

Abstract
Toll-like receptor (TLR) engagement by pathogen-associated molecular patterns (PAMPs) is an important mechanism for optimal cellular immune responses. APC TLR engagement indirectly enhances activated CD4+ T cell proliferation, differentiation, and survival by promoting the up-regulation of costimulatory molecules and the secretion of proinflammatory cytokines. However, TLRs are also expressed on CD4+ T cells, suggesting that PAMPs may also act directly on activated CD4+ T cells to mediate functional responses. In this study, we show that activated mouse CD4+ T cells express TLR-3 and TLR-9 but not TLR-2 and TLR-4. Treatment of highly purified activated CD4+ T cells with the dsRNA synthetic analog poly(I:C) and CpG oligodeoxynucleotides (CpG DNA), respective ligands for TLR-3 and TLR-9, directly enhanced their survival without augmenting proliferation. In contrast, peptidoglycan and LPS, respective ligands for TLR-2 and TLR-4 had no effect. Enhanced survival mediated by either poly(I:C) or CpG DNA required NF-κB activation and was associated with Bcl-xL up-regulation. However, only CpG DNA, but not poly(I:C)-mediated effects on activated CD4+ T cells required the TLR/IL-1R domain containing adaptor molecule myeloid differentiation factor 88. Collectively, our results demonstrate that PAMPs can directly promote activated CD4+ T cell survival, suggesting that TLRs on T cells can directly modulate adaptive immune responses.
Toll-like receptors (TLRs)2 mediate the recognition of pathogen-associated molecular patterns (PAMPs) by cells of the innate immune system allowing the detection of infection and inflammation (1). On APCs, PAMP engagement of TLRs promotes maturation, a process characterized by the up-regulation of MHC and costimulatory molecules and the secretion of proinflammatory cytokines, which in turn leads to the induction of proliferation and survival pathways in Ag-specific CD4+ T cells (2). Several distinct molecular pathways contribute to these effects. For example, TCR engagement activates NF-κB, a transcription factor that mediates many inflammatory responses and is important in maintaining activated CD4+ T cell survival (3). TCR survival signals are further enhanced by costimulation through CD28 that promotes the synthesis of the prosurvival molecule Bcl-xL and the cytokine IL-2 (4). IL-2 in turn provides survival signals to activated CD4+ T cells through induction of Bcl-2 (5). Furthermore, PAMP-stimulated APCs also secrete type I IFNs and IL-15, both of which enhance activated CD4+ T cell survival following cessation of APC TCR engagement (6, 7). Thus, PAMPs clearly promote activated CD4+ T cell survival indirectly by initiating maturation responses from APCs.
Interestingly, CD4+ T cells also express TLRs suggesting that PAMPs may directly induce activated CD4+ T cell survival (8, 9). TLR expression has been reported on γδ T cells and regulatory CD4+CD25+ T cells. However, the function of TLRs on CD4+ T cells remains poorly understood. It has been recently reported that stimulation of TLR-4 on regulatory T cells increases the suppressive activity and proliferation of these cells (9). However, whether PAMPs are capable of inducing direct functional responses in activated nonregulatory CD4+ T cells or whether TLR-mediated responses in CD4+ T cells use the same signaling pathways that have previously been described in APCs is not known.
TLR signaling is initiated through at least two pathways: one dependent on the adaptor molecule myeloid differentiation factor 88 (MyD88) and the other that is MyD88 independent (10). All TLRs utilize the MyD88 pathway but not all TLRs are dependent on it to mediate all functional responses to PAMPs (11). For example, TLR-4-mediated IL-6 and TNF-α synthesis by dendritic cells (DCs) is dependent on MyD88 but maturation responses such as costimulatory molecule up-regulation are relatively independent (12, 13). In contrast, all TLR-9-mediated functional responses are dependent on MyD88 (14). Nevertheless, both pathways lead to the activation of NF-κB and the mitogen-activated protein (MAP) kinases (15).
In this study, we demonstrate that activated CD4+ T cells express RNA from TLR-3, TLR-5, and TLR-9 but not TLR-2 and TLR-4. To assess the functional significance of this pattern of TLR expression, we added TLR ligands to highly purified preparations of activated CD4+ T cells. Both poly(I:C) or CpG DNA, respective ligands for TLR-3 and TLR-9, but not PGN or LPS, respective ligands for TLR-2 and TLR-4, directly enhanced the survival of activated CD4+ T cells in vitro and in vivo. CpG DNA, but not poly(I:C)-mediated survival was dependent on MyD88. However, both CpG DNA or poly(I:C) were dependent on NF-κB activation to mediate survival in activated CD4+ T cells. We conclude that PAMPs directly promote the survival of activated CD4+ T cells and present evidence that this effect is mediated directly through CD4+ T cell expressed TLRs.
Materials and Methods
Mice
BALB/c mice were purchased from The Jackson Laboratory (Bar Harbor, ME). DO11.10 mice on the BALB/c background have been described previously (16). MyD88−/− mice were a generous gift from S. Akira (17). For these experiments MyD88+/− mice were backcrossed at least five times onto a C57BL/6 background and intercrossed to generate MyD88−/− and MyD88+/+ wild-type control littermates.
CD4+ T cell purification
In experiments using BALB/c CD4+ T cells, splenocytes and lymph node cells were pooled, erythrocyte-depleted by hypotonic lysis, and labeled with CD4-FITC mAb (GK1.5; BD Biosciences, Mountain View, CA) and CD25-PE mAb (PC61; BD Biosciences). Labeled cells were sorted by a FACSVantage high-speed sorter (BD Biosciences) into CD25−CD4+ populations and then incubated with CD44-biotin mAb (IM7; BD Biosciences) and the following mixture of biotinylated mAbs from the MACS CD4+ T cell isolation kit (Miltenyi Biotec, Auburn, CA): CD8a (Ly-2), CD11b (Mac-1), CD45R (B220), pan NK (DX5), and Ly-76 (TER-119). These cells were then further incubated with anti-biotin magnetic beads (Miltenyi Biotec) and purified over LS columns (Miltenyi Biotec) in accordance with the manufacturer’s recommendations to obtain the naive CD44lowCD25−CD4+ T cell fraction (purity >99%). In experiments using DO11.10, MyD88−/− or MyD88+/+ wild-type littermate control mice, CD4+ T cells were directly purified from erythrocyte-depleted splenocyte and lymph node cells with the MACS CD4+ T cell isolation kit. Purity in these fractions exceeded 96%. The remainder of the cells were CD8+ T cells. APC contamination could not be detected by FACS analysis. However, the presence of APCs was routinely assessed by RT-PCR for MHC class II IAβ message (see Materials and Methods). By the limits of detection in our RT-PCR assay, exceeding one APC in 1000 CD4+ T cells, the purified CD4+ T cell preparations used in all of these studies have <0.1% APC contamination (data not shown).
CD4+ T cell activation
All CD4+ T cell activation was conducted in complete culture medium composed of RPMI 1640 (Life Technologies, Grand Island, NY), 1.5 μM 2-ME (Sigma-Aldrich, St. Louis, MO), 50 μg/ml gentamicin (Life Technologies), and 10% FCS (Mediatech, Washington, DC) at 37° C in 5% CO2. Purified CD4+ T cells from either BALB/c, MyD88−/−, or MyD88+/+ wild-type control littermates were activated on 24-well plates (Costar, Cambridge, MA) coated with 1.0 μg/ml CD3ε mAb (2C11; BD Biosciences) and 1.0 μg/ml CD28 mAb (37.51; BD Biosciences) for 16 h. In experiments with DO11.10 mice, 1 μg/ml of pOVA, a peptide derived from chicken albumin amino acid residues 322–332 and synthesized at the Protein Chemistry Facility of the University of Pennsylvania, was added to 2 × 106/ml erythrocyte-depleted splenocyte and lymph node cell pools for 16 h. Following pOVA-induced activation, CD4+ T cell APC complexes were disrupted with 5 mM EDTA/PBS for 10 min at 25°C, washed twice in PBS, and purified with magnetic beads using the MACS CD4+ T cell isolation kit as described above. Purity of activated DO11.10 CD4+ T cells exceeded 96%. As described above, the remainder of the contaminants by FACS analysis were CD8+ T cells, with APCs at <0.1% by RT-PCR.
Semiquantitative RT-PCR
APCs were prepared from BALB/c pooled splenocyte and lymph node cells that were T cell depleted with MACS anti-CD90.2 beads (Miltenyi Biotec). APC, naïve, and activated CD4+ T cell total RNA was prepared by lysis with RLT buffer (Qiagen, Valencia, CA) and with buffers and columns supplied from the RNAeasykit with DNase I (Qiagen) in accordance with the manufacturer’s instructions. RNA was then reversed transcribed using and amplified with the TITANIUM One Step RT-PCR kit (Clontech Laboratories, Palo Alto, CA) under nonsaturating conditions. The following PCR cycling conditions were used: one cycle at 95°C for 3 min followed by 25–28 cycles of 94.5°C for 30 s and 60°C for 1 min and a final cycle at 72°C for 20 min. Specific primer sequences were as follows: 5′ TLR-2, TGCATCACCGGTCAGAAAACAACT; 3′ TLR-2, GGCCC GAACCAGGAGGAAGATAAA; 5′ TLR-3, CCCCTCGCTCTTTTTATG GAC; 3′ TLR-3, CCTGGCCGCTGAGTTTTTGTTC; 5′ TLR-4, GCCCCG CTTTCACCTCTG; 3′ TLR-4, TGCCGTTTCTTGTTCTTCCTCT; 5′ TLR-5, CAGCCCCGTGTTGGTAATA; 3′ TLR-5, CCCGGAATGAAGA ATGGAG; 5′ TLR-9, CTATACAGCCTGCGCGTT-CTCTTC; 3′ TLR-9, AGCTTGCGCAGGCGGGTTAGGTTC; 5′ I-Aβd, ACGC-GGGCCGAGGT GGACA; 3′ I-Aβd, GCCCCCGATGCGGGCTCAAC; 5′ G3PDH, ACCA CAGTCCATGCCATCAC; and 3′ G3PDH, TCCACCACCCTGTTGCT GTA. PCR products were resolved by 2% agarose gel electrophoresis, stained with ethidium bromide, and imaged with a Gel Doc analyzer (Bio-Rad, Hercules, CA).
TLR ligand and inhibitor reagents
The CpG oligonucleotide TCCATGACGTTCCTGACGTT (CpG DNA) and non-CpG oligonucleotide TCCATGAGCTTCCTGAGCTT (non-CpG DNA) have been described previously (18) and were synthesized on a phosphorothioate backbone and purified by HPLC (Life Technologies). Poly(I:C), poly(C), and poly(dI:dC) were purchased from Amersham Biosciences (Arlington Heights, IL) and LPS, derived from the O55:B5 Escherichia coli strain, was purchased from Sigma-Aldrich. PGN was purchased from Invivogen (Carlsbad, CA). TLR ligands used in all experiments were dissolved in PBS except for PGN, which was solubilized in PBS with 0.02% ethanol. SB203580, U0126, NEMO-binding domain peptide (NBD), and NBD-C were all dissolved in DMSO and purchased from Calbiochem (La Jolla, CA).
NF-κB and MAP kinase signaling analysis
BALB/c CD44lowCD25−CD4+ T cells were activated with plate-bound 1.0 μg/ml anti-CD3 and 1.0 μg/ml anti-CD28 mAbs for 16 h, washed, and rested for 8 h at 37°C. Activated (1.5 × 106) CD4+ T cells were then treated with TLR ligands for the indicated times, lysed in 1× SDS loading buffer (Bio-Rad Life Sciences), resolved on a 12% bis-Tris SDS-PAGE gel (Life Technologies), transferred to nitrocellulose filters (Life Technologies), and either probed with rabbit anti-mouse phospho-specific Abs for p-IκBα, p-p38, p-extracellular signal-regulated kinase (ERK) 1/2, or p-C-Jun N-terminal kinase (JNK)/stress-activated protein kinase (SAPK). To assess total amounts of signaling molecules, filters were also probed with either rabbit anti-mouse Iκβα, p-38, ERK-1/2, or JNK/SAPK Abs. Detection was conducted with HRP-conjugated goat ant-rabbit Abs, ECL reagent (Amersham), and X-OMAT Film (Kodak, Rochester, NY). All Abs were purchased from Cell Signal Technologies (Beverly, MA).
Survival and proliferation analysis
Following activation, purified CD4+ T cells were washed twice in PBS and replated in culture medium at 106/ml. Cultures were left untreated or treated with either TLR ligands and/or inhibitors for indicated times and concentrations. Following incubation, CD4+ T cells were washed twice in PBS/2% FBS and stained with CD4-allophycocyanin mAb and 7-aminoactinomycin D (7-AAD; BD PharMingen) or annexin (BD PharMingen) and survival was assessed by exclusion of either of these two stains. For absolute live cell counts 50,000 CD45.1+ splenocytes labeled with anti-CD45.1-PE mAb (A20; BD PharMingen) were also added to stained CD4+ T cell sample FACS tubes just before FACS analysis. Live CD4+ T cell counts were calculated by taking the ratio of the number of CD4+CD45.1− 7-ADD− events collected to the number CD45.1+ events collected and multiplying by 50,000. Proliferation was measured by CFSE dye dilution as previously described (19).
Adoptive transfer and ex vivo proliferation analysis
Five million purified DO11.10 CD4+ T cells were activated by pOVA-pulsed APCs (1 μg/ml), purified with magnetic beads, treated with poly(I:C) (90 μg/ml), CpG DNA (30 μM), LPS (100 ng/ml), or left untreated for 16 h, washed in PBS twice, and then adoptively transferred into BALB/c hosts. At day 30, spleen and peripheral lymph nodes were harvested and stained with anti-CD4 mAb and the DO11.10 clonotypic mAb KJI-26. Survival of activated DO11.10 CD4+ T cells in each host was determined by FACS analysis and is expressed as a percent with respect to the total number of CD4+ T cells found in either the host spleen or lymph nodes along with a mean (thick line) for each treatment group. To measure ex vivo proliferative responses, 72-h quadruplicate cultures were prepared in 96-well plates with 50,000 irradiated T cell-depleted pOVA-pulsed (1 μg/ml) splenocytes and 150,000 CD4+ T cells purified with the MACS CD4+ T cell isolation kit from day 30 peripheral lymph nodes or spleen pooled from each treatment group. [3H]Thymidine was added to cultures for an additional 8 h and read on a beta scintillation counter and is expressed as a mean for each treatment group ± SEM.
Analysis of antiapoptotic molecules
For Bcl-2 evaluation, CD4+ T cells were permeabilized with 0.1% saponin/0.2% FBS/PBS and stained with anti-Bcl-2 PE mAb (3F-11; BD PharMingen) or an isotype hamster IgG-PE control (A19–3;BD PharMingen) and staining was analyzed by FACS. Bcl-xL, Bcl-3, and β-actin were analyzed by Western blotting (see above) using rabbit anti-mouse Abs specific for Bcl-xL (BD Transduction Laboratories, Lexington, KY), Bcl-3 (Santa Cruz Biotechnology, Santa Cruz, CA), and β-actin (Accurate Labs, Westbury, NY).
Results
CD4+ T cells modulate TLR expression in response to TCR stimulation
To examine TLR expression patterns in activated CD4+ T cells, highly purified naive CD44lowCD25− CD4+ T cells were either left to rest for 8 h or activated for 16 h with plate-bound anti-CD3 and anti-CD28 mAbs and RT-PCR was performed under nonsaturating conditions for TLRs that are known to have naturally occurring ligands (Fig. 1). TLR-2, -3, -4, -5, and -9 expression was detected in naive CD4+ T cells before activation. However, after stimulation, TLR-4 and TLR-2 RNA expression was undetectable while TLR-3 and TLR-9 message was up-regulated. To exclude the possibility that this pattern of TLR expression was partially a result of APC contamination, we also examined I-Aβ chain expression. By the limits of detection of this RT-PCR measurement, which is sensitive to <0.1% APC contamination, no I-Aβ chain RNA expression was detected in CD4+ T cells before or after activation (for further details, please see Materials and Methods). Therefore our results show that CD4+ T cells express TLR RNA and modulate its expression following TCR stimulation.
FIGURE 1.
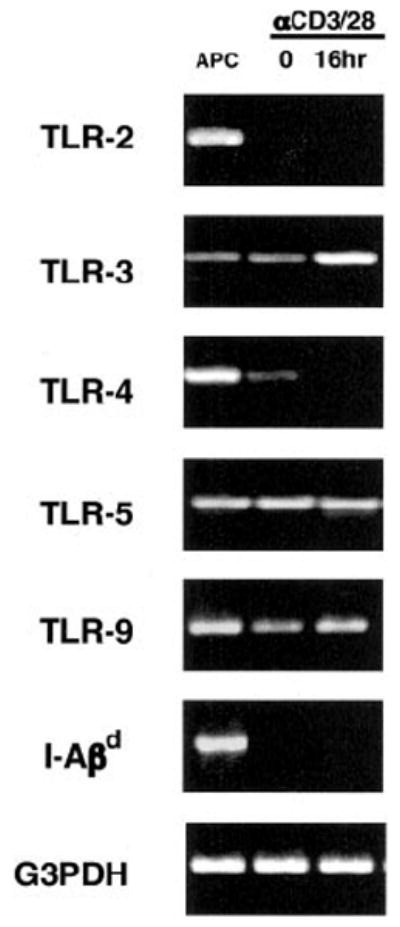
TLR RNA expression patterns in activated CD4+ T cells. T cell-depleted pooled BALB/c spleen and lymph node cells (APC), BALB/c CD44lowCD25−CD4+ T cells, or BALB/c CD44lowCD25−CD4+ T cells activated for 16 h with plate-bound anti-CD3 plus anti-CD28 mAbs were harvested and lysed for total RNA. RNA was reverse transcribed and amplified under nonsaturating conditions with TLR-specific primers, G3PDH primers, or I-Aβd primers to monitor APC contamination. PCR products were then resolved by agarose gel electrophoresis and stained with ethidium bromide. The result is representative of three independently performed experiments.
Poly(I:C) and CpG DNA but not LPS induce NF-κB and MAP kinase activity in activated CD4+ T cells
TLR ligands induce the activation of nuclear factor NF-κB and MAP kinases in APCs (15, 20). We therefore asked whether TLR ligands could also induce NF-κB and MAP kinase activity in activated CD4+ T cells (Fig. 2). Both the TLR-3 ligand poly(I:C) and the TLR-9 ligand CpG DNA were able to induce rapid NF-κB activity as evident by phosphorylation of IκBα. In a likewise manner, both ligands were also able to effect phosphorylation of p38 MAP kinase (MAPK), ERK 1/2, and JNK/SAPK. In contrast, LPS did not induce detectable IκBα or MAP family kinase activity consistent with the absence of TLR-4 in activated CD4+ T cells. Thus, TLR ligands are able to activate downstream signaling pathways in a manner concordant with the cognate TLR expression pattern in activated CD4+ T cells.
FIGURE 2.
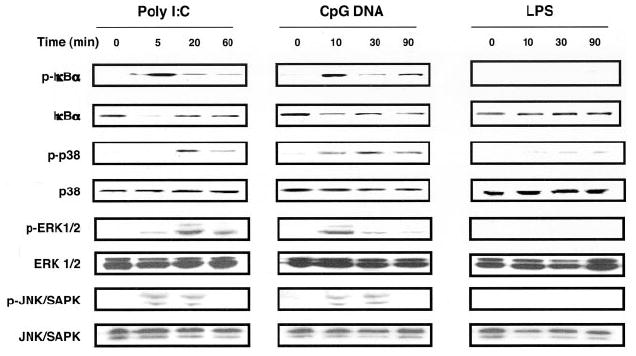
Poly(I:C) or CpG DNA but not LPS induce NF-κB and MAPK signaling in activated CD4+ T cells. BALB/c CD44lowCD25−CD4+ T cells were activated with plate-bound anti-CD3 plus anti-CD28 mAbs for 16 h, washed, replated, and rested for an additional 8 h. Following rest, CD4+ T cells were treated with either CpG DNA (30 μM), poly(I:C) (90 μg/ml), or LPS (1 μg/ml) for the indicated times, lysed, and analyzed by Western blot with Abs specific for total or phosphorylated forms of IκBα, p38 MAPK, ERK1/2, and JNK/SAPK. The result is representative of three independently performed experiments.
Poly(I:C) or CpG DNA directly enhance activated CD4+ T cell survival
TLR ligands directly promote the survival of several cell types including neutrophils, DC, and B cells (21-23). Since we observed that activated CD4+ T cells also express TLRs and signal in response to TLR ligands, we asked whether TLR ligands could directly enhance survival of these cells. DO11.10 CD4+ T cells, that encode a transgenic TCR specific for a peptide derived from chicken OVA (pOVA), were activated with pOVA-pulsed APCs for 16 h ex vivo, purified by magnetic beads to remove all non-CD4+ T cells, and replated in unsupplemented culture medium in the absence or presence of TLR ligands. Survival was then assessed 72 h following activation (Fig. 3A). Poly(I:C) or CpG DNA induced increases of activated CD4+ T cell survival from 38% to 71 and 73%, respectively. By contrast, PGN or LPS did not significantly enhance activated CD4+ T cell survival promoting only marginal increases from 38 to 41% and 43%, respectively. TLR ligand enhanced mediated survival was comparable to IFN-α, which has been previously reported to enhance the survival for activated T cells (24), improving survival from 38 to 67%. We also observed dose responsive increases of activated CD4+ T cell survival in poly(I:C) and CpG DNA-treated cultures (Fig. 3B). Enhanced survival was not a nonspecific response to nucleic acids since the addition poly(C), poly(dI:dC), and a control non-CpG DNA had no significant effect. Moreover, LPS and PGN treatment also did not produce significant increments in survival consistent with the absence of detectable TLR-2 and TLR-4 RNA expression on activated CD4+ T cells.
FIGURE 3.
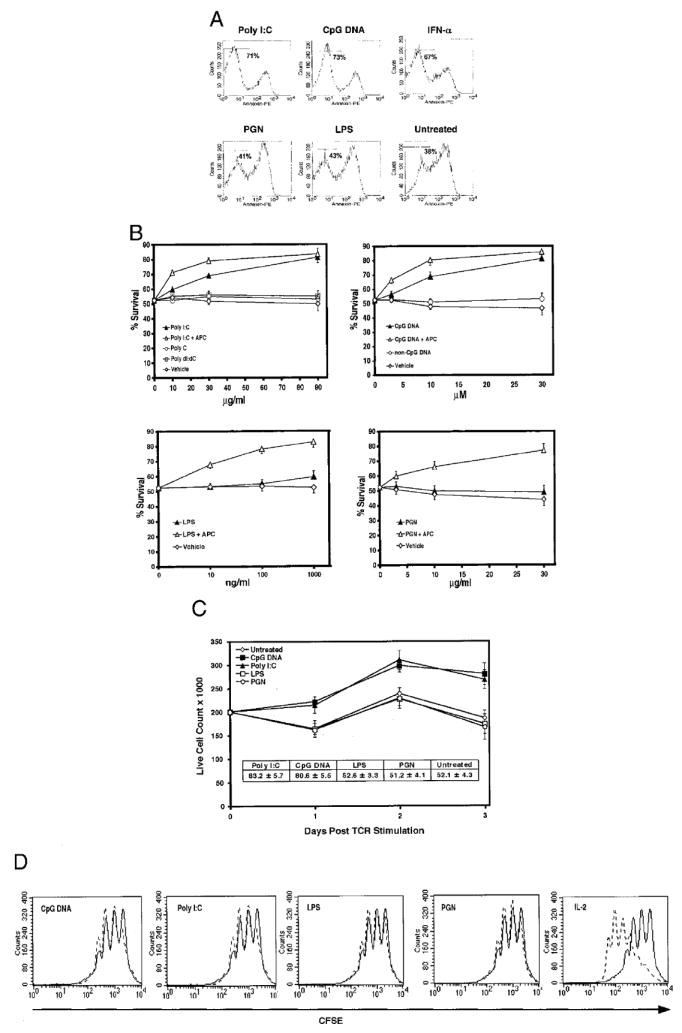
Poly(I:C) or CpG DNA directly enhances the survival but not the proliferation of activated CD4+ T cells. A, Purified DO11.10 CD4+ T cells, first activated with pOVA-pulsed APCs for 16 h and then purified by magnetic beads, were treated with either poly(I:C) (90 μg/ml), CpG DNA (30 μM), LPS (1 μg/ml), PGN (30 μg/ml), IFN-α (1000 U/ml), or left untreated for 72 h and stained with annexin. The result is representative of three independently performed experiments. B, Purified activated DO11.10 CD4+ T cells, as prepared in A, were treated with graded amounts of TLR ligands or associated controls in the presence or absence of APCs. Survival was assessed by the exclusion of 7-AAD for four independently performed experiments ± SEM. C, BALB/c CD44lowCD25−CD4+ T cells were activated with plate-bound anti-CD3 plus anti-CD28 mAbs, washed, replated at 200,000 cells/well, and treated with either poly(I:C) (90 μg/ml), CpG DNA (30 μM), LPS (1 μg/ml), PGN (30 μg/ml), or left untreated. At the end of 24-, 48-, and 72-h periods, absolute live cell counts were performed. Seventy-two-hour percentage survival is represented in the inset. Results are from four independently performed experiments ± SEM. D, CFSE labeled purified activated DO11.10 CD4+ T cells, prepared as in A, were cultured in either medium alone (solid lines), IL-2 (50 U/ml; dotted line), or with TLR ligands (dotted lines), poly(I:C) (90 μg/ml), CpG DNA (30 μM), LPS (100 ng/ml), or PGN (30 μg/ml) for 72 h and assessed for proliferation by CFSE dilution.
To exclude the possibility that poly(I:C) or CpG DNA enhanced survival was an indirect effect arising from contamination with APCs or other non-T cell TLR-bearing cells in our purified CD4+ T cell preparations, TLR ligand-treated cultures were also spiked with a T cell-depleted preparation of pooled lymph node cells and splenocytes (APC). The addition of APCs to poly(I:C) and CpG DNA-treated cultures significantly increased dose-responsive survival over TLR ligand-treated cultures alone, suggesting synergy between indirect and direct mechanisms of activated CD4+ T cell survival. However, in LPS- or PGN-treated cultures only the addition of APCs could enhance the survival of activated CD4+ T cells, thus indicating the functional absence of other TLR-responsive cells in our purified CD4+ T cell preparations.
Since percentage survival measurements may not be reflective of viable CD4+ T cell numbers in vitro, we also quantified absolute live cell number counts for periods up to 72 h following activation of FACS sorted CD4+ T cells with plate-bound anti-CD3 plus anti-CD28 mAbs (Fig. 3C). Consistent with increases in percentage cell survival, concordant increases in live CD4+ T cell numbers were observed in poly(I:C) or CpG DNA-treated cultures. By comparison, there were no significant increases in live cell numbers in LPS- or PGN-treated cultures relative to untreated cultures.
In addition to reflecting enhanced cell survival, the increases in live cells numbers we observed could be explained by TLR ligand-mediated increases in CD4+ T cell proliferation. In B cells, LPS, CpG DNA, and poly(I:C) can induce proliferation independently of the B cell receptor (25-27). Moreover, LPS has been recently shown to augment regulatory CD4+ T cell proliferation (9). To address this issue, CFSE-labeled DO11.10 CD4+ T cells were first activated by pOVA-pulsed APCs for 16 h, purified by magnetic beads, and then treated with TLR ligands and assessed for proliferation 72 h following activation (Fig. 3D). As expected LPS and PGN treatment, which did not promote direct enhancement of survival of activated CD4+ T cells, did not induce more robust proliferative responses in comparison to untreated activated controls. However, and unlike the cytokine IL-2, poly(I:C) and CpG DNA also did not enhance proliferation relative to untreated controls, indicating that the observed increases in viable CD4+ T cell numbers were not due to proliferation differences across cultures but solely reflected the enhancement of cell survival.
Poly(I:C) and CpG DNA-mediated survival of activated CD4+ T cells is dependent on NF-κB activation
Activation of NF-κB is known to be associated with survival responses in activated CD4+ T cells (3, 28). To determine whether TLR ligand augmented survival responses in activated CD4+ T cells was also dependent on NF-κB activation, we inhibited IκB phosphorylation by using a lipid-soluble peptide (NBD) that selectively binds to the NF-κB essential modifier (NEMO) and blocks its association with the IκB kinases IKKα and IKKβ (IKKαβ) (29) (Fig. 4A). NEMO-IKKαβ interaction is necessary for IκB signal-induced phosphorylation and thus inhibiting this association prevents subsequent IκB degradation and NF-κB translocation to the nucleus (30). Previous work has shown that NBD does not modulate JNK activity unlike peptides that directly inhibit NF-κB translocation (29). We found that blockade of NF-κB activation by NBD inhibited the ability of both poly(I:C) or CpG DNA to enhance activated CD4+ T cell survival. These effects were dose dependent. At 20 μM NBD, TLR ligand augmentation of activated CD4+ T cell was nearly completely reversed. As a control, we also treated cultures with a closely related but inactive lipid-soluble form of the peptide NBD-C and observed no significant loss of TLR ligand-mediated survival.
FIGURE 4.
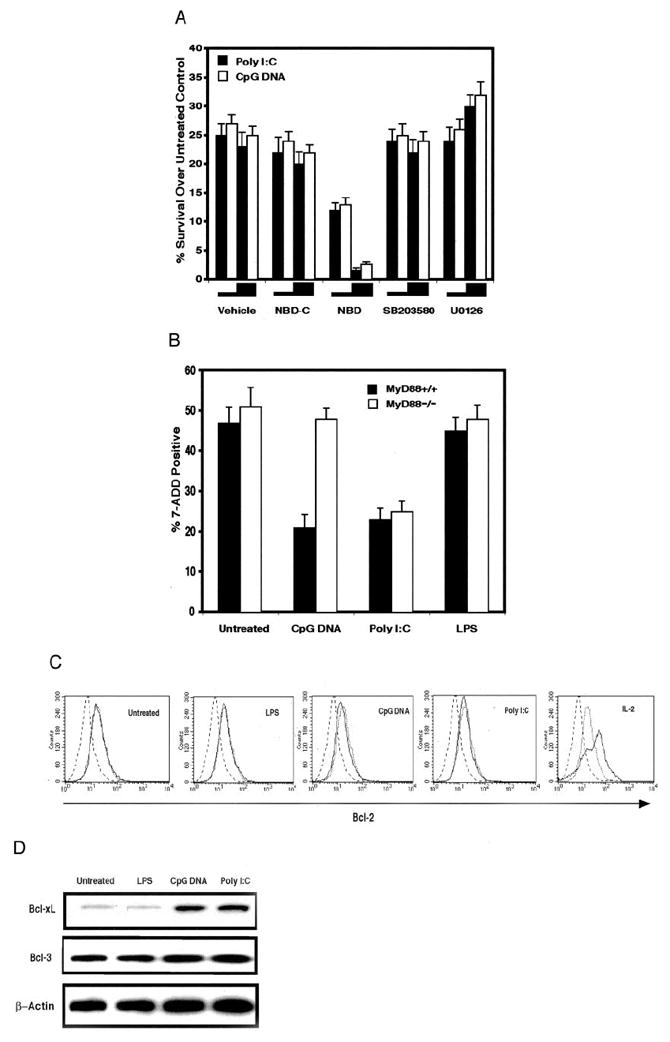
Poly(I:C) or CpG DNA-mediated survival requires NF-κB activation and is associated with Bcl-xL up-regulation but only CpG DNA-mediated survival is MyD88 dependent. A, Purified DO11.10 CD4+ T cells, first activated with pOVA-pulsed APCs for 16 h and then purified by magnetic beads, were treated with poly(I:C) (90 μg/ml), CpG DNA (30 μM), or left untreated and coincubated with NBD (5 or 20 μM), NBD-C (5 or 20 μM), SB203580 (2.5 or 10 μM), U0126 (2.5 or 10 μM), or vehicle control for 48 h. Viability is expressed as the percentage difference of 7-AAD excluding cells between TLR ligand-treated and untreated cells. Results are from four independently performed experiments ± SEM. B, Magnetic bead-purified CD4+ T cells from MyD88−/− and wild-type control littermates (MyD88+/+) were activated with plate-bound anti-CD3 plus anti-CD28 mAbs, washed, and replated with either poly(I:C) (30 μg/ml), CpG DNA (10 μM), LPS (100 ng/ml), or left untreated for 72 h. Death was assessed as the percentage of 7-AAD positively stained cells. Results are from three independently performed experiments ± SEM. C, Purified activated DO11.10 CD4+ T cells, prepared as in A, were treated with either poly(I:C) (90 μg/ml), CpG DNA (30 μM), LPS(1 μg/ml), PGN (30 μg/ml), IL-2 (50 U/ml), or left untreated for 48 h. TLR ligand-treated cells (solid lines), IL-2-treated cells (solid line), or untreated cells (dotted line) were stained for Bcl-2 or an isotype control (dashed line). The result is representative of three independently performed experiments. D, Purified activated DO11.10 CD4+ T cells, prepared as in A, were treated with either poly(I:C) (90 μg/ml), CpG DNA (30 μM), LPS (1 μg/ml), or left untreated 48 h, lysed, and analyzed by Western blot with Abs specific for mouse Bcl-xL, Bcl-3, or β-actin.
MAPK p38 and ERK 1/2 are also activated by TLR ligands and their function has previously been shown to be important in controlling T cell-mediated inflammatory responses including survival (31). Therefore, we asked whether MAPK p38 or ERK 1/2 activation is necessary for TLR ligand-mediated survival in activated CD4+ T cells. We added the ERK1/2 activation inhibitor U0126 or the MAPK p38 inhibitor SB203580 to TLR ligand-treated activated CD4+ T cells and assessed viability. Neither U0126 or SB203580 treatment decreased poly(I:C) and CpG DNA enhanced survival although we noted that a concentration of 10 μM U0126 induced a small increase in overall survival rates of TLR-treated activated CD4+ T cells. Thus, NF-κB but not MAPK p38 or ERK1/2 activation is required to mediate TLR ligand-induced survival of activated CD4+ T cells.
CpG DNA but not poly(I:C)-mediated survival of activated CD4+ T cells is dependent on MyD88
MyD88 is an adaptor molecule recruited to TLRs by TLR ligand engagement and is known to mediate inflammatory responses to many PAMPs (32). The absence of MyD88 in APCs makes them completely unresponsive to CpG DNA and is therefore thought to be essential in all TLR-9-mediated responses (14). In contrast, deficiency in MyD88 APCs partially eliminates TLR-3-mediated cytokine synthesis but leaves NF-κB, MAPK, and DC maturation responses intact (27). Therefore to examine the role of MyD88 in TLR ligand-mediated survival responses in activated CD4+ T cells, we treated MyD88−/− activated CD4+ T cells with CpG DNA and poly(I:C) and assessed survival (Fig. 4B). We found that MyD88 was required to mediate CpG DNA augmented survival of activated CD4+ T cells. In contrast, poly(I:C)-enhanced survival responses were left intact in MyD88−/− activated CD4+ T cells. Therefore, our results demonstrate that at least two signaling pathways, MyD88 dependent and MyD88 independent, are capable of mediating direct TLR ligand augmented survival in activated CD4+ T cells.
Poly(I:C) or CpG DNA treatment of activated CD4+ T cells up-regulates Bcl-xL but not Bcl-2 or Bcl-3
Members of the Bcl family are mediators of activated CD4+ T cell survival. Bcl-2 and Bcl-xL are both up-regulated in CD4+ T cells following Ag priming (33). Bcl-3 has been reported to be up-regulated in activated CD4+ T cells isolated from adjuvant-treated mice and in overexpression studies has been reported to increase survival (34). We therefore measured levels of each of these molecules following TLR ligand treatment of activated CD4+ T cells (Fig. 4, C and D). Bcl-2 protein levels were not changed by TLR ligand treatment relative to untreated activated CD4+ T cell controls. Additionally, Bcl-3 protein levels were also left unaffected despite the fact that all of the TLR ligands used in the experiment are also used as adjuvants (35). However, we observed significant increases in Bcl-xL protein in CpG DNA and poly(I:C)- treated activated CD4+ T cells over LPS-treated and untreated activated CD4+ T cells. Thus, directly mediated activated CD4+ T cell survival is associated with specific Bcl-xL up-regulation.
Poly(I:C) or CpG DNA treatment of activated CD4+ T cells enhances their survival in vivo
Although we could induce TLR ligand-mediated survival of activated CD4+ T cells in vitro, it still remained uncertain whether these cells had a preferential survival advantage in vivo. To address this question, DO11.10 CD4+ T cells were first activated with pOVA-pulsed APCs, purified by magnetic beads, treated with TLR ligands for 16 h, washed, and then adoptively transferred into congenic BALB/c hosts. Survival and ex vivo proliferative responses were assessed 30 days later (Fig. 5). Consistent with what has been previously reported for activated effector CD4+ T cells, adoptively transferred activated DO11.10 CD4+ T cells seem to preferentially home to the spleen rather than to the peripheral lymph nodes (36). Importantly, poly(I:C) or CpG DNA treatment of activated DO11.10 CD4+ T cells increased the percentage of recovered T cells in the host spleen by nearly 2-fold in comparison to spleens from mice that received either LPS-treated or untreated DO11.10 CD4+ T cells. Likewise, this 2-fold increase in the percentage of splenic DO11.10 CD4+ T cells from hosts that received either poly(I:C)- or CpG DNA-treated activated DO11.10 CD4+ T cells was mirrored by a 2-fold increase in the proliferative response to pOVA in comparison to pOVA-induced proliferative responses of splenic CD4+ T cells from hosts that received either LPS-treated or untreated activated DO11.10 CD4+ T cells. These data suggest that TLR ligands improve recall responses by increasing the number of Ag-specific CD4+ T cells in vivo following activation.
FIGURE 5.
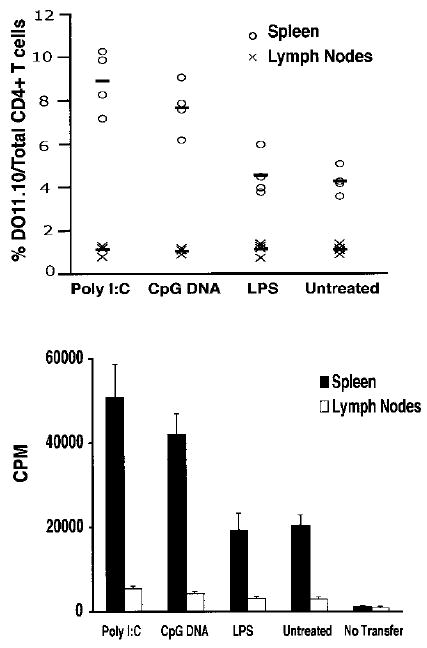
Activated CD4+ T cell survival in vivo is enhanced by either poly(I:C) or CpG DNA treatment before adoptive transfer into naive hosts. Five × 106 DO11.10 CD4+ T cells, first activated by pOVA-pulsed APCs for 16 h and then purified by magnetic beads, were treated with poly(I:C) (90 μg/ml), CpG DNA (30 μM), LPS (100 ng/ml), or left untreated for 16 h, washed, and then adoptively transferred into naive syngeneic BALB/c hosts. Following a 30-day rest, spleen and lymph nodes were harvested and stained with anti-CD4 mAb and the DO11.10 clonotypic mAb KJI-26 or fractionated for CD4+ T cells with magnetic beads and stimulated with pOVA-loaded APCs for 72 h and pulsed with [3H]thymidine for an additional 8 h. Survival of activated DO11.10 CD4+ T cells is shown for each mouse and is expressed as a percent with respect to the total number of CD4+ T cells found in either the host spleen or lymph nodes along with a mean (thick line) for each treatment group. DO11.10 CD4+ T cell proliferative responses are expressed as a mean for each treatment group ± SEM. Both survival and proliferation measurement data are from the same experiment and are representative of three independently performed experiments.
Discussion
In this report, we first examined TLR message levels in naive CD44lowCD25−CD4+T cells and activated CD4+ T cells. We found that activated CD4+ T cells, in contrast to naive CD4+T cells, do not express TLR-4 and TLR-2 and increase the expression of TLR-3 and TLR-9 in response to TCR engagement. RNA message levels were used as a proxy for expression due to a lack of Abs that recognize mouse TLRs. Recent studies have presented an incomplete picture regarding TCR expression in T cells. Nevertheless, both TLR-3 and TLR-9 messages have been found in resting CD4+ T cell preparations where naive and activated cells have not been fractionated and in mouse T cell lines (37-39). In a single report where the TLR-4 message was specifically assessed in plate-bound anti-CD3 plus anti-CD28 mAb-activated regulatory and nonregulatory CD4+ T cells, TLR-4 expression was detected in the regulatory but not in the nonregulatory population (9). However, in contrast to our work and previous studies, TLR-3 and TLR-9 expression was not found on naive CD4+ T cells although activated CD4+ T cells were not specifically investigated.
As previous studies conducted with TLR ligand-treated APCs from TLR knockout mice demonstrated that NF-κB and MAPK induction requires the expression of cognate TLRs, we used TLR ligands to validate our observed pattern of TLR expression in activated CD4+ T cells (18, 27, 40). Poly(I:C) and CpG DNA, but not LPS, induced phosphorylation of I-κB, p38 MAPK, ERK ½, and JNK/SAPK. Thus, TLR-associated downstream activation pathways are activated by TLR ligands in a manner that matches the observed pattern of TLR expression in activated CD4+ T cells.
To further investigate the possible involvement of TLRs in these responses, we used activated CD4+ T cells from mice that are MyD88 deficient. Our observed requirement for MyD88 to promote CpG DNA-mediated survival in activated CD4+ T cells strongly indicates that TLR-9 is mediating these responses since all functional responses mediated by TLR-9 have been reported to be MyD88 dependent (14). In contrast, most TLR-3-mediated poly(I:C) responses are MyD88 independent including NF-κB activation (27). Poly(I:C) can also directly activate two intracellular pattern recognition receptors, dsRNA-dependent protein kinase (PKR) and 2′-oligoadenylate synthetase/RNase L (41, 42). Both of these factors when functioning coordinately in virally infected cells inhibit protein translation leading to apoptosis, thus making them unlikely targets to mediate poly(I:C)-induced survival. However, intracellular PKR activation does induce NF-κB and MAPK responses, which still raises the possibility that survival responses may be initiated by this manner (43). We argue that this is quite unlikely since intracellular poly(I:C) PKR-mediated TLR-3-independent responses requires liposomal encapsulation of poly(I:C) (44). If PKR activation does play a role in survival, it may be via an indirect mechanism through its recruitment to the TLR-3 proximal signaling complex following poly(I:C) stimulation (45).
Since TLR ligand-mediated survival in several cell types is NF-κB dependent, we asked whether the same were true in activated CD4+ T cells. We chose to inhibit NF-κB activation with NBD, a peptide that prevents IκB phosphorylation through selectively preventing the association of IKKαβ with its regulatory protein NEMO. In the MyD88-dependent TLR signaling pathway, IKKαβ activation has been shown to be requisite for IκB phosphorylation (46). Moreover, LPS-mediated B cell survival requires the presence of IKKβ and IKKα (47, 48). For some functional responses, MyD88-independent IκB phosphorylation may be controlled by two other IKK homologues, IKKε and TANK-binding kinase 1 (TBK-1), both of which have been shown to control poly(I:C)-induced IFN-β synthesis (49, 50). Our observations of MyD88-independent poly(I:C)-mediated survival in activated CD4+ T cells raises the question of whether IKKε and TBK-1 could also be playing a role in mediating survival responses. We believe this is not likely, since NBD which blocks IKKαβ, but not IKKε/TBK-1 activity, was able to inhibit poly(I:C)-mediated survival of activated CD4+ T cells. Moreover, poly(I:C) signaling through TLR-3 has been previously reported to activate IKKαβ, and catalytically inactive IKKβ mutants inhibit poly(I:C)-mediated NF-κB-dependent transcription (50, 51). Thus, our data in activated CD4+ T cells indicates that the IKKαβ/NEMO complex promotes TLR ligand-mediated NF-κB activation to enhance survival. In contrast, we present evidence that MAPK p38 or ERK 1/2 activation is not necessary for TLR ligand-activated CD4+ T cell survival.
We also examined the effects of TLR ligands on the expression levels of prosurvival molecules. Recognizing studies that suggest that PAMP-mediated survival may be dependent on Bcl-3 levels (34), we first assessed levels of this molecule in TLR ligand-treated activated CD4+ T cells. We did not observe significant differences in Bcl-3 expression in poly(I:C) or CpG DNA-treated activated CD4+ T cells when compared with untreated activated CD4+ T cell controls. Our observations may be explained by the dependence on CD40 costimulation to promote Bcl-3 up-regulation in activated CD4+ T cells in these previous studies (52). Bcl-2 levels and Bcl-xL levels were also examined in TLR ligand-treated activated CD4+ T cells and we found that Bcl-xL but not Bcl-2 is up-regulated following TLR ligand treatment. This result is in agreement with previous work on PAMP-stimulated B cells and DCs (21, 23). Since the Bcl-xL gene is known to be a downstream target of NF-κB, we suggest that Bcl-xL is promoting TLR ligand-mediated survival (53).
In conclusion, we present evidence that TLR ligands directly enhance the survival of activated CD4+ T cells. TLR-mediated survival had the net effect of increasing expansion and slowing contraction rates of activated CD4+ T cells without accentuating proliferation. It has been hypothesized that adjuvant-induced activated CD4+ T survival can be mediated through APCs by the secretion of proinflammatory cytokines (28). Interestingly, our studies indicate that for two such PAMPs that are effective adjuvants, poly(I:C) and CpG DNA, adjuvant-mediated survival of activated CD4+ T cells may not require APCs. Moreover, in contrast to the indirect means by which TLR ligands control CD4+ T cell responses through APCs, direct effects may allow Ag-specific CD4+ T cells to respond to PAMPs in situations where APC function is suboptimal, perhaps due to infection (54). For example, some viruses which use dsRNA intermediates in their own life cycle, also encode products that inhibit DC maturation and cytokine synthesis and thereby promote infection by attenuating appropriate CD4+ T cell responses (55, 56). This effect could be counteracted by augmented CD4+ T cell survival responses driven by the release of PAMPs such as dsRNA. Thus, like cells in the innate immune system, activated CD4+ T cells may also retain the capability to sense the inflammatory environment by directly responding to PAMPs. This may represent a novel mechanism by which PAMPs promote adaptive immune responses.
Acknowledgments
We thank Shizuo Akira for providing us with the MyD88-deficient mice. We also thank Amer Beg, Patrick Walsh, Steve Bensinger, Oriol Sunyer, Adam Schrum, and Katlin Kariko for helpful discussions.
Footnotes
Abbreviations used in this paper: TLR, Toll-like receptor; PAMP, pathogen-associated molecular pattern; MyD88, myeloid differentiation factor 88; DC, dendritic cell; MAP, mitogen-activated protein; PGN, peptidoglycan; ERK, extracellular signal-regulated kinase; JNK, c-Jun N-terminal kinase; SAPK, stress-activated protein kinase; 7-AAD, 7-aminoactinomycin D; MAPK, MAP kinase; NEMO, NF-κB essential modifier; NBD, NEMO-binding domain peptide; PKR, dsRNA-dependent protein kinase; TBK-1, TANK-binding kinase 1.
References
- 1.Medzhitov R, Preston-Hurlburt P, Janeway CA., Jr A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 2.Kaisho T, Akira S. Dendritic-cell function in Toll-like receptor- and MyD88-knockout mice. Trends Immunol. 2001;22:78. doi: 10.1016/s1471-4906(00)01811-1. [DOI] [PubMed] [Google Scholar]
- 3.Zheng Y, Vig M, Lyons J, Van Parijs L, Beg AA. Combined deficiency of p50 and cRel in CD4+ T cells reveals an essential requirement for nuclear factor κB in regulating mature T cell survival and in vivo function. J Exp Med. 2003;197:861. doi: 10.1084/jem.20021610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boise LH, Minn AJ, Noel PJ, June CH, Accavitti MA, Lindsten T, Thompson CB. CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-XL. Immunity. 1995;3:87. doi: 10.1016/1074-7613(95)90161-2. [DOI] [PubMed] [Google Scholar]
- 5.Mueller DL, Seiffert S, Fang W, Behrens TW. Differential regulation of bcl-2 and bcl-x by CD3, CD28, and the IL-2 receptor in cloned CD4+ helper T cells: a model for the long-term survival of memory cells. J Immunol. 1996;156:1764. [PubMed] [Google Scholar]
- 6.Oshiumi H, Matsumoto M, Funami K, Akazawa T, Seya T. TI-CAM-1, an adaptor molecule that participates in Toll-like receptor 3-mediated interferon-β induction. Nat Immunol. 2003;4:161. doi: 10.1038/ni886. [DOI] [PubMed] [Google Scholar]
- 7.Mattei F, Schiavoni G, Belardelli F, Tough DF. IL-15 is expressed by dendritic cells in response to type I IFN, double-stranded RNA, or lipopolysaccharide and promotes dendritic cell activation. J Immunol. 2001;167:1179. doi: 10.4049/jimmunol.167.3.1179. [DOI] [PubMed] [Google Scholar]
- 8.Mokuno Y, Matsuguchi T, Takano M, Nishimura H, Washizu J, Ogawa T, Takeuchi O, Akira S, Nimura Y, Yoshikai Y. Expression of Toll-like receptor 2 on γδ T cells bearing invariant Vγ6/Vδ1 induced by Escherichia coli infection in mice. J Immunol. 2000;165:931. doi: 10.4049/jimmunol.165.2.931. [DOI] [PubMed] [Google Scholar]
- 9.Caramalho I, Lopes-Carvalho T, Ostler D, Zelenay S, Haury M, Demengeot J. Regulatory T cells selectively express Toll-like receptors and are activated by lipopolysaccharide. J Exp Med. 2003;197:403. doi: 10.1084/jem.20021633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takeuchi O, Akira S. MyD88 as a bottle neck in Toll/IL-1 signaling. Curr Top Microbiol Immunol. 2002;270:155. doi: 10.1007/978-3-642-59430-4_10. [DOI] [PubMed] [Google Scholar]
- 11.Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S. Role of adaptor TRIF in the MyD88-independent Toll-like receptor signaling pathway. Science. 2003;301:640. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 12.Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 1999;11:115. doi: 10.1016/s1074-7613(00)80086-2. [DOI] [PubMed] [Google Scholar]
- 13.Akira S, Hoshino K, Kaisho T. The role of Toll-like receptors and MyD88 in innate immune responses. J Endotoxin Res. 2000;6:383. [PubMed] [Google Scholar]
- 14.Schnare M, Holt AC, Takeda K, Akira S, Medzhitov R. Recognition of CpG DNA is mediated by signaling pathways dependent on the adaptor protein MyD88. Curr Biol. 2000;10:1139. doi: 10.1016/s0960-9822(00)00700-4. [DOI] [PubMed] [Google Scholar]
- 15.Akira S. Toll-like receptor signaling. J Biol Chem. 2003;278:38105. doi: 10.1074/jbc.R300028200. [DOI] [PubMed] [Google Scholar]
- 16.Hsieh CS, Heimberger AB, Gold JS, O’Garra A, Murphy KM. Differential regulation of T helper phenotype development by interleukins 4 and 10 in an αβ T-cell-receptor transgenic system. Proc Natl Acad Sci USA. 1992;89:6065. doi: 10.1073/pnas.89.13.6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, Nakanishi K, Akira S. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity. 1998;9:143. doi: 10.1016/s1074-7613(00)80596-8. [DOI] [PubMed] [Google Scholar]
- 18.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 19.Wells AD, Gudmundsdottir H, Turka LA. Following the fate of individual T cells throughout activation and clonal expansion: signals from T cell receptor and CD28 differentially regulate the induction and duration of a proliferative response. J Clin Invest. 1997;100:3173. doi: 10.1172/JCI119873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martin MU, Wesche H. Summary and comparison of the signaling mechanisms of the Toll/interleukin-1 receptor family. Biochim Biophys Acta. 2002;1592:265. doi: 10.1016/s0167-4889(02)00320-8. [DOI] [PubMed] [Google Scholar]
- 21.Lundqvist A, Nagata T, Kiessling R, Pisa P. Mature dendritic cells are protected from Fas/CD95-mediated apoptosis by upregulation of Bcl-XL. Cancer Immunol Immunother. 2002;51:139. doi: 10.1007/s00262-002-0265-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sabroe I, Prince LR, Jones EC, Horsburgh MJ, Foster SJ, Vogel SN, Dower SK, Whyte MK. Selective roles for Toll-like receptor (TLR) 2 and TLR4 in the regulation of neutrophil activation and life span. J Immunol. 2003;170:5268. doi: 10.4049/jimmunol.170.10.5268. [DOI] [PubMed] [Google Scholar]
- 23.Grillot DA, Merino R, Pena JC, Fanslow WC, Finkelman FD, Thompson CB, Nunez G. bcl-x exhibits regulated expression during B cell development and activation and modulates lymphocyte survival in transgenic mice. J Exp Med. 1996;183:381. doi: 10.1084/jem.183.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marrack P, Kappler J, Mitchell T. Type I interferons keep activated T cells alive. J Exp Med. 1999;189:521. doi: 10.1084/jem.189.3.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Andersson J, Sjoberg O, Moller G. Induction of immunoglobulin and antibody synthesis in vitro by lipopolysaccharides. Eur J Immunol. 1972;2:349. doi: 10.1002/eji.1830020410. [DOI] [PubMed] [Google Scholar]
- 26.Sun S, Beard C, Jaenisch R, Jones P, Sprent J. Mitogenicity of DNA from different organisms for murine B cells. J Immunol. 1997;159:3119. [PubMed] [Google Scholar]
- 27.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature. 2001;413:732. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 28.Hildeman DA, Zhu Y, Mitchell TC, Kappler J, Marrack P. Molecular mechanisms of activated T cell death in vivo. Curr Opin Immunol. 2002;14:354. doi: 10.1016/s0952-7915(02)00335-7. [DOI] [PubMed] [Google Scholar]
- 29.May MJ, D’Acquisto F, Madge LA, Glockner J, Pober JS, Ghosh S. Selective inhibition of NF-κB activation by a peptide that blocks the interaction of NEMO with the IκB kinase complex. Science. 2000;289:1550. doi: 10.1126/science.289.5484.1550. [DOI] [PubMed] [Google Scholar]
- 30.Yamaoka S, Courtois G, Bessia C, Whiteside ST, Weil R, Agou F, Kirk HE, Kay RJ, Israel A. Complementation cloning of NEMO, a component of the IκB kinase complex essential for NF-κB activation. Cell. 1998;93:1231. doi: 10.1016/s0092-8674(00)81466-x. [DOI] [PubMed] [Google Scholar]
- 31.Schafer PH, Wang L, Wadsworth SA, Davis JE, Siekierka JJ. T cell activation signals up-regulate p38 mitogen-activated protein kinase activity and induce TNF-α production in a manner distinct from LPS activation of monocytes. J Immunol. 1999;162:659. [PubMed] [Google Scholar]
- 32.Akira S, Yamamoto M, Takeda K. Role of adapters in Toll-like receptor signalling. Biochem Soc Trans. 2003;31:637. doi: 10.1042/bst0310637. [DOI] [PubMed] [Google Scholar]
- 33.Boise LH, Noel PJ, Thompson CB. CD28 and apoptosis. Curr Opin Immunol. 1995;7:620. doi: 10.1016/0952-7915(95)80067-0. [DOI] [PubMed] [Google Scholar]
- 34.Mitchell TC, Hildeman D, Kedl RM, Teague TK, Schaefer BC, White J, Zhu Y, Kappler J, Marrack P. Immunological adjuvants promote activated T cell survival via induction of Bcl-3. Nat Immunol. 2001;2:397. doi: 10.1038/87692. [DOI] [PubMed] [Google Scholar]
- 35.Kaisho T, Akira S. Toll-like receptors as adjuvant receptors. Biochim Biophys Acta. 2002;1589:1. doi: 10.1016/s0167-4889(01)00182-3. [DOI] [PubMed] [Google Scholar]
- 36.Bradley LM, Watson SR, Swain SL. Entry of naive CD4 T cells into peripheral lymph nodes requires L-selectin. J Exp Med. 1994;180:2401. doi: 10.1084/jem.180.6.2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Applequist SE, Wallin RP, Ljunggren HG. Variable expression of Toll-like receptor in murine innate and adaptive immune cell lines. Int Immunol. 2002;14:1065. doi: 10.1093/intimm/dxf069. [DOI] [PubMed] [Google Scholar]
- 38.Zarember KA, Godowski PJ. Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J Immunol. 2002;168:554. doi: 10.4049/jimmunol.168.2.554. [DOI] [PubMed] [Google Scholar]
- 39.Hornung V, Rothenfusser S, Britsch S, Krug A, Jahrsdorfer B, Giese T, Endres S, Hartmann G. Quantitative expression of Toll-like receptor 1–10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J Immunol. 2002;168:4531. doi: 10.4049/jimmunol.168.9.4531. [DOI] [PubMed] [Google Scholar]
- 40.Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol. 1999;162:3749. [PubMed] [Google Scholar]
- 41.Diaz-Guerra M, Rivas C, Esteban M. Activation of the IFN-inducible enzyme RNase L causes apoptosis of animal cells. Virology. 1997;236:354. doi: 10.1006/viro.1997.8719. [DOI] [PubMed] [Google Scholar]
- 42.Gil J, Alcami J, Esteban M. Induction of apoptosis by double-stranded-RNA-dependent protein kinase (PKR) involves the α subunit of eukaryotic translation initiation factor 2 and NF-κB. Mol Cell Biol. 1999;19:4653. doi: 10.1128/mcb.19.7.4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Iordanov MS, Wong J, Bell JC, Magun BE. Activation of NF-κB by double-stranded RNA (dsRNA) in the absence of protein kinase R and RNase L demonstrates the existence of two separate dsRNA-triggered antiviral programs. Mol Cell Biol. 2001;21:61. doi: 10.1128/MCB.21.1.61-72.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Diebold SS, Montoya M, Unger H, Alexopoulou L, Roy P, Haswell LE, Al-Shamkhani A, Flavell R, Borrow P, Reis e Sousa C. Viral infection switches non-plasmacytoid dendritic cells into high interferon producers. Nature. 2003;424:324. doi: 10.1038/nature01783. [DOI] [PubMed] [Google Scholar]
- 45.Jiang Z, Zamanian-Daryoush M, Nie H, Silva AM, Williams BR, Li X. Poly(I-C)-induced Toll-like receptor 3 (TLR3)-mediated activation of NFκB and MAP kinase is through an interleukin-1 receptor-associated kinase (IRAK)-independent pathway employing the signaling components TLR3-TRAF6-TAK1-TAB2-PKR. J Biol Chem. 2003;278:16713. doi: 10.1074/jbc.M300562200. [DOI] [PubMed] [Google Scholar]
- 46.Wang Q, Dziarski R, Kirschning CJ, Muzio M, Gupta D. Micrococci and peptidoglycan activate TLR2→MyD88→IRAK→TRAF→NIK→IKK→NF-κB signal transduction pathway that induces transcription of interleukin-8. Infect Immun. 2001;69:2270. doi: 10.1128/IAI.69.4.2270-2276.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li ZW, Omori SA, Labuda T, Karin M, Rickert RC. IKKβ is required for peripheral B cell survival and proliferation. J Immunol. 2003;170:4630. doi: 10.4049/jimmunol.170.9.4630. [DOI] [PubMed] [Google Scholar]
- 48.Kaisho T, Takeda K, Tsujimura T, Kawai T, Nomura F, Terada N, Akira S. IκB kinase α is essential for mature B cell development and function. J Exp Med. 2001;193:417. doi: 10.1084/jem.193.4.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tojima Y, Fujimoto A, Delhase M, Chen Y, Hatakeyama S, Nakayama K, Kaneko Y, Nimura Y, Motoyama N, Ikeda K, et al. NAK is an IκB kinase-activating kinase. Nature. 2000;404:778. doi: 10.1038/35008109. [DOI] [PubMed] [Google Scholar]
- 50.Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, Maniatis T. IKKε and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. 2003;4:491. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- 51.Gil J, Rullas J, Garcia MA, Alcami J, Esteban M. The catalytic activity of dsRNA-dependent protein kinase, PKR, is required for NF-κB activation. Oncogene. 2001;20:385. doi: 10.1038/sj.onc.1204109. [DOI] [PubMed] [Google Scholar]
- 52.Mitchell TC, Teague TK, Hildeman DA, Bender J, Rees WA, Kedl RM, Swanson B, Kappler JW, Marrack P. Stronger correlation of bcl-3 than bcl-2, bcl-xL, costimulation, or antioxidants with adjuvant-induced T cell survival. Ann NY Acad Sci. 2002;975:114. doi: 10.1111/j.1749-6632.2002.tb05946.x. [DOI] [PubMed] [Google Scholar]
- 53.Caamano J, Hunter CA. NF-κB family of transcription factors: central regulators of innate and adaptive immune functions. Clin Microbiol Rev. 2002;15:414. doi: 10.1128/CMR.15.3.414-429.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Arrode G, Davrinche C. Dendritic cells and HCMV cross-presentation. Curr Top Microbiol Immunol. 2003;276:277. doi: 10.1007/978-3-662-06508-2_13. [DOI] [PubMed] [Google Scholar]
- 55.Jude BA, Pobezinskaya Y, Bishop J, Parke S, Medzhitov RM, Chervonsky AV, Golovkina TV. Subversion of the innate immune system by a retrovirus. Nat Immunol. 2003;4:573. doi: 10.1038/ni926. [DOI] [PubMed] [Google Scholar]
- 56.Engelmayer J, Larsson M, Subklewe M, Chahroudi A, Cox WI, Steinman RM, Bhardwaj N. Vaccinia virus inhibits the maturation of human dendritic cells: a novel mechanism of immune evasion. J Immunol. 1999;163:6762. [PubMed] [Google Scholar]