

Abstract
Charcot-Marie-Tooth 1A (CMT1A) neuropathy, the most common inherited peripheral neuropathy, is primarily caused by a gene duplication for the peripheral myelin protein-22 (PMP22). In an accordant mouse model, we investigated the role of monocyte chemoattractant protein-1 (MCP-1/CCL2) as a regulator of nerve macrophages and neural damage including axonopathy and demyelination. By generating PMP22tg mice with reduced levels or lack of MCP-1/CCL2, we found that MCP-1/CCL2 is involved in the increase of macrophages in mutant nerves. PMP22tg mice with wild-type levels of MCP-1/CCL2 showed strong macrophage increase in the diseased nerves, whereas either 50% reduction or total absence of MCP-1/CCL2 led to a moderate or a strong reduction of nerve macrophages, respectively. Interestingly, MCP-1/CCL2 expression level and macrophage numbers were correlated with features indicative of axon damage, such as maldistribution of K+ channels, reduced compound muscle action potentials, and muscle weakness. Demyelinating features, however, were most highly reduced when MCP-1/CCL2 was diminished by 50%, whereas complete lack of MCP-1/CCL2 showed an intermediate demyelinating phenotype. We also identified the MEK1/2-ERK1/2-pathway as being involved in MCP-1/CCL2 expression in the Schwann cells of the CMT1A model. Our data show that, in a CMT1A model, MCP-1/CCL2 activates nerve macrophages, mediates both axon damage and demyelination, and may thus be a promising target for therapeutic approaches.
Inherited peripheral neuropathies are incurable disabling disorders of the peripheral nervous system. The majorities of these disorders belong to the Charcot-Marie-Tooth (CMT) type 1 neuropathies and are primarily caused by mutations in genes for myelin-related components. Clinically, they are characterized by muscle wasting, weakness, and sensory dysfunction. The exact de- or dysmyelinating mechanisms are only partially understood in most cases and may comprise impaired stability of protein interactions among myelin components, the impaired interaction between Schwann cell molecules, extracellular matrix components, as well as different intracellular pathways of Schwann cell stress and injury.1–4 In the most common form, CMT1A, a duplication of the peripheral myelin protein-22 (PMP22) gene leads to the demyelinating disorder, possibly due to intrinsic cell stress related to protein overexpression. Based on the observation that progesterone regulates myelin genes in vitro, administration of the progesterone receptor antagonist onapristone reduced overexpression of PMP22 and ameliorated neuropathy in a rat model of CMT1A in both young and young adult animals.5,6 Alternatively, observations in a transgenic mouse model overexpressing PMP22 led to a therapeutical approach with ascorbic acid.7 In one CMT1B model carrying a S63del mutation in the gene for the myelin component P0, an unfolded protein response has been shown of being of pathological relevance.8 In other CMT1 models, we focused on the role of immune cells, which are involved in the primarily genetically-induced neuropathies and may be a common pathway for distinct CMT entities.9 In this context, macrophage activation by the chemokine monocyte chemoattractant protein-1 (MCP-1/CCL2) plays a key role in the pathogenesis.10 Moreover, MCP-1/CCL2 upregulation is mediated by the MEK1/2-ERK1/2-pathway in the CMT1B model.11 MCP-1/CCL2 has not only been found upregulated in CMT1B models but also in a mouse model for CMT1A where macrophages phagocytose myelin within endoneurial tubes, suggesting an active role in demyelinating neuropathy.12 In the present study we identified MCP-1/CCL2 not only as being involved in macrophage activation but also as mediator of axon damage and demyelination in a model of the most common form of CMT.
Materials and Methods
Mice and Determination of Genotypes
Transgenic (tg) PMP22-overexpressing mice of the C61 strain carrying four copies of a human YAC clone encompassing the complete hPMP2213,14 were kept in our animal facilities. Mice were maintained on a mixed C57BL/6xCBA/Ca background and crossbred with MCP-1/CCL2 mutant mice15 to receive immune-modulated double mutants. Additionally, the single PMP22 mutants were backcrossed to a C57Bl/6 background for six to eight generations. For all investigations only heterozygous PMP22-overexpressing mice and their wild-type littermates were used. Genotyping was performed for PMP22 mutants by PCR reaction using primers specific for human Pmp22 (forward primer, 5′-TCAGGATATCTATCTGATTCTC-3′; reverse primer, 5′-AAGCTCATGGAGCACAAAACC-3′) and mouse β-actin genes (forward primer, 5′-AACCGTGAAAAGATGACCC-3′; reverse primer, 5′-TCGTTGCCAATAGTGATGACC-3′). Cycling conditions were 95°C for 15 minutes, followed by 38 cycles of 95°C for 30 seconds, 55°C for 30 seconds, 72°C for 1 minute, with a final cycle at 72°C for 10 minutes. Genotyping for the ccl2 wild-type and the ccl2 knockout allele was performed as described previously.10,11
All mouse strains used in this study were kept under specific pathogen-free conditions at the Department of Neurology, Julius-Maximilians-University, Wuerzburg, Germany. Animal experiments were approved by the local authorities (Regierung von Unterfranken).
Tissue Preparation for Immunohistochemistry
Quantification of F4/80-positive macrophages was performed on fresh frozen cross-sections (10 μm) at different ages for all mice groups as previously described.16,17 For localization of the phospho-ERK1/2 signal, teased fibers of femoral quadriceps nerves were used, stained and evaluated on a confocal microscope (DM RE-7 SDK, Leica, Germany) as previously described.11
To localize MCP-1/CCL2 protein, we used teased fiber preparations of femoral quadriceps nerves of wild-type (n = 3), PMP22tg mice (n = 3), and PMP22tg/MCP-1−/− (n = 2) at the age of two to three months. Briefly, mice were deeply anesthetized and trans-cardially perfused with 2% paraformaldehyd. Femoral quadriceps nerves were excised and nerve fibers were separated (“teased”) and fixed in ice-cold acetone (10 minutes). Teased fibers were then blocked with 5% BSA in PBS with 0.3% TritonX-100 and incubated with primary antibody (rabbit anti-mouse MCP-1, 4 μg/ml, Peprotech, Hamburg, Germany) overnight at 4°C. To visualize the primary antibody, biotinylated secondary antibody to rabbit Ig was incubated for 1 hour, followed by avidin/biotin reagent (Vector Labs, Burlingame, CA) and staining with diaminobenzidine-HCl and H2O2. In addition, in some experiments, we used tyramide signal amplification (1:100, in amplification reagent for 5 minutes) to enhance the resulting immune signal according to manufacturer's guidelines (Perkin Elmer, Waltham, MA). For negative controls, either the primary antibody was omitted or the primary antibody was preincubated with 10-fold M rMCP-1 Peptid (Peprotech, Hamburg, Germany) for 1 hour at 37°C. Before incubation with the primary antibody, immune complexes were centrifuged for 5 minutes at 13,500g.
Furthermore, teased fiber preparations of femoral quadriceps nerves from six-month-old PMP22wt/MCP-1+/+ (wild-type, n = 3), PMP22tg/MCP-1+/+ (n = 4), PMP22tg/MCP-1+/− (n = 3), and PMP22tg/MCP-1−/− (n = 3) mice were used to examine the distribution of Na+ channels (Nav1.6), K+ channels (Kv1.2), and contactin associated protein (Caspr) by immunohistochemistry as previously described.18 Expression of K+ channels was quantified by the presence or absence of Kv1.2, asymmetric and diffuse expression by localization with Caspr. For this quantification, at least 120 nodes of Ranvier per mouse were examined.
Morphometric Analysis by Electron Microscopy
Peripheral femoral nerves of adult mice were prepared for electron microscopy as described elsewhere.12 PMP22/MCP-1 mice at the age of six months (n = 3 to 4) were analyzed with regard to the occurrence of axons devoid of myelin, thinly myelinated axons, hypermyelinated axons, supernumerary Schwann cells (onion bulb formations), and foamy macrophages. Additionally, g-ratios of myelinated fibers were determined. In all experiments, the investigator (B.K.) was not aware of the genotype of the mice.
Tissue Purification for RNA and Protein Lysates
Peripheral nerve tissue for RNA and protein extraction was prepared as previously described.11 After photometric measurement of the RNA concentration, 0.5 μg RNA were transcribed into cDNA using reverse transcription reagents (Applied Biosystems, Foster City, CA). Protein concentration was determined by Lowry assay (Sigma Aldrich, Taufkirchen, Germany), and equal amounts of proteins were used for SDS-Page and Western blotting.
Western Blot Analyses
Western blot technique was applied for detection of different signaling proteins and their activation status as previously described.11 Successful protein transfer onto a nitrocellulose membrane was controlled with Ponceau-S staining for a maximum of five minutes. Stainings with antibodies against the nonphosphorylated total proteins were used as loading controls for the phosphorylated form of the proteins.
Semiquantitative Real-Time PCR
Peripheral nerve lysates of PMP22tg mice and their wild-type littermates were studied for MCP-1 mRNA expression at the age of two and six months by quantitative real-time PCR (qRT-PCR) with TaqMan universal PCR master mix (Applied Biosystems, Foster City, CA) and predeveloped TaqMan assays (18s, 4319413E; MCP-1, Mm99999056_m1). All samples were measured in triplicate and finally analyzed in relation to 18s RNA content. All indicated values are standardized according to samples of wild-type nerves.
Neurographic Recordings
Six-month-old PMP22/MCP-1 double mutants were examined regarding their conduction properties of the sciatic nerves as described previously.19 Proximal and distal supramaximal stimulation led to the determination of durations and amplitudes of compound muscle action potentials (CMAP) and of nerve conduction velocity. The investigator of this experiment (C.W.) was not aware of the genotype of the mice.
Motor Performance Test
To examine the grip strength of PMP22/MCP-1 double mutants at the age of five to six months, grip tests of forelimbs as well as of hindlimbs were performed. An automated grip strength meter (Columbus Instruments, Columbus, OH) was used as described previously.20,21 Briefly, mice were trained to hold the grip bar and were dragged off with constant strength. Ten measurements per mouse and day were performed on three consecutive days. In addition, mice were weighed at the first day of testing.
In Vivo Inhibition of MEK1/2-Signaling Cascade by Injection of CI-1040
To test the functional relevance of the MEK1/2/ERK1/2 cascade in vivo, we performed inhibition studies with the MEK1/2-inhibitor CI-1040 (PD184352), which was kindly provided by Pfizer (New York, NY). Two-month-old PMP22tg mice and their wild-type littermates were treated for 21 consecutive days with intraperitoneal injection of either 100 mg/kg CI-1040 in dimethylsulfoxid (DMSO; PMP22tg: n = 5, wild-type mice n = 3) or DMSO only as control (PMP22tg: n = 5, wild-type mice n = 4) as previously described.11,22 A maximal injection volume of 50 μl was used, and the body weight was monitored daily before injection. Mice that showed an impaired health status were sacrificed earlier and not included in the results. After 21 days of treatment, mice were sacrificed and sciatic and femoral nerves were dissected and stored for RNA and protein purification at −80°C.
Statistical Analyses
Statistical analyses were performed by using the unpaired two-tailed Student t test and the nonparametric Mann–Whitney U test. All data are reported as mean ± SD.
Results
MCP-1/CCL2 mRNA Is Upregulated in Peripheral Nerves of PMP22tg Mice
Corroborating our previous findings that MCP-1/CCL2 mRNA is upregulated in sciatic nerve and macrophages are increased in femoral nerve of PMP22tg mice,12 we were interested in the expression of MCP-1/CCL2 in femoral nerves. MCP-1/CCL2 mRNA expression was significantly increased in sciatic nerves (Figure 1A) as well as in femoral quadriceps nerves (Figure 1B) of PMP22tg mice compared with their wild-type littermates. As a next step, we scored the cellular localization of MCP-1/CCL2 in peripheral nerves of PMP22tg mice. Teased fiber preparations of femoral quadriceps nerves from two-month-old PMP22tg mice revealed a characteristic pattern of MCP-1-positivity reminiscent of Schmidt-Lanterman incisures (Figure 1C). It is of note that MCP-1–positive structures were predominantly detected in larger caliber fibers (Figure 1C). The characteristic immunopositivity was not observed in preparations that were treated with antibodies that had been preincubated with rMCP-1 (Figure 1D) or in fiber preparations from PMP22tg/MCP-1−/− mice (data not shown). No staining was visible in the complete absence of the primary antibody (Figure 1E). Wild-type mice did only reveal a weak MCP-1 immunolabeling (data not shown), corroborating the low mRNA levels (see above).
Figure 1.
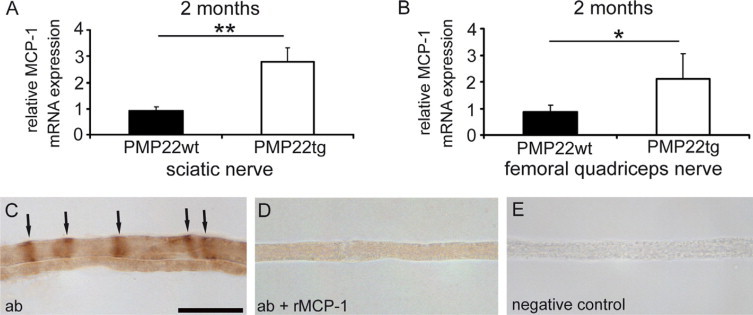
MCP-1/CCL2 is increased in peripheral nerves of PMP22tg mice. A and B: MCP-1/CCL2-expression in peripheral nerves of two-month-old PMP22-overexpressing mice. Semiquantitative RT-PCR (mean values ± SD) in sciatic (A) and femoral quadriceps nerves (B) of PMP22tg (n = 4) revealed a significantly increased expression of MCP-1/CCL2 mRNA in comparison with nerves of PMP22wt (n = 3). Representative data from two to three different experiments are shown. *P < 0.05, **P < 0.01, Student t test. C–E: Immunohistochemical detection of MCP-1 on teased fiber preparations of femoral quadriceps nerves from two-month-old PMP22tg mice. Teased fiber preparations of PMP22tg mice incubated with primary MCP-1 antibody (ab) showed a characteristic pattern indicative for immunopositive Schmidt-Lanterman incisures (arrows, C). When the primary antibody was preincubated with rMCP-1, the labeling was weak and diffuse (D) and completely absent, when the primary antibody was omitted (E). Scale bar = 30 μm.
The Number of Macrophages Is Less Elevated in Femoral Nerves of PMP22tg Mice with Reduced or Absent MCP-1/CCL2-Expression
Because MCP-1/CCL2 is significantly upregulated in Schwann cells of mutant nerves, we investigated the pathogenic role of the chemokine by crossbreeding PMP22 myelin mutants with MCP-1/CCL2-deficient (MCP-1−/−) mice. First, we examined the MCP-1/CCL2 mRNA expression in femoral quadriceps nerves of PMP22 mutant mice with different MCP-1/CCL2 genotypes (see Supplemental Figure 1 at http://ajp.amjpathol.org). MCP-1/CCL2 mRNA expression was diminished in PMP22wt/MCP-1+/− and completely absent in PMP22wt/MCP-1−/− mice. The increased MCP-1/CCL2-expression in PMP22tg was reduced in PMP22tg/MCP-1+/− and absent in PMP22tg/MCP-1−/− mutants.
Next, we determined the number of F4/80-positive macrophages in femoral nerves of mutant mice for time points at which MCP-1/CCL2 is up-regulated to correlate MCP-1/CCL2 expression and macrophage increase. PMP22tg mice with complete MCP-1/CCL2 (MCP-1+/+) or with reduced MCP-1/CCL2 genes (MCP-1+/−) showed an elevated number of macrophages in femoral quadriceps nerves already at the age of two months (Figure 2A). Interestingly, there was also a higher macrophage number in the complete absence of MCP-1/CCL2 (PMP22tg/MCP-1−/−) in comparison with wild-type mice (Figure 2A). The high number persisted in femoral quadriceps nerves of six-month-old mice in PMP22tg/MCP-1+/+, whereas macrophages were significantly less elevated in nerves of PMP22tg/MCP-1+/− and PMP22tg/MCP-1−/− mice (Figure 2B). Peripheral nerves of PMP22wt/MCP-1+/+, PMP22wt/MCP-1+/− and PMP22wt/MCP-1−/− did not show an increase in macrophage number.
Figure 2.
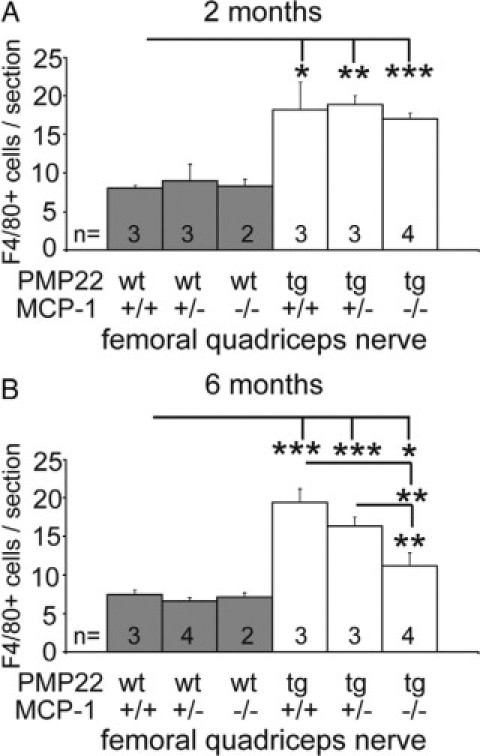
The extent of MCP-1/CCL2-expression determines the number of nerve-macrophages in PMP22tg mice. Quantification of F4/80-positive macrophages in femoral quadriceps nerves of wild-type and PMP22 mutant mice at the age of two (A) and six months (B). Note that all PMP22wt exhibit comparable numbers of macrophages independent of age and MCP-1/CCL2-expression. The increase of macrophages in PMP22tg mice is diminished by the reduction and complete absence of MCP-1/CCL2 at six months of age. *P < 0.05, **P < 0.01, ***P < 0.001, Student t test.
Reduced MCP-1/CCL2-Expression in Femoral Nerves Causes Less Demyelination
Based on our previous observation that in P0-heterozygous knockout mice the demyelinating phenotype is ameliorated when MCP-1/CCL2 is reduced,10 we focused on the pathological alterations in femoral quadriceps nerves of PMP22tg/MCP-1+/+, PMP22tg/MCP-1+/−, and PMP22tg/MCP-1−/− mutants in comparison with wild-type mice at the age of six months by electron microscopy. Ultrathin sections of PMP22/MCP-1 double mutant mice (Figure 3, A–D) displayed differences in the pathological phenotype. The number of demyelinated and thinly myelinated axons were increased in femoral quadriceps nerves of PMP22tg/MCP-1+/+ mice, whereas nerves of PMP22tg/MCP-1+/− and PMP22tg/MCP-1−/− revealed an amelioration of the pathological phenotype (Figure 3, E and F). The lowest number of pathological alterations was found in femoral quadriceps nerve of PMP22tg/MCP-1+/− mice. Interestingly, hypermyelinated fibers as a typical hallmark of PMP22tg mice were not affected by the MCP-1 genotype (Figure 3F). Foamy macrophages, reflecting macrophage phagocytosis, showed a tendency to lower numbers in femoral quadriceps nerves of PMP22tg/MCP-1+/− mice compared with nerves of PMP22tg/MCP-1+/+ and PMP22tg/MCP-1−/− mice (Figure 3G). In addition, no differences were found in the number of supernumerary Schwann cells between nerves of the double mutants (data not shown). Peripheral nerves of PMP22wt/MCP-1+/− and PMP22wt/MCP-1−/− mice exhibited no alterations compared with nerves of PMP22wt/MCP-1+/+ mice (data not shown).
Figure 3.
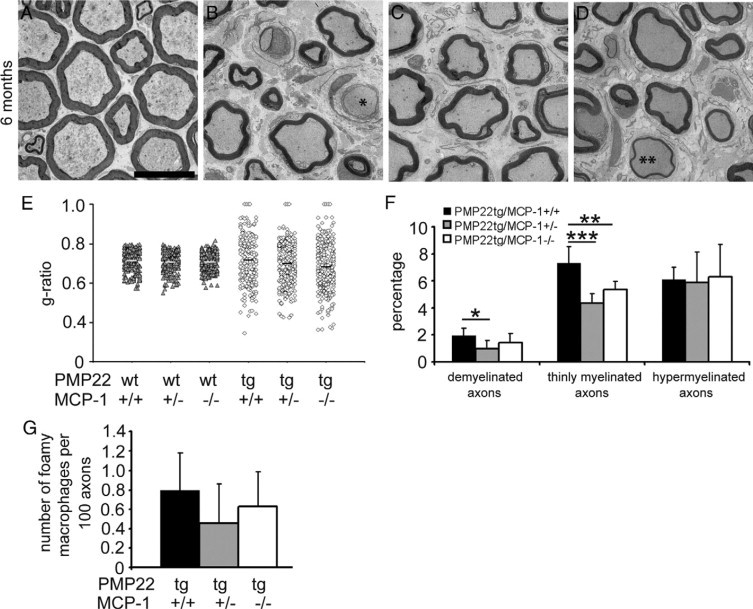
Demyelination is ameliorated in peripheral nerves of PMP22tg/MCP-1+/− and PMP22tg/MCP-1−/− mice. Morphometric analysis of femoral nerves from wild-type and PMP22 mutant mice (n = 3 to 4). Electron microscopy of femoral quadriceps nerves of six-month-old PMP22wt/MCP-1+/+ (A), PMP22tg/MCP-1+/+ (B), PMP22tg/MCP-1+/− (C), and PMP22tg/MCP-1−/− mutants (D). Analysis of femoral quadriceps nerves revealed fewer features indicative of demyelination in PMP22tg/MCP-1+/− and PMP22tg/MCP-1−/− mice, such as demyelinated axons (asterisk) and thinly myelinated axons (double asterisk). Scale bar = 10 μm. E: G-ratios reflect the milder demyelinating neuropathy in nerves of PMP22tg/MCP-1 double mutant mice. F: Quantification of pathological alterations confirms the ameliorated demyelination in femoral quadriceps nerves of PMP22tg/MCP-1+/− and PMP22tg/MCP-1−/− mice, whereas hypermyelination is independent from MCP-1/CCL2-expression. G: Foamy macrophages containing myelin debris showed a tendency toward lower numbers in femoral quadriceps nerves of PMP22tg/MCP-1+/− mice compared with nerves of PMP22tg/MCP-1+/+ and PMP22tg/MCP-1−/− mice. *P < 0.05, **P < 0.01, ***P < 0.001, Mann–Whitney U test.
Lack of MCP-1/CCL2-Expression in PMP22tg Mice Leads to Improved Neurographic Properties and Increased Grip Strength
To correlate demyelinating phenotypes with nerve function and motor performance, we performed neurographic recordings and grip strength tests, respectively.
Electrophysiological recordings were performed with six-month-old PMP22wt/MCP-1+/+, PMP22tg/MCP-1+/+, and PMP22tg/MCP-1−/− mice. Distal stimulation in PMP22wt/MCP-1+/+ mice led to CMAP with values of approximately 15 mV (Table 1). PMP22tg/MCP-1+/+ mutants showed a reduced amplitude (3.0 ± 0.1 mV), a prolonged duration (3.4 ± 0.7 ms) of CMAP, and a slowed nerve conduction velocity (25.2 ± 3.5 m/s). Whereas the nerve conduction velocity was similar in all PMP22tg/MCP-1 mutants, the amplitude and the duration was altered in PMP22tg/MCP-1−/− compared with PMP22tg/MCP-1+/+ mutants. Interestingly, the absence of MCP-1/CCL2 in PMP22tg mice causes a significantly elevated CMAP and a reduced duration in contrast to PMP22tg/MCP-1+/+ at the age of six months. As a functional correlation to CMAPs, we also investigated strengths of distal muscles. For this purpose, we performed grip tests in fore- and hindlimbs of PMP22/MCP-1 double mutant mice. The grip strengths of the forelimbs were not significantly altered in all genotypes (data not shown), likely because of the relative shortness and thus reduced general susceptibility for damage of the corresponding nerves. However, the strengths of the hindlimbs showed obvious differences in the PMP22/MCP-1 double mutants (Figure 4). The grip strength of six-month-old PMP22tg/MCP-1+/+ was significantly reduced compared with PMP22wt/MCP-1+/+ mice. The two PMP22tg/MCP-1+/− mutant mice we examined showed a grip strength comparable with PMP22tg/MCP-1+/+ mutant mice (not shown). Interestingly, PMP22tg/MCP-1−/− mutant mice exhibited a stronger grip strength of the hindlimbs compared with PMP22tg/MCP-1+/+ and PMP22tg/MCP-1+/− mice, corroborating our neurographic recordings (CMAP).
Table 1.
Neurographic Recordings of Six-Month-Old PMP22 Mutant Mice in the Presence or Absence of MCP-1/CCL2
| PMP22wt/MCP-1+/+ | PMP22tg/MCP-1+/+ | PMP22tg/MCP-1−/− | |
|---|---|---|---|
| CMAP/proximal stimulation, mV | 13.8 ± 3.2 | 2.6 ± 0.4* | 6.5 ± 1.7*† |
| CMAP/distal stimulation, mV | 15.1 ± 3.3 | 3.0 ± 0.1* | 6.9 ± 2.1*† |
| duration of CMAP/proximal stimulation, ms | 1.7 ± 0 | 3.0 ± 0.5* | 2.4 ± 0.2*† |
| duration of CMAP/distal stimulation, ms | 1.7 ± 0.1 | 3.4 ± 0.7* | 2.3 ± 0.1*† |
| NCV, m/s | 43.9 ± 1.7 | 25.2 ± 3.5* | 29.1 ± 4.3* |
| n-numbers | 3 | 4 | 4 |
Neurographic recordings reveal an increase in the amplitude and a reduction in duration of CMAP in PMP22tg/MCP-1−/− mice compared to PMP22tg/MCP-1+/+ mice.
P < 0.05, Student t test.
Significant differences compared with PMP22wt/MCP-1+/+ mice.
Significant differences compared with PMP22tg/MCP-1+/+ mice.
Figure 4.
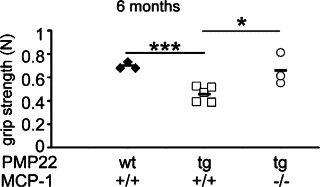
Complete absence of MCP-1/CCL2 increases muscle strength in PMP22tg mice. Muscle (grip) strength of hindlimbs of wild-type and PMP22 mutants at the age of six months. PMP22tg/MCP-1+/+ mice show an impaired muscle strength compared with PMP22wt/MCP-1+/+ mutants, whereas PMP22tg/MCP-1−/− mice exhibit an improvement of grip strength to a level similar to that of PMP22wt/MCP-1+/+ mice. Each indicated value represents a tested individual. *P < 0.05, ***P < 0.001, Student t test.
The Distribution of K+ Channels Is Altered in PMP22tg Mice in the Presence of MCP-1/CCL2
To examine “axonal” parameters that may explain the improved motor functions of the PMP22tg/MCP-1−/− mutant mice, we focused our attention on the nodes of Ranvier using markers for nodal, para-, and juxtaparanodal domains. In teased fiber preparations of femoral quadriceps nerves, the distribution of voltage-gated sodium channels of the Nav-type and Caspr as nodal and paranodal markers, respectively, was always normal in both wild-type and PMP22tg mice, independently of whether MCP-1/CCL2 was expressed or not (Figure 5, A and B). Similarly, voltage-gated K+ channels (Kv1.2) were found in their normal (ie, juxtaparanodal) location and symmetrically flanked the paranodal aspects in wild-type mice (Figure 5C). Rarely in wild-type mice did we also find juxtaparanodal K+ channels asymmetrically distributed or juxtaparanodes completely lacking the K+ channels. These abnormal features were substantially elevated in PMP22tg mutants and diffusely distributed channels were also seen (Figure 5, D–G). Quantification of these structures revealed that PMP22tg/MCP-1+/+ mice exhibited only 20% of nodes of Ranvier with normal distribution of K+ channels (Figure 5H). Comparable situation was found in femoral quadriceps nerves of PMP22tg/MCP-1+/− mice. Importantly, in the complete absence of MCP-1/CCL2 (PMP22tg/MCP-1−/− mice) more than half (52%) of the juxtaparanodes showed normal K+ channel distribution. Whereas we found no striking difference for the asymmetric distribution of the K+ channels in PMP22tg/MCP-1+/+, PMP22tg/MCP-1+/−, and PMP22tg/MCP-1−/− mice, the diffuse expression and the percentage of absent K+ channels were clearly reduced in PMP22tg/MCP-1−/− mice (Figure 5H).
Figure 5.
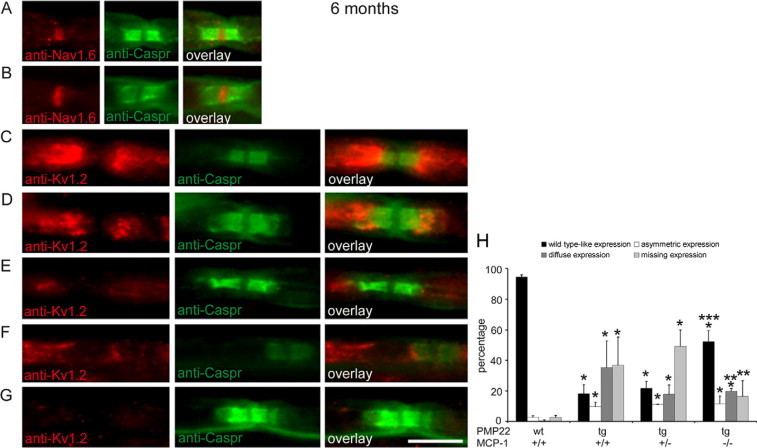
PMP22tg mice show a maldistribution of Kv1.2 channels, which is ameliorated in the absence of MCP-1/CCL2. Ion channels (anti-Nav1.6 and anti-Kv1.2) in teased fiber preparations of femoral quadriceps nerves from six-month-old PMP22/MCP-1 mutant mice were visualized with Cy3, paranodal Caspr with FITC. (A–G, scale bar = 10 μm). Nav1.6 expression is comparable in wild-type (A, red) and PMP22tg/MCP-1+/+ mice (B, red). Kv1.2 is symmetrically distributed in juxtaparanodal regions of fibers from wild-type mice (C, red). Whereas in addition to wild-type-like expression (D, red), an asymmetric distribution (E, red) and a diffuse expression (F, red) is also found in teased fibers of PMP22tg/MCP-1+/+ mice. Note that some fibers do not show any Kv1.2 expression (G). The distribution of Caspr-positive profiles (green, FITC) are not altered in wild-type and PMP22tg/MCP-1+/+ mutant mice. Quantification of Kv1.2 distribution in teased fiber preparations of MCP-1 mutant mice (H). In the complete absence of MCP-1/CCL2, PMP22tg mice (PMP22tg/MCP-1−/−) showed a more wild-type–like expression of Kv1.2 in peripheral nerves. *Significant differences compared with PMP22wt/MCP-1+/+ mice. **Significant differences compared with PMP22tg/MCP-1+/+ mice. ***Significant differences compared with PMP22tg/MCP-1+/+ and PMP22tg/MCP-1+/− mice. P < 0.05, Mann–Whitney U test.
Peripheral Nerves of PMP22tg Mice Exhibit an Activation of the ERK1/2 Signaling Cascade
Because MCP-1/CCL2 appears to be a crucial disease modulator in an animal model for CMT1A, we were interested in the Schwann cell intrinsic regulation of the chemokine. In another myelin mutant, the P0-heterozygous (P0+/−) mice, the MEK1/2/ERK1/2 signaling cascade could be identified as an important pathway for MCP-1/CCL2 expression.11 Here, we performed the corresponding examination in nerves of PMP22 mutant mice at the age of one, two, and six months regarding the phosphorylation of ERK1/2 and its upstream signaling protein, MEK1/2. We further tested by Western blot analyses whether other signaling cascades could have regulatory function for MCP-1/CCL2 in PMP22tg mice like JNK, p38, IKBα, and STAT1α.23–27 Analyses of protein lysates from two-month-old mice revealed no altered activation status of JNK and IKBα (Figure 6, A and B). Furthermore, higher activation of STAT1α could not be observed in nerves of PMP22tg, whereas the phosphorylated protein of p38 was not detectable in femoral quadriceps nerves of PMP22tg or wild types (data not shown).
Figure 6.
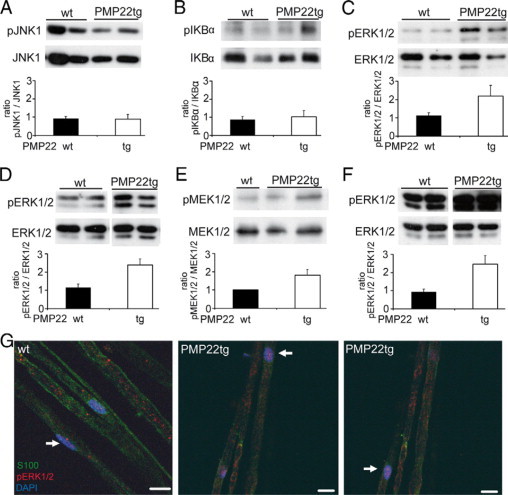
The MEK1/2/ERK1/2 signaling cascade is activated in nerves of PMP22tg mice. Western blot analyses of peripheral nerve lysates from two-month-old (A–E) and of six-month-old (F) wild-type (wt) and PMP22tg mutant mice. The diagrams below the immunoblots illustrate the corresponding densitometric analyses of three experiments comprising two to five mice. JNK1 (A, 46 kDa) and IKBα (B, 37 kDa) do not show any increased phosphorylation in femoral quadriceps nerves in PMP22tg mice relative to wild-type. In contrast, stronger phosphorylation of ERK1/2 (pERK1/2, 44/42 kDa) is visible in femoral quadriceps (C) and sciatic nerves (D) of PMP22tg mice compared with wild-types. E: The upstream kinase of ERK1/2, MEK1/2 (45 kDa), is stronger phosphorylated in femoral quadriceps nerve of PMP22tg mice. F: The stronger ERK1/2-phosphorylation is still visible in femoral quadriceps nerves of six-month-old PMP22tg mice compared with wild-types. In all blots, staining with antibodies against the corresponding total protein serves as loading controls. G: Localization of phospho-ERK1/2 in single fiber preparations of two-month-old mice. In comparison with a more non-nuclear and faint labeling in PMP22wt (n = 3), femoral quadriceps nerves of PMP22tg mice (n = 3) reveal an additional phospho-ERK1/2 immunoreactivity (red, Cy3, arrow) in nuclei (DAPI, blue) of S100β-positive Schwann cells (green, FITC). Scale bar = 10 μm.
In one-month old mice, the phosphorylation of ERK1/2 in peripheral nerves of PMP22tg mice was comparable with wild types (data not shown). In contrast, in femoral quadriceps and sciatic nerves of two-month-old PMP22tg mice a stronger phosphorylation of ERK1/2 was detectable (Figure 6, C and D). Also, MEK1/2 was stronger phosphorylated in femoral quadriceps nerves in two-month-old PMP22tg mice compared with wild-type littermates (Figure 6E). The higher activation of ERK1/2 in nerves of PMP22tg mice was detected until six months of age (Figure 6F).
To localize the phospho-ERK1/2-signal, we performed immunohistochemistry on teased fiber preparations. Single fiber preparations of femoral quadriceps nerves of PMP22wt revealed a predominantly cytoplasmic phospho-ERK1/2-positive signal (Figure 6G). Only a weak phospho-ERK1/2 staining was located within nuclei of Schwann cells. In contrast, single fiber preparations of femoral quadriceps nerves from PMP22tg mice exhibited a strong nuclear phospho-ERK1/2 labeling and a less intensive cytoplasmic staining (Figure 6G).
In Vivo Inhibition of the MEK1/2/ERK1/2 Signaling Cascade Leads to a Reduced MCP-1/CCL2 mRNA Expression in Peripheral Nerves of PMP22tg Mice
To investigate the relevance of ERK1/2-phosphorylation for the expression of MCP-1/CCL2, we used the MEK1/2-inhibitor CI-1040 (PD184352) in vivo. For this, we injected CI-1040 in DMSO (100 mg/kg, i.p.) daily for a period of 21 days and analyzed peripheral nerves regarding the phosphorylation of ERK1/2 and MCP-1/CCL2 mRNA expression.
Corroborative our findings above, sham-treated PMP22tg mice revealed a stronger ERK1/2-phosphorylation in femoral quadriceps nerves compared with sham-treated wild-type littermates. In femoral quadriceps nerves of CI-1040–treated PMP22tg mice, the phospho-ERK1/2 signal was reduced in comparison with sham-treated mutants (Figure 7A). A similar reduction was seen in lysates from sciatic nerves of PMP22tg mutants (data not shown). Next, we performed quantitative real-time PCR to examine the level of MCP-1/CCL2 mRNA in peripheral nerves of treated mice. MCP-1/CCL2 mRNA was reduced by approximately 50% in nerves of CI-1040–treated PMP22tg mutants compared with nerves of PMP22tg mutants treated with the vehicle DMSO (Figure 7B).
Figure 7.
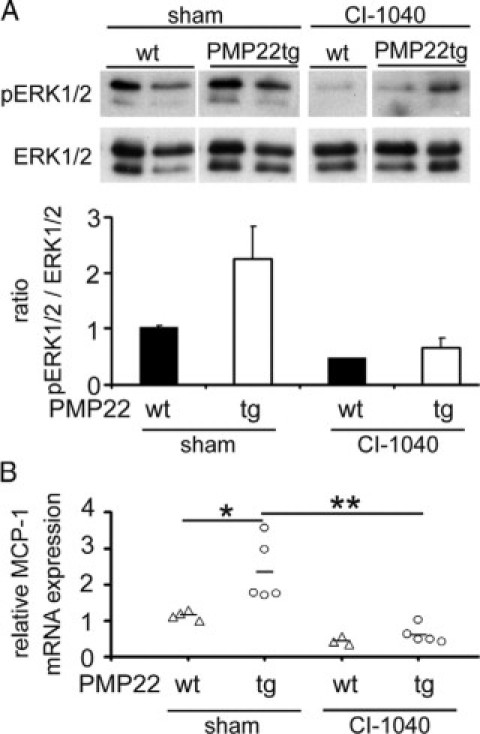
Increased MCP-1/CCL2-expression in peripheral nerves of PMP22tg mice is mediated by the MEK1/2/ERK1/2 signaling cascade. In vivo inhibition of the MEK1/2/ERK1/2 cascade and MCP-1/CCL2-expression by daily treatment with CI-1040 for three weeks. A: Western blot analyses and the corresponding densitometric measurement of femoral quadriceps nerve lysates of CI-1040 and sham-treated mice at the age of two months. Data are shown as a mean value of two experiments with three to five mice in each group. Note the decreased phosphorylation of ERK1/2 (44/42 kDa) in nerves of mice with daily CI-1040 injection. B: MCP-1/CCL2 mRNA is reduced in nerves of CI-1040–treated PMP22tg mice in comparison with sham-treated mutants. Treatment of wild-type mice does not significantly change the MCP-1/CCL2-expression. *P < 0.05, Student t test, **P < 0.01.
Discussion
In the present study, we demonstrated that, in peripheral nerves of PMP22tg mice, a model for the most common form of CMT, MCP-1/CCL2, was upregulated. We found that the chemokine is Schwann cell–derived and plays a substantial role in the increase of macrophage numbers in the myelin mutant nerves. This was established by cross-breeding experiments with the corresponding MCP-1/CCL2 knockout mutants. Moreover, our experiments revealed that, in the myelin mutants, not only macrophage numbers are correlated with MCP-1/CCL2 expression, but also with features indicative of axon damage: At high (wt) doses of MCP-1/CCL2, K+ channels were maldistributed and axon function was poor, as reflected by CMAP and grip test strengths, whereas in the complete absence of MCP-1/CCL2, axon integrity was substantially ameliorated. Accordingly, intermediate levels of axon function were detected in PMP22tg mutants with intermediate levels of MCP-1/CCL2. Because of these tight correlations between macrophage activation and axon damage, it is plausible to assume that macrophages activated by MCP-1/CCL2 might be detrimental for axon integrity. A possible candidate molecule in axon damage is TNFα, because it is highly expressed in PMP22tg/MCP-1+/+ mutants (which show axonopathy) but substantially less expressed in MCP-1/CCL2-deficient mice with reduced axonopathy (data not shown). Indeed, TNFα is involved in axon damage in inflammatory neuropathies (28 for review). However, because the cognate receptor of MCP-1/CCL2, CCR2, is amply expressed on neurons, it is also possible that the chemokine itself—in addition to effects on macrophages—may have direct effects on neuronal features.29–31
We found a strong correlation between the CMAP amplitude, the grip strength, and the distribution of K+ channels in PMP22tg/MCP-1+/+ versus PMP22tg/MCP-1−/− mice. At first, we considered the possibility that reduced axon numbers may explain these changes. However, this could not be confirmed by counting myelin-competent axons by electron microscopy in different peripheral nerves, including the plantar nerve (Kohl and Martini, unpublished). Furthermore, using pre- and postsynaptic markers at the light microscopic level, we found no substantial alterations in the neuromuscular junctions of the different genotypes (Kohl and Martini, unpublished). However, quantitative electron microscopy of neuromuscular junctions of the various mutants has not yet been performed to identify more subtle, yet functionally relevant alterations. Thus, the only axonal alterations we identified so far were the abnormal distribution of K+ channels. Because K+ channels are known to be involved in the restoration of the resting potential, functional abnormalities might become particularly obvious under tonic stimulation conditions, as might occur in the course of our grip strength investigations. It is, therefore, possible that the reduced muscle strength is directly related to the abnormal K+ channel distribution.
With regard of features typical of demyelination (as opposed to axonal features), reduced and null levels of MCP-1/CCL2-expression lead to a significantly improved phenotype, with a more robust trend to amelioration in the MCP-1+/− genotypes. An even stronger ameliorating effect in MCP-1+/− versus MCP-1−/− genotypes has been seen in P0+/− mice where demyelinating features were only ameliorated when MCP-1/CCL2-expression was reduced by 50%.10 In both P0 and PMP22 mutants, the null-level of MCP-1/CCL2 was associated with an increase of pro-inflammatory cytokines. Whereas in PMP22tg/MCP-1−/− mice predominantly IL-6, LIF, and M-CSF were upregulated (data not shown), in the corresponding P0+/−/MCP-1−/− mutant M-CSF and TNFα were elevated. Additionally, the number of foamy macrophages was highest in nerves of P0+/−/MCP-1−/− mice. It is, therefore, possible that genetic loss of MCP-1/CCL2 leads to a compensatory reaction in the mutant Schwann cells resulting in an upregulation of cytokines and increase of myelin damage.
It is of note that one myelin-related feature—hypermyelination—was not dependent on MCP-1/CCL2-expression. It is conceivable that a primary hypermyelinating neuropathy is one of the first pathological hallmarks related to PMP22 overexpression, which might be directly or indirectly related to the upregulation of another chemokine, CXCL14.32 Pathological features being independent of otherwise pathogenetically-relevant immune reactions in CMT models have previously been identified in Cx32-deficient mutants where, in this case, the number of periaxonal swellings was independent of immune cells.33 Thus, it is tempting to postulate that distinct mutants might be characterized by individual “primary” (immune-independent) and more common “secondary” (immune-dependent) features.
The moderate upregulation of MCP-1/CCL2 mRNA by a factor of two to three in PMP22tg mice is a typical feature of a chronic demyelinating disorder. Comparable levels are found in other models for CMT, the P0+/− mice10 and the Connexin-32-mutant mice (Groh, Fischer and Martini, unpublished observations). By contrast, acute lesions in peripheral nerves leading to Wallerian degeneration show a much stronger and timely restricted upregulation of MCP-1/CCL2 that at least partially mediates the well-established and robust infiltration of phagocytic macrophages.34,35 Whereas in Wallerian degeneration the majority of macrophages is infiltrating,35 the situation in models for CMT appears to be more complex, implicating both the increase of resident macrophages and infiltration from an external pool.10,36
Interestingly, axonopathic features appeared to be ameliorated independently from demyelinating features, as reflected by the different degrees of axon- or myelin-related improvement at reduced or absent levels of MCP-1/CCL2. It is of note in this context that a similar uncoupling of axonopathic changes and demyelination has been detected in the course of a treatment approach of another CMT1A model, the CMT1A rat.6 Because axon damage is the determinating clinical culprit in demyelinating disorders,37,38 it is most challenging to develop treatment strategies focusing on axon preservation. Our study suggests that one possible approach might be the inhibition of MCP-1 synthesis in mutant Schwann cells, possibly via inactivation of the identified MEK1/2-ERK1/2 pathways.
Acknowledgements
The authors are grateful to Heinrich Blazyca, Carolin Kiesel, Nadine Weckesser, Silke Loserth, and Bettina Meyer for expert technical assistance, and to Helga Brünner and Karl-Heinz Aulenbach for excellent animal care. Valuable discussions with Jakob Troppmair (Innsbruck) and Hans-Werner Müller (Düsseldorf) are also acknowledged.
Footnotes
Supported by the German Research Foundation (SFB 581 and MA1053/5-1/2).
The MEK-inhibitor CI-1040 was generously provided by Pfizer (Groton, CT).
Supplemental material for this article can be found on http://ajp.amjpathol.org.
Web Extra Material
{kind=link}
References
- 1.Scherer SS, Wrabetz L. Molecular mechanisms of inherited demyelinating neuropathies. Glia. 2008;56:1578–1589. doi: 10.1002/glia.20751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berger P, Niemann A, Suter U. Schwann cells and the pathogenesis of inherited motor and sensory neuropathies (Charcot-Marie-Tooth disease) Glia. 2006;54:243–257. doi: 10.1002/glia.20386. [DOI] [PubMed] [Google Scholar]
- 3.Nave KA, Sereda MW, Ehrenreich H. Mechanisms of disease: inherited demyelinating neuropathies–from basic to clinical research. Nat Clin Pract Neurol. 2007;3:453–464. doi: 10.1038/ncpneuro0583. [DOI] [PubMed] [Google Scholar]
- 4.Niemann A, Berger P, Suter U. Pathomechanisms of mutant proteins in Charcot-Marie-Tooth disease. Neuromolecular Med. 2006;8:217–242. doi: 10.1385/nmm:8:1-2:217. [DOI] [PubMed] [Google Scholar]
- 5.Sereda MW, Meyer zu Horste G, Suter U, Uzma N, Nave KA. Therapeutic administration of progesterone antagonist in a model of Charcot-Marie-Tooth disease (CMT-1A) Nat Med. 2003;9:1533–1537. doi: 10.1038/nm957. [DOI] [PubMed] [Google Scholar]
- 6.Meyer zu Horste G, Prukop T, Liebetanz D, Mobius W, Nave KA, Sereda MW. Antiprogesterone therapy uncouples axonal loss from demyelination in a transgenic rat model of CMT1A neuropathy. Ann Neurol. 2007;61:61–72. doi: 10.1002/ana.21026. [DOI] [PubMed] [Google Scholar]
- 7.Passage E, Norreel JC, Noack-Fraissignes P, Sanguedolce V, Pizant J, Thirion X, Robaglia-Schlupp A, Pellissier JF, Fontes M. Ascorbic acid treatment corrects the phenotype of a mouse model of Charcot-Marie-Tooth disease. Nat Med. 2004;10:396–401. doi: 10.1038/nm1023. [DOI] [PubMed] [Google Scholar]
- 8.Pennuto M, Tinelli E, Malaguti M, Del Carro U, D'Antonio M, Ron D, Quattrini A, Feltri ML, Wrabetz L. Ablation of the UPR-mediator CHOP restores motor function and reduces demyelination in Charcot-Marie-Tooth 1B Mice. Neuron. 2008;57:393–405. doi: 10.1016/j.neuron.2007.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ip CW, Kroner A, Fischer S, Berghoff M, Kobsar I, Mäurer M, Martini R. Role of immune cells in animal models for inherited peripheral neuropathies. Neuromol Med. 2006;8:175–189. doi: 10.1385/nmm:8:1-2:175. [DOI] [PubMed] [Google Scholar]
- 10.Fischer S, Kleinschnitz C, Müller M, Kobsar I, Ip CW, Rollins BJ, Martini R. Monocyte chemoattractant protein-1 is a pathogenic component in a model for a hereditary peripheral neuropathy. Mol Cell Neurosci. 2008;37:359–366. doi: 10.1016/j.mcn.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 11.Fischer S, Weishaupt A, Troppmair J, Martini R. Increase of MCP-1 (CCL2) in myelin mutant Schwann cells is mediated by MEK-ERK signaling pathway. Glia. 2008;56:836–843. doi: 10.1002/glia.20657. [DOI] [PubMed] [Google Scholar]
- 12.Kobsar I, Hasenpusch-Theil K, Wessig C, Muller HW, Martini R. Evidence for macrophage-mediated myelin disruption in an animal model for Charcot-Marie-Tooth neuropathy type 1A. J Neurosci Res. 2005;81:857–864. doi: 10.1002/jnr.20601. [DOI] [PubMed] [Google Scholar]
- 13.Huxley C, Passage E, Manson A, Putzu G, Figarella-Branger D, Pellisier JF, Fontes M. Construction of a mouse model of Charcot-Marie-Tooth disease type 1A by pronuclear injection of human YAC DNA. Hum Mol Gent. 1996;5:563–569. doi: 10.1093/hmg/5.5.563. [DOI] [PubMed] [Google Scholar]
- 14.Huxley C, Passage E, Robertson AM, Youl B, Huston S, Manson A, Sabéran-Djoniedi D, Figarella-Branger D, Pellisier JF, Thomas PK, Fontes M. Correlation between varying levels and the degree of demyelination and reduction in nerve conduction velocity in transgenic mice. Hum Mol Genet. 1998;7:449–458. doi: 10.1093/hmg/7.3.449. [DOI] [PubMed] [Google Scholar]
- 15.Lu B, Rutledge BJ, Gu L, Fiorillo J, Lukacs NW, Kunkel SL, North R, Gerard C, Rollins BJ. Abnormalities in monocyte recruitment and cytokine expression in monocyte chemoattractant protein 1-deficient mice. J Exp Med. 1998;187:601–608. doi: 10.1084/jem.187.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carenini S, Mäurer M, Werner A, Blazyca H, Toyka KV, Schmid CD, Raivich G, Martini R. The role of macrophages in demyelinating peripheral nervous system of mice heterozygously deficient in P0. J Cell Biol. 2001;152:301–308. doi: 10.1083/jcb.152.2.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schmid CD, Stienekemeier M, Oehen S, Bootz F, Zielasek J, Gold R, Toyka KV, Schachner M, Martini R. Immune deficiency in mouse models for inherited peripheral neuropathies leads to improved myelin maintenance. J Neurosci. 2000;20:729–735. doi: 10.1523/JNEUROSCI.20-02-00729.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ulzheimer JC, Peles E, Levinson SR, Martini R. Altered expression of ion channel isoforms at the node of Ranvier in P0-deficient myelin mutants. Mol Cell Neurosci. 2004;25:83–94. doi: 10.1016/j.mcn.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 19.Samsam M, Mi W, Wessig C, Zielasek J, Toyka KV, Coleman MP, Martini R. The wlds mutation delays robust loss of motor and sensory axons in a genetic model for myelin-related axonopathy. J Neurosci. 2003;23:2833–2839. doi: 10.1523/JNEUROSCI.23-07-02833.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Masu Y, Wolf E, Holtmann B, Sendtner M, Brem G, Thoenen H. Disruption of the CNTF gene results in motor neuron degeneration. Nature. 1993;365:27–32. doi: 10.1038/365027a0. [DOI] [PubMed] [Google Scholar]
- 21.Holtmann B, Wiese S, Samsam M, Grohmann K, Pennica D, Martini R, Sendtner M. Triple knock-out of CNTF. LIF, and CT-1 defines cooperative and distinct roles of these neurotrophic factors for motoneuron maintenance and function. J Neurosci. 2005;25:1778–1787. doi: 10.1523/JNEUROSCI.4249-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sebolt-Leopold JS, Dudley DT, Herrera R, Van Becelaere K, Wiland A, Gowan RC, Tecle H, Barrett SD, Bridges A, Przybranowski S, Leopold WR, Saltiel AR. Blockade of the MAP kinase pathway suppresses growth of colon tumors in vivo. Nat Med. 1999;5:810–816. doi: 10.1038/10533. [DOI] [PubMed] [Google Scholar]
- 23.Goebeler M, Gillitzer R, Kilian K, Utzel K, Brocker EB, Rapp UR, Ludwig S. Multiple signaling pathways regulate NF-kappaB-dependent transcription of the monocyte chemoattractant protein-1 gene in primary endothelial cells. Blood. 2001;97:46–55. doi: 10.1182/blood.v97.1.46. [DOI] [PubMed] [Google Scholar]
- 24.Boekhoudt GH, Guo Z, Beresford GW, Boss JM. Communication between NF-kappa B and Sp1 controls histone acetylation within the proximal promoter of the monocyte chemoattractant protein 1 gene. J Immunol. 2003;170:4139–4147. doi: 10.4049/jimmunol.170.8.4139. [DOI] [PubMed] [Google Scholar]
- 25.Sheng WS, Hu S, Ni HT, Rowen TN, Lokensgard JR, Peterson PK. TNF-alpha-induced chemokine production and apoptosis in human neural precursor cells. J Leukoc Biol. 2005;78:1233–1241. doi: 10.1189/jlb.0405221. [DOI] [PubMed] [Google Scholar]
- 26.Waetzig V, Czeloth K, Hidding U, Mielke K, Kanzow M, Brecht S, Goetz M, Lucius R, Herdegen T, Hanisch UK. c-Jun N-terminal kinases (JNKs) mediate pro-inflammatory actions of microglia. Glia. 2005;50:235–246. doi: 10.1002/glia.20173. [DOI] [PubMed] [Google Scholar]
- 27.Yoo JK, Kwon H, Khil LY, Zhang L, Jun HS, Yoon JW. IL-18 induces monocyte chemotactic protein-1 production in macrophages through the phosphatidylinositol 3-kinase/Akt and MEK/ERK1/2 pathways. J Immunol. 2005;175:8280–8286. doi: 10.4049/jimmunol.175.12.8280. [DOI] [PubMed] [Google Scholar]
- 28.Stubgen JP. Tumor necrosis factor-alpha antagonists and neuropathy. Muscle Nerve. 2008;37:281–292. doi: 10.1002/mus.20924. [DOI] [PubMed] [Google Scholar]
- 29.Melik-Parsadaniantz S, Rostene W. Chemokines and neuromodulation. J Neuroimmunol. 2008;198:62–68. doi: 10.1016/j.jneuroim.2008.04.022. [DOI] [PubMed] [Google Scholar]
- 30.Jung H, Toth PT, White FA, Miller RJ. Monocyte chemoattractant protein-1 functions as a neuromodulator in dorsal root ganglia neurons. J Neurochem. 2008;104:254–263. doi: 10.1111/j.1471-4159.2007.04969.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Madrigal JLM, Leza JC, Polak P, Kalinin S, Feinstein DL. Astrocyte-derived MCP-1 mediates neuroprotective effects of noradrenaline. J Neurosci. 2009;29:263–267. doi: 10.1523/JNEUROSCI.4926-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barbaria EM, Kohl B, Buhren BA, Hasenpusch-Theil K, Kruse F, Kury P, Martini R, Muller HW. The alpha-chemokine CXCL14 is up-regulated in the sciatic nerve of a mouse model of Charcot-Marie-Tooth disease type 1A and alters myelin gene expression in cultured Schwann cells. Neurobiol Dis. 2009;33:448–458. doi: 10.1016/j.nbd.2008.11.014. [DOI] [PubMed] [Google Scholar]
- 33.Kobsar I, Berghoff M, Samsam M, Wessig C, Maurer M, Toyka KV, Martini R. Preserved myelin integrity and reduced axonopathy in connexin32-deficient mice lacking the recombination activating gene-1. Brain. 2003;126:804–813. doi: 10.1093/brain/awg072. [DOI] [PubMed] [Google Scholar]
- 34.Kleinschnitz C, Brinkhoff J, Zelenka M, Sommer C, Stoll G. The extent of cytokine induction in peripheral nerve lesions depends on the mode of injury and NMDA receptor signaling. J Neuroimmunol. 2004;149:77–83. doi: 10.1016/j.jneuroim.2003.12.013. [DOI] [PubMed] [Google Scholar]
- 35.Martini R, Fischer S, Lopez-Vales R, David S. Interactions between Schwann cells and macrophages in injury and inherited demyelinating disease. Glia. 2008;56:1566–1577. doi: 10.1002/glia.20766. [DOI] [PubMed] [Google Scholar]
- 36.Muller M, Berghoff M, Kobsar I, Kiefer R, Martini R. Macrophage colony stimulating factor is a crucial factor for the intrinsic macrophage response in mice heterozygously deficient for the myelin protein P0. Exp Neurol. 2007;203:55–62. doi: 10.1016/j.expneurol.2006.07.018. [DOI] [PubMed] [Google Scholar]
- 37.Berciano J, Garcia A, Calleja J, Combarros O. Clinico-electrophysiological correlation of extensor digitorum brevis muscle atrophy in children with Charcot-Marie-Tooth disease 1A duplication. Neuromusc Disord. 2000;10:419–424. doi: 10.1016/s0960-8966(99)00114-5. [DOI] [PubMed] [Google Scholar]
- 38.Berciano J, Combarros O. Hereditary neuropathies. Curr Opin Neurol. 2003;16:613–622. doi: 10.1097/01.wco.0000093105.34793.dd. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.