. [DOI] [PubMed] [Google Scholar]
- 12.Hensley P, Yang YR, Schachman HK. On the detection of homotropic effects in enzymes of low co-operativity. Application to modified aspartate transcarbamoylase. J. Mol. Biol. 1981;152:131–152. doi: 10.1016/0022-2836(81)90098-x. [DOI] [PubMed] [Google Scholar]
- 13.Lorenz M. Visualizing protein-RNA interactions inside cells by fluorescence resonance energy transfer. RNA. 2009;15:97–103. doi: 10.1261/rna.1307809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takamura Y, Nomura G. Changes in the intracellular concentration of acetyl-CoA and malonyl-CoA in relation to the carbon and energy metabolism of Escherichia coli K12. J. Gen. Microbiol. 1988;134:2249–2253. doi: 10.1099/00221287-134-8-2249. [DOI] [PubMed] [Google Scholar]
- 15.Sundararaj S, Guo A, Habibi-Nazhad B, Rouani M, Stothard P, Ellison M, Wishart DS. The CyberCell Database (CCDB): a comprehensive, self-updating, relational database to coordinate and facilitate in silico modeling of Escherichia coli. Nucleic Acids Res. 2004;32(Database issue):D293–D295. doi: 10.1093/nar/gkh108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Selinger DW, Cheung KJ, Mei R, Johansson EM, Richmond CS, Blattner FR, Lockhart DJ, Church GM. RNA expression analysis using a 30 base pair resolution Escherichia coli genome array. Nat. Biotechnol. 2000;18:1262–1268. doi: 10.1038/82367. [DOI] [PubMed] [Google Scholar]
- 17.Voeller DM, Changchien L, Maley GF, Maley F, Takechi T, Turner RE, Montfort WR, Allegra CJ, Chu E. Characterization of a specific interaction between Escherichia coli thymidylate synthase and Escherichia coli thymidylate synthase mRNA. Nucleic Acids Res. 1995;23:869–875. doi: 10.1093/nar/23.5.869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chu E, Voeller D, Koeller DM, Drake JC, Takimoto CH, Maley GF, Maley F, Allegra CJ. Identification of an RNA binding site for human thymidylate synthase. Proc. Natl Acad. Sci. USA. 1993;90:517–521. doi: 10.1073/pnas.90.2.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chu E, Koeller DM, Casey JL, Drake JC, Chabner BA, Elwood PC, Zinn S, Allegra CJ. Autoregulation of human thymidylate synthase messenger RNA translation by thymidylate synthase. Proc. Natl Acad. Sci. USA. 1991;88:8977–8981. doi: 10.1073/pnas.88.20.8977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bai DH, Pape ME, Lopez-Casillas F, Luo XC, Dixon JE, Kim KH. Molecular cloning of cDNA for acetyl-coenzyme A carboxylase. J. Biol. Chem. 1986;261:12395–12399. [PubMed] [Google Scholar]
- 21.Nikolau BJ, Ohlrogge JB, Wurtele ES. Plant biotin-containing carboxylases. Arch. Biochem. Biophys. 2003;414:211–222. doi: 10.1016/s0003-9861(03)00156-5. [DOI] [PubMed] [Google Scholar]
- 22.Bao X, Shorrosh BS, Ohlrogge JB. Isolation and characterization of an Arabidopsis biotin carboxylase gene and its promoter. Plant Mol. Biol. 1997;35:539–550. doi: 10.1023/a:1005881006620. [DOI] [PubMed] [Google Scholar]
- 23.Sun J, Ke J, Johnson JL, Nikolau BJ, Wurtele ES. Biochemical and molecular biological characterization of CAC2: the Arabidopsis thaliana gene coding for the biotin carboxylase subunit of plastidic acetyl-coenzyme A carboxylase. Plant Physiol. 1997;115:1371–1383. doi: 10.1104/pp.115.4.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ke J, Wen T, Nikolau BJ, Wurtele ES. Coordinate regulation of the nuclear and plastidic genes coding for the subunits of the heteromeric acetyl-coenzyme A carboxylase. Plant Physiol. 2000;122:1057–1071. doi: 10.1104/pp.122.4.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holden HM, Benning MM, Haller T, Gerlt JA. The Crotonase superfamily: divergently related enzymes that catalyze different reactions involving acyl coenzyme A thioesters. Acc. Chem. Res. 2001;34:145–157. doi: 10.1021/ar000053l. [DOI] [PubMed] [Google Scholar]
- 26.Hamed RB, Batchelar ET, Clifton IJ, Schofield CJ. Mechanisms and structures of crotonase superfamily enzymes – How nature controls enolate and oxyanion reactivity. Cell. Mol. Life Sci. 2008;65:2507–2527. doi: 10.1007/s00018-008-8082-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.