

Abstract
Modulation of host immune responses is a common strategy for promoting virus persistence and avoiding clearance. Human cytomegalovirus (HCMV) is known to encode numerous immunomodulatory genes, including a homolog of the cytokine human interleukin-10 (hIL-10). While having limited sequence homology to hIL-10, cytomegalovirus IL-10 (cmvIL-10) shares many functional characteristics with the human cytokine and acts as a potent suppressor of the inflammatory immune response. The mechanism by which hIL-10 inhibits inflammatory cytokines involves a transcriptional block via inhibition of nuclear factor-κB (NF-κB) activity. To determine whether cmvIL-10 employs the same mechanism to inhibit cytokine production, the effect of cmvIL-10 on NF-κB signaling in monocytes was investigated. The results demonstrate that cmvIL-10 does inhibit NF-κB activation, as evidenced by reduced degradation of the NF-κB inhibitor IκB-α, and decreased transcription of the NF-κB–responsive genes tumor necrosis factor-α (TNF-α) and IL-1β. These studies confirm that cmvIL-10 mediates cytokine suppression by blocking NF-κB transcriptional activity in human monocytes.
Human cytomegalovirus (HCMV) is a member of the Herpesviridae family and is ubiquitous in the population (2). HCMV infection is generally asymptomatic in immunocompetent individuals; however, severe and often fatal conditions may occur in the immunocompromised, including the developing fetus, transplant recipients, or HIV-infected individuals (9,14,32). Although the immune system has numerous mechanisms for clearing virus infection, HCMV has countered with its own immune evasion adaptations. In particular, the virus encodes a homolog of the immune suppressive cytokine human interleukin-10 (hIL-10). The viral cytokine cytomegalovirus IL-10 (cmvIL-10) binds to the cellular IL-10 receptor (18,19), triggering effects that include reduced cell proliferation, inhibition of cytokine synthesis, downregulation of MHC molecules (39), and impairment of dendritic cell maturation (7,30). When secreted from infected cells, cmvIL-10 has the potential to cause widespread suppressive effects on multiple types of uninfected immune cells.
The ability of hIL-10 to decrease cytokine expression has been found to involve inhibition of the transcription factor nuclear factor-κB (NF-κB) (12,20,40). The NF-κB family of dimeric transcription factors mediates cellular responses to a wide variety of stimuli. The functional transcription factors consist of homo- and heterodimers from the NF-κB protein family, which includes five proteins: RelA/p65, c-Rel, RelB, p50, and p52 (36). Each of these proteins contains a Rel homology domain, which contains a DNA binding domain, a dimerization domain, and a nuclear localization signal. While any combination of subunits is possible, the predominant form of NF-κB mediating transcriptional activation in most cell types is the p65:p50 heterodimer (36). In unstimulated cells, NF-κB dimers are inactivated by the binding of an inhibitor, IκB-α, which masks the nuclear localization signal, blocks DNA binding, and promotes nuclear export of bound dimers (21,41). Most immune stimuli activate NF-κB by inducing the degradation of the inhibitor IκB-α. This degradation is triggered when IκB-α is phosphorylated by IκB kinase (IKK), and immediately after phosphorlyation, IκB-α undergoes rapid polyubiquitylation and is destroyed by the proteasome. The degradation of IκB-α frees NF-κB, enabling the dimer to enter into the nucleus, bind to DNA, and induce gene expression associated with inflammatory immune responses.
Previous studies have shown that hIL-10 treatment of monocytes leads to inhibition of NF-κB activity (12). cmvIL-10 has also been shown to inhibit cytokine production by monocytes (39), but the complete mechanism for this inhibition is unknown. Although previous studies have identified a role for phosphoinositide-3 kinase signaling in cmvIL-10–mediated cytokine suppression, inhibition of phosphoinositide-3 kinase activity only partially restored the cytokine levels (37), indicating that cmvIL-10 must stimulate other pathways or use multiple mechanisms to modulate cytokine expression. In order to determine whether cmvIL-10 might modulate NF-κB activity, monocytes were treated with the viral cytokine and levels of IκB-α were analyzed. THP-1 monocytes were first incubated with lipopolysaccharide (LPS) for 3 h, and then cell lysates were analyzed by Western blot. As shown in Fig. 1A, the 41-kD IκB-α inhibitor protein is present in untreated cells, but the amount of this protein is greatly reduced in cells that have been exposed to a potent immune stimulus such as LPS. The lack of this protein in lysates of LPS-treated cells is consistent with the rapid degradation of IκB-α known to accompany NF-κB activation. When THP-1 cells were pretreated with hIL-10 for 1 h prior to LPS exposure, no degradation of IκB-α was observed. Likewise, cmvIL-10 also functioned to inhibit IκB-α degradation. When THP-1 monocytes were treated with cmvIL-10 prior to LPS stimulation, levels of the inhibitor IκB-α remained high and NF-κB remained inactive. Stat3, which has previously been found to be present in THP-1 cells at relatively constant levels (37), was also detected via immunoblotting and served as a protein loading control. Although the accumulation of IκB-α was most pronounced when cells were treated with 100 ng/mL of cmvIL-10, increased IκB-α protein concentrations were also noted in lysates from cells treated with 1 or 20 ng/mL of cmvIL-10, suggesting that the inhibitory effect of the viral cytokine on NF-κB activity was dose-dependent.
FIG. 1.
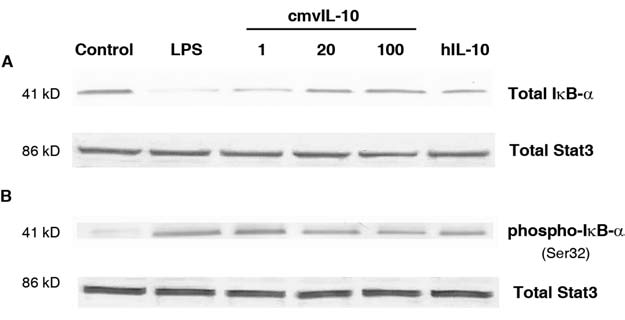
cmvIL-10 prevents degradation of IκB-α. THP-1 cells were treated with purified recombinant cmvIL-10 (1, 20, or 100 ng/mL; R&D Systems, Minneapolis, MN) or hIL-10 (100 ng/mL; R&D Systems) for 1 h prior to stimulation with 10 ng/mL LPS (Sigma, St. Louis, MO). The total protein in each lysate was quantified using the Bio-Rad Protein Assay Kit (Hercules, CA), and 10 μg total protein was loaded into each well. The lysates were resolved by SDS-PAGE and analyzed by immunoblotting with polyclonal antiserum to either (A) total IκB-α or (B) phosphorylated IκB-α (phospho-IκB-α) (Cell Signaling Technologies, Danvers, MA). In each case, total Stat3 protein was detected via blotting with polyclonal antiserum to Stat3 (Cell Signaling Technologies) to ensure equal sample loading. When detecting phosphorylated IκB-α, cell treatments were performed in the presence of 20 mM lactacystin (Sigma) to inhibit proteasome-mediated degradation. Alkaline phosphatase-conjugated goat anti-rabbit secondary antibody (Anaspec, San Jose, CA) and Western Blue Substrate (Promega, Madison, WI) were utilized for detection. The blots shown are representative of three independent experiments.
Although these studies were conducted in THP-1 cells, the same result was observed with primary monocytes. Monocytes were isolated from human blood of two different donors by magnetic separation with anti-CD14+ microbeads (37). The primary monocytes were then exposed to LPS with or without pre-incubation with cmvIL-10. Immunoblotting revealed that IκB-α degradation was inhibited in these cells (data not shown), suggesting that cmvIL-10 suppresses NF-κB activation in both primary monocytes and monocytic cell lines.
Because cmvIL-10 treatment prevented the degradation of IκB-α, it was expected that levels of phosphorylated IκB-α would also be lower in cells exposed to the viral cytokine. To examine this, Western blot analysis of cell lysates was performed with an antibody to detect IκB-α that had been phosphorylated on serine residue 32, the site frequently modified by IKK (22). Detection of phosphorylated IκB-α protein required treatment of the cells with lactacystin, a proteosome inhibitor, in order to prevent rapid degradation of the phosphorylated form of the inhibitor. THP-1 monocytes were incubated with cmvIL-10 and lactacystin for 1 h prior to LPS stimulation, and then cell lysates were immunoblotted. As expected, there was no phosphorylated IκB-α present in unstimulated cells (Fig. 1B). In contrast, LPS treatment induced phosphorylation of IκB-α, which accumulated in the cytoplasm and was not degraded in the presence of the proteasome inhibitor. Pre-incubation with cmvIL-10 inhibited LPS-induced phosphorylation of IκB-α on serine 32, and this inhibition occurred in a dose-dependent manner. hIL-10 also inhibited phosphorylation of IκB-α, suggesting that the viral cytokine mimics the ability of the human cytokine to modulate this signaling pathway. The reduction in phosphorylated IκB-α levels suggests that hIL-10 and cmvIL-10 prevent NF-κB activation by inhibiting the activity of the IκB kinase, IKK. This result is consistent with previous reports that suppression of IKK function was the mechanism by which hIL-10 blocked NF-κB transcriptional activity (34).
Having determined that cmvIL-10 impacts NF-κB by preventing degradation of the inhibitor IκB-α, evidence of reduced NF-κB activity was investigated next. To this end, transcription of NF-κB–regulated cytokine genes was evaluated at the mRNA level using reverse transcriptase PCR. The inflammatory genes TNF-α and IL-1β were investigated because they are known to contain NF-κB response elements in their promoters (8,16,35). In order to determine whether cmvIL-10 altered NF-κB–regulated gene expression of TNF-α and IL-1β, THP-1 cells were incubated with 100 ng/mL cmvIL-10 or hIL-10 for 1 h prior to LPS stimulation. Total RNA was harvested from the treated monocytes and then reverse transcribed to cDNA. The cDNA template was then amplified with gene-specific primers and resulting bands were resolved via gel electrophoresis. As shown in Fig. 2A, only faint bands were observed in samples from untreated cells, indicating that THP-1 monocytes had very low basal levels of expression of TNF-α and IL-1β. Treatment of THP-1 cells with LPS resulted in a dramatic increase in mRNA levels for both TNF-α and IL-1β, as evidenced by the intense bands in the second lane of Fig. 2A. The LPS-induced TNF-α and IL-1β mRNA expression in THP-1 monocytes was inhibited by both cmvIL-10 and hIL-10, as indicated by decreased band intensity. β-Actin served as a loading control, and equivalent bands were observed for all samples.
FIG. 2.
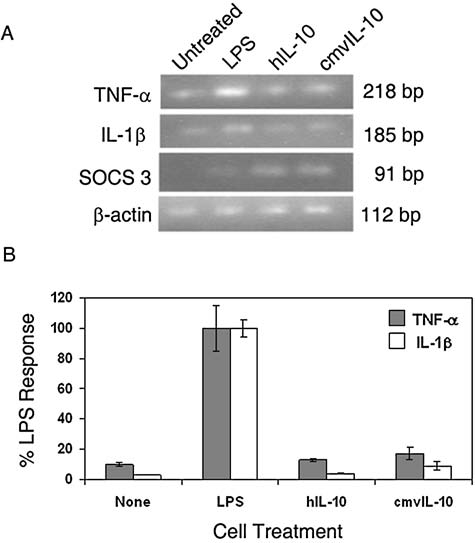
cmvIL-10 inhibits transcription of inflammatory cytokines. (A) THP-1 monocytes were treated with 100 ng/mL cmvIL-10 or hIL-10 prior to stimulation with 10 ng/mL LPS for 3 h. Total cellular RNA was isolated (RNEasy Midi Kit; Qiagen, Valencia, CA), reverse transcribed, and amplified with gene-specific primers. PCR conditions included an initial denaturation at 94°C for 1 min and then 30 cycles of 94, 60, and 72°C (30 sec each). PCR products were resolved by gel electrophoresis using a 2% agarose gel. The gel shown is representative of two independent experiments (SOCS 3, suppressor of cytokine signaling protein 3). (B) Real time quantitative PCR analysis was performed using prepared cDNA templates, gene-specific primers, and SYBR Green Buffer (BioRad, Hercules, CA) in a 20-μL reaction mixture. The forward and reverse primers were as follows: for β-actin: AAGAGAGGCATCCTCACCCT (f), TACATGGCTGGGGTGTTGAA (r); for TNF-α: AACCTCCTCTCTGCCATCAA (f), CCAAAGTAGACCTGCCCAGA (r); for IL-1β: TCCCCAGCCCTTTTGGA (f), TTAGAACCAAATGTGGCCGTG (r). The primers for SOCS 3 were obtained from Qiagen (QuantiTect Primer Assay). The Livak comparative 2−ΔC(t) method was used to calculate relative gene expression and measure fold differences in target nucleic acid between samples. The C(t) values of target genes TNF-α and IL-1β were normalized to the housekeeping gene β-actin and the ΔC(t)Average was calculated from three replicates, then expressed as a percentage of total LPS-induced levels of cytokine. Error bars represent standard error among replicates. These data are representative of three independent experiments.
In order to determine the extent of transcriptional repression mediated by cmvIL-10, real time quantitative PCR (RT-qPCR) was employed. For this analysis, cDNA templates were amplified in reactions containing SYBR Green Buffer, and the expression levels were normalized to the housekeeping gene β-actin. Compared to untreated control cells, LPS stimulation caused an 80-fold increase in expression of TNF-α. Treatment with cmvIL-10 led to an 88% decrease in LPS-induced TNF-α gene expression, and treatment with hIL-10 led to an 82% decrease (Fig. 2B). No statistically significant difference was observed between cmvIL-10 and hIL-10 with regard to suppression of TNF-α gene expression, as measured by the Student-Neuman-Keuls test.
Likewise, IL-1β levels were analyzed via RT-qPCR, and LPS induced a 200-fold increase in IL-1β expression in comparison with control cells. LPS-induced IL-1β gene expression was found to be inhibited by 87% by cmvIL-10 treatment, and by 99% by treatment with hIL-10 (Fig. 2B). Again, this difference was not found to be statistically significant, suggesting that both cytokines effectively inhibit IL-1β transcription. These results show that cmvIL-10 employs the same mechanism for immune suppression as hIL-10, and that the viral cytokine is equal to the cellular cytokine in repression of TNF-α and IL-1β gene expression. These results are also in agreement with previous studies that analyzed inflammatory cytokine protein levels by ELISA, and indicated that the cmvIL-10 and hIL-10 both inhibit inflammatory cytokine secretion to the same extent (37,39).
Because cmvIL-10 has been documented to have primarily inhibitory effects, the study focused on reduction of LPS-stimulated cytokine expression. However, there was also the possibility that cmvIL-10 treatment alone might directly induce gene expression. SOCS 3 (suppressor of cytokine signaling protein 3) expression is known to be upregulated by hIL-10 (27–29), and so it seemed likely that this gene might also be affected by cmvIL-10. As shown in Fig. 2A, no expression of SOCS 3 was detected in untreated THP-1 monocytes, and LPS stimulation did not induce a change in SOCS 3 gene expression. In contrast, treatment with either hIL-10 or cmvIL-10 did result in significant upregulation of SOCS3 expression in THP-1 cells, as evidenced by the appearance of the 91-bp band in the third and fourth lanes of Fig. 2A. These results demonstrate that not only does cmvIL-10 suppress transcription of pro-inflammatory cytokines, but the viral cytokine can directly enhance the transcription of negative immune regulators like SOCS 3. The SOCS protein family is involved in regulation of cytokine signaling via negative feedback regulation of JAK-STAT pathways. Upon binding to the cellular IL-10 receptor, cmvIL-10 has been shown to activate JAK1, which then phosphorylates and activates Stat3 (31,37). Several studies have found that SOCS 3 controls the kinetics of decay of STAT3 phosphorylation (13,26). This supports the notion that SOCS 3 may play a role in determining the duration and intensity of cmvIL-10–mediated immune suppression.
HCMV infection is characterized by latent lifelong infection, and manipulation of host immunity permits the virus to avoid complete clearance. One immune evasion strategy utilized by HCMV is hijacking of the robust anti-inflammatory effects of hIL-10. This immune suppression is accomplished by cmvIL-10, the virally-encoded homolog of the cellular cytokine. While the potent anti-inflammatory effects of cmvIL-10 have been established (7,19,30,37,39), this is the first report showing that these effects are mediated through direct inhibition of NF-κB transcriptional activity. Our results demonstrate that cmvIL-10 treatment prevents the phosphorylation and degradation of the inhibitor IκB-α, which remains bound to NF-κB subunits in the cytoplasm and prevents their entry into the nucleus. This effect is most likely mediated by inhibition of IKK activity, which is consistent with previous reports that hIL-10 blocks IKK activation and inhibits DNA binding of NF-κB p65:p50 heterodimers (34). Interestingly, subsequent investigations also showed that hIL-10 actually promotes nuclear translocation of p50 subunits (12). In contrast to other NF-κB family members, p50 subunits lack a transactivation domain. As a result, p50 heterodimers, which retain the ability to bind NF-κB sites, are considered to be transcriptional repressors (23), further explaining the inhibitory effects of hIL-10 on NF-κB–regulated gene expression. Ironically, recent studies have shown that p50 homodimers can actually promote transcription of IL-10 in mice, and a novel p50-responsive cis-element was identified in the promoter of the murine IL-10 gene (5). Future investigations are necessary to determine whether cmvIL-10 also promotes the translocation of p50 homodimers to the nucleus; however, it is notable that cmvIL-10 was found to induce expression of hIL-10 in dendritic cells (6) and B lymphocytes (38).
It is important to note that cmvIL-10 inhibition was not specific for LPS-induced NF-κB activation. THP-1 cells stimulated with either PMA (phorbol 12-myristate 13-acetate; Sigma) or TNF-α (R&D Systems) also exhibited degradation of IκB-α, and this degradation was significantly decreased if the cells were pre-incubated with cmvIL-10 (data not shown). Thus, cmvIL-10 prevents NF-κB from stimulating transcription induced by a variety of stimuli by enabling the inhibitor to accumulate in the cytoplasm, rendering NF-κB subunits unable to enter the nucleus.
In vivo, the inhibition of NF-κB activity by cmvIL-10 may affect infected cells secreting cmvIL-10 via autocrine signaling, as well as impair the function of uninfected immune cells and contribute to the overall immunosuppressive environment that enables virus persistence and the establishment of latency. Paradoxically, activation of the NF-κB pathway is essential for productive HCMV infection (10,11). When HCMV productively infects a cell, three classes of genes are transcribed: immediate-early (IE), early, and late (25). IE genes are essential for virus replication, and the expression of these genes is regulated by the major immediate-early promoter (MIEP) (1,42). The MIEP, which is critical for efficient virus replication (17,24), contains four consensus NF-κB binding sites (25,33). Induction of NF-κB following HCMV infection drives the transactivation of the MIEP and is a crucial step in initiation of viral gene expression and productive virus replication. Thus, virus infection leads to NF-κB activation in cells harboring the viral genome, while the secreted viral cytokine cmvIL-10 suppresses NF-κB signaling in uninfected bystander cells. Indeed, IL-10 has been identified as one of the single most important factors that impede virus clearance, and in some experimental systems, blocking IL-10 results in rapid elimination of virus infection (3,4). It has also been suggested that HCMV strains be sub-typed by the amount of cmvIL-10 produced, and that higher producers are the ones more likely to reactivate and cause disease (15). It is not yet known what the impact of blocking cmvIL-10 during the course of infection in vivo might be.
HCMV remains a major cause of morbidity and mortality among immunocompromised individuals. We show here that cmvIL-10–mediated immune suppression occurs through inhibition of NF-κB activity. Continued definition of the molecular processes that HCMV uses to modulate immune function is a necessary and important step in the development of effective treatment strategies.
Acknowledgments
This work was supported by National Institutes of Health grant AI063529 and University of San Francisco Faculty Development Funds to J.V.S. We thank Tori Manning for technical assistance and critical reading of this manuscript.
Disclosure Statement
No conflicting financial interests exist.
References
- 1.Boshart M. Weber F. Jahn G. Dorsch-Hasler K. Fleckenstein B. Schaffner W. A very strong enhancer is located upstream of an immediate early gene of human cytomegalovirus. Cell. 1985;41:521–530. doi: 10.1016/s0092-8674(85)80025-8. [DOI] [PubMed] [Google Scholar]
- 2.Britt WJ. Alford CA. Cytomegalovirus. In: Fields DMKBN, editor; Howley PM, et al., editors. Fields Virology. 3rd. Lippincott-Raven; Philadelphia: 1996. pp. 2493–2523. [Google Scholar]
- 3.Brooks DG. Lee AM. Elsaesser H. McGavern DB. Old-stone MB. IL-10 blockade facilitates DNA vaccine-induced T cell responses and enhances clearance of persistent virus infection. J Exp Med. 2008;205:533–541. doi: 10.1084/jem.20071948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brooks DG. Trifilo MJ. Edelmann KH. Teyton L. McGavern DB. Oldstone MB. Interleukin-10 determines viral clearance or persistence in vivo. Nat Med. 2006;12:1301–1309. doi: 10.1038/nm1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cao S. Zhang X. Edwards JP. Mosser DM. NF-kappaB1 (p50) homodimers differentially regulate pro- and anti-inflammatory cytokines in macrophages. J Biol Chem. 2006;281:26041–26050. doi: 10.1074/jbc.M602222200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang W. Baumgarth N. Greg JP. Baron CA. Barry PA. HCMV-encoded IL-10 alters dendritic cell functionality: a comprehensive comparison with cellular IL-10; 29th International Herpesvirus Workshop; Reno NV. 2004. [Google Scholar]
- 7.Chang WL. Baumgarth N. Yu D. Barry PA. Human cytomegalovirus-encoded interleukin-10 homolog inhibits maturation of dendritic cells and alters their functionality. J Virol. 2004;78:8720–8731. doi: 10.1128/JVI.78.16.8720-8731.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Collart MA. Baeuerle P. Vassalli P. Regulation of tumor necrosis factor alpha transcription in macrophages: involvement of four kappa B-like motifs and of constitutive and inducible forms of NF-kappa B. Mol Cell Biol. 1990;10:1498–1506. doi: 10.1128/mcb.10.4.1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de la Hoz RE. Stephens G. Sherlock C. Diagnosis and treatment approaches of CMV infections in adult patients. J Clin Virol. 2002;25(Suppl 2):S1–12. doi: 10.1016/s1386-6532(02)00091-4. [DOI] [PubMed] [Google Scholar]
- 10.DeMeritt IB. Milford LE. Yurochko AD. Activation of the NF-kappaB pathway in human cytomegalovirus-infected cells is necessary for efficient transactivation of the major immediate-early promoter. J Virol. 2004;78:4498–4507. doi: 10.1128/JVI.78.9.4498-4507.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DeMeritt IB. Podduturi JP. Tilley AM. Nogalski MT. Yurochko AD. Prolonged activation of NF-kappaB by human cytomegalovirus promotes efficient viral replication and late gene expression. Virology. 2006;346:15–31. doi: 10.1016/j.virol.2005.09.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Driessler F. Venstrom K. Sabat R. Asadullah K. Schottelius AJ. Molecular mechanisms of interleukin-10-mediated inhibition of NF-kappaB activity: a role for p50. Clin Exp Immunol. 2004;135:64–73. doi: 10.1111/j.1365-2249.2004.02342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.El Kasmi KC. Holst J. Coffre M, et al. General nature of the STAT3-activated anti-inflammatory response. J Immunol. 2006;177:7880–7888. doi: 10.4049/jimmunol.177.11.7880. [DOI] [PubMed] [Google Scholar]
- 14.Emery VC. Cytomegalovirus and the aging population. Drugs Aging. 2001;18:927–933. doi: 10.2165/00002512-200118120-00004. [DOI] [PubMed] [Google Scholar]
- 15.Geist LJ. The self defense of cytomegalovirus: in this case the lawyer is no fool. Transplantation. 2008;86:202–203. doi: 10.1097/TP.0b013e31817b06e1. [DOI] [PubMed] [Google Scholar]
- 16.Hiscott J. Marois J. Garoufalis J, et al. Characterization of a functional NF-kappa B site in the human interleukin 1 beta promoter: evidence for a positive autoregulatory loop. Mol Cell Biol. 1993;13:6231–6240. doi: 10.1128/mcb.13.10.6231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Isomura H. Stinski MF. The human cytomegalovirus major immediate-early enhancer determines the efficiency of immediate-early gene transcription and viral replication in permissive cells at low multiplicity of infection. J Virol. 2003;77:3602–3614. doi: 10.1128/JVI.77.6.3602-3614.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jones BC. Logsdon NJ. Josephson K. Cook J. Barry PA. Walter MR. Crystal structure of human cytomegalovirus IL-10 bound to soluble human IL-10R1. Proc Natl Acad Sci USA. 2002;99:9404–9409. doi: 10.1073/pnas.152147499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kotenko SV. Saccani S. Izotova LS. Mirochnitchenko OV. Pestka S. Human cytomegalovirus harbors its own unique IL-10 homolog (cmvIL-10) Proc Natl Acad Sci USA. 2000;97:1695–1700. doi: 10.1073/pnas.97.4.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuwata H. Watanabe Y. Miyoshi H. Yamamoto M. Kaisho T. Takeda K. Akira S. IL-10-inducible Bcl-3 negatively regulates LPS-induced TNF-alpha production in macrophages. Blood. 2003;102:4123–4129. doi: 10.1182/blood-2003-04-1228. [DOI] [PubMed] [Google Scholar]
- 21.Lee SH. Hannink M. The N-terminal nuclear export sequence of IkappaBalpha is required for RanGTP-dependent binding to CRM1. J Biol Chem. 2001;276:23599–23606. doi: 10.1074/jbc.M011197200. [DOI] [PubMed] [Google Scholar]
- 22.Li Q. Verma IM. NF-kappaB regulation in the immune system. Nat Rev Immunol. 2002;2:725–734. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- 23.May MJ. Ghosh S. Rel/NF-kappa B and I kappa B proteins: an overview. Semin Cancer Biol. 1997;8:63–73. doi: 10.1006/scbi.1997.0057. [DOI] [PubMed] [Google Scholar]
- 24.Meier JL. Pruessner JA. The human cytomegalovirus major immediate-early distal enhancer region is required for efficient viral replication and immediate-early gene expression. J Virol. 2000;74:1602–1613. doi: 10.1128/jvi.74.4.1602-1613.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mocarski ES. Cytomegaloviruses and their replication. In: Fields DMKBN, editor; Howley PM, et al., editors. Fields Virology. 3rd. Lippincott-Raven Publishers; Philadelphia: 1996. pp. 2447–2492. [Google Scholar]
- 26.Murray PJ. The JAK-STAT signaling pathway: input and output integration. J Immunol. 2007;178:2623–2629. doi: 10.4049/jimmunol.178.5.2623. [DOI] [PubMed] [Google Scholar]
- 27.Niemand C. Nimmesgern A. Haan S, et al. Activation of STAT3 by IL-6 and IL-10 in primary human macrophages is differentially modulated by suppressor of cytokine signaling 3. J Immunol. 2003;170:3263–3672. doi: 10.4049/jimmunol.170.6.3263. [DOI] [PubMed] [Google Scholar]
- 28.Qin H. Roberts KL. Niyongere SA. Cong Y. Elson CO. Benveniste EN. Molecular mechanism of lipopolysaccharide-induced SOCS-3 gene expression in macrophages and microglia. J Immunol. 2007;179:5966–5976. doi: 10.4049/jimmunol.179.9.5966. [DOI] [PubMed] [Google Scholar]
- 29.Qin H. Wilson CA. Roberts KL. Baker BJ. Zhao X. Benveniste EN. IL-10 inhibits lipopolysaccharide-induced CD40 gene expression through induction of suppressor of cytokine signaling-3. J Immunol. 2006;177:7761–7771. doi: 10.4049/jimmunol.177.11.7761. [DOI] [PubMed] [Google Scholar]
- 30.Raftery MJ. Wieland D. Gronewald S. Kraus AA. Giese T. Schonrich G. Shaping phenotype, function, and survival of dendritic cells by cytomegalovirus-encoded IL-10. J Immunol. 2004;173:3383–3391. doi: 10.4049/jimmunol.173.5.3383. [DOI] [PubMed] [Google Scholar]
- 31.Riley JK. Takeda K. Akira S. Schreiber RD. Interleukin-10 receptor signaling through the JAK-STAT pathway. Requirement for two distinct receptor-derived signals for anti-inflammatory action. J Biol Chem. 1999;274:16513–16521. doi: 10.1074/jbc.274.23.16513. [DOI] [PubMed] [Google Scholar]
- 32.Rivera LB. Boppana SB. Fowler KB. Britt WJ. Stagno S. Pass RF. Predictors of hearing loss in children with symptomatic congenital cytomegalovirus infection. Pediatrics. 2002;110:762–767. doi: 10.1542/peds.110.4.762. [DOI] [PubMed] [Google Scholar]
- 33.Sambucetti LC. Cherrington JM. Wilkinson GW. Mocarski ES. NF-kappa B activation of the cytomegalovirus enhancer is mediated by a viral transactivator and by T cell stimulation. EMBO J. 1989;8:4251–4258. doi: 10.1002/j.1460-2075.1989.tb08610.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schottelius AJ. Mayo MW. Sartor RB. Baldwin AS., Jr Interleukin-10 signaling blocks inhibitor of kappaB kinase activity and nuclear factor kappaB DNA binding. J Biol Chem. 1999;274:31868–31874. doi: 10.1074/jbc.274.45.31868. [DOI] [PubMed] [Google Scholar]
- 35.Shakhov AN. Collart MA. Vassalli P. Nedospasov SA. Jongeneel CV. Kappa B-type enhancers are involved in lipopolysaccharide-mediated transcriptional activation of the tumor necrosis factor alpha gene in primary macrophages. J Exp Med. 1990;171:35–47. doi: 10.1084/jem.171.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Siebenlist U. Brown K. Claudio E. Control of lymphocyte development by nuclear factor-kappaB. Nat Rev Immunol. 2005;5:435–445. doi: 10.1038/nri1629. [DOI] [PubMed] [Google Scholar]
- 37.Spencer JV. The cytomegalovirus homolog of interleukin-10 requires phosphatidylinositol 3-kinase activity for inhibition of cytokine synthesis in monocytes. J Virol. 2007;81:2083–2086. doi: 10.1128/JVI.01655-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Spencer JV. Cadaoas J. Castillo PR. Saini V. Slobedman B. Stimulation of B lymphocytes by cmvIL-10 but not LAcmvIL-10. Virology. 2008;374:164–169. doi: 10.1016/j.virol.2007.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Spencer JV. Lockridge KM. Barry PA. Lin G. Tsang M. Penfold ME. Schall TJ. Potent immunosuppressive activities of cytomegalovirus-encoded interleukin-10. J Virol. 2002;76:1285–1292. doi: 10.1128/JVI.76.3.1285-1292.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tabary O. Muselet C. Escotte S. Antonicelli F. Hubert D. Dusser D. Jacquot J. Interleukin-10 inhibits elevated chemokine interleukin-8 and regulated on activation normal T cell expressed and secreted production in cystic fibrosis bronchial epithelial cells by targeting the I(k)B kinase alpha/beta complex. Am J Pathol. 2003;162:293–302. doi: 10.1016/s0002-9440(10)63820-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tam WF. Lee LH. Davis L. Sen R. Cytoplasmic sequestration of rel proteins by IkappaBalpha requires CRM1-dependent nuclear export. Mol Cell Biol. 2000;20:2269–2284. doi: 10.1128/mcb.20.6.2269-2284.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thomsen DR. Stenberg RM. Goins WF. Stinski MF. Promoter-regulatory region of the major immediate early gene of human cytomegalovirus. Proc Natl Acad Sci USA. 1984;81:659–663. doi: 10.1073/pnas.81.3.659. [DOI] [PMC free article] [PubMed] [Google Scholar]