

Abstract
The terbenzimidazoles are a class of synthetic ligands that poison the human topoisomerase I (TOP1) enzyme and promote cancer cell death. It has been proposed that drugs of this class act as TOP1 poisons by binding to the minor groove of the DNA substrate of TOP1 and altering its structure in a manner that results in enzyme-mediated DNA cleavage. To test this hypothesis, we characterize and compare the binding properties of a 5-phenylterbenzimidazole derivative (5PTB) to the d(GA4T4C)2 and d(GT4A4C)2 duplexes. The d(GA4T4C)2 duplex contains an uninterrupted 8-bp A⋅T domain, which, on the basis of x-ray crystallographic data, should induce a highly hydrated “A-tract” conformation. This duplex also exhibits anomalously slow migration in a polyacrylamide gel, a feature characteristic of a noncanonical global conformational state frequently described as “bent.” By contrast, the d(GT4A4C)2 duplex contains two 4-bp A⋅T tracts separated by a TpA dinucleotide step, which should induce a less hydrated “B-like” conformation. This duplex also migrates normally in a polyacrylamide gel, a feature further characteristic of a global, canonical B-form duplex. Our data reveal that, at 20°C, 5PTB exhibits an ≈2.3 kcal/mol greater affinity for the d(GA4T4C)2 duplex than for the d(GT4A4C)2 duplex. Significantly, we find this sequence/conformational binding specificity of 5PTB to be entropic in origin, an observation consistent with a greater degree of drug binding-induced dehydration of the more solvated d(GA4T4C)2 duplex. By contrast with the differential duplex affinity exhibited by 5PTB, netropsin and 4′,6-diamidino-2-phenylindole (DAPI), two AT-specific minor groove binding ligands that are inactive as human TOP1 poisons, bind to both duplexes with similar affinities. The electrophoretic behaviors of the ligand-free and ligand-bound duplexes are consistent with 5PTB-induced bending and/or unwinding of both duplexes, which, for the d(GA4T4C)2 duplex, is synergistic with the endogenous sequence-directed electrophoretic properties of the ligand-free duplex state. By contrast, the binding to either duplex of netropsin or DAPI induces little or no change in the electrophoretic mobilities of the duplexes. Our results demonstrate that the TOP1 poison 5PTB binds differentially to and alters the structures of the two duplexes, in contrast to netropsin and DAPI, which bind with similar affinities to the two duplexes and do not significantly alter their structures. These results are consistent with a mechanism for TOP1 poisoning in which drugs such as 5PTB differentially target conformationally distinct DNA sites and induce structural changes that promote enzyme-mediated DNA cleavage.
Keywords: isothermal calorimetry, minor groove width and hydration, adenine⋅thymine tracts, DNA bending, ligand-induced stimulation of topoisomerase 1-mediated DNA cleavage
The DNA topoisomerases (TOP1 and TOP2) comprise a unique class of enzymes that alter the topological state of DNA by catalyzing the breakage and subsequent religation of the DNA strands (1). These enzymes recently have been recognized as appealing targets for novel cancer chemotherapeutic agents (2–6). Liu and coworkers recently have identified the bibenzimidazoles Hoechst 33342 and 33258 as representatives of a class of human TOP1 poisons that exhibit cytotoxic behavior in a number of cancer cell lines (7, 8). Crystallographic studies originally reported by the Dickerson group (9) and later by the Wang (10) and Neidle (11) groups, as well as subsequent NMR studies (12–14), reveal these drugs to bind to the minor groove of AT-tract duplex DNA. However, this binding mode alone does not appear sufficient to impart biological activity as a TOP1 poison because other AT-specific, minor groove-directed ligands, such as netropsin, 4′,6-diamidino-2-phenylindole (DAPI), berenil, and distamycin A, are weak or inactive poisons of human TOP1 (8). Thus, minor groove binding alone, although perhaps necessary, is not sufficient to poison TOP1.
Upon DNA binding, Hoechst 33258 and 33342 stimulate TOP1-mediated DNA cleavage by stabilizing the covalent intermediate of the enzyme reaction (the “reversible cleavable complex”) (5–8). The resulting enhancement of enzyme-mediated DNA cleavage ultimately leads to cell death (5, 6). However, the actual mechanism by which the bibenzimidazoles and related minor groove-directed drugs achieve these biochemical/biological effects remains to be determined. The study reported here is designed to further our understanding of the mechanism of TOP1 poisoning by minor groove binding ligands, including defining the underlying molecular forces that control this mechanism.
As noted above, the bibenzimidazole Hoechst 33342 is able to kill a number of different cancer cell types. However, this drug is much less toxic to cancer cells that overexpress multidrug resistance (MDR1) (8). These observations have prompted the synthesis by the LaVoie group of mono-, bi-, and terbenzimidazole derivatives, and evaluation by the Liu group of the abilities of these derivatives to poison TOP1 and promote cancer cell death (15–18). A promising compound to emerge from these studies is the terbenzimidazole derivative 5-phenyl-2-[2′-(benzimidazol-5"-yl)benzimidazol-5′-yl]benzimidazole (5PTB) (Fig. 1), which poisons human TOP1 and is toxic to tumor cells that overexpress MDR1 (16). We already have shown that 5PTB, like Hoechst 33258, exhibits linear dichroism properties consistent with minor groove binding to duplex DNA (19). To evaluate the duplex properties, if any, that 5PTB selectively recognizes, we employ in this study a broad range of spectroscopic, calorimetric, and electrophoretic techniques to characterize the binding of 5PTB to two decameric DNA duplexes that previously have been shown to exhibit differential thermodynamic (20), dynamic (21), and electrophoretic (22) properties. One of the duplexes, d(GA4T4C)2, contains an uninterrupted 8-bp A⋅T tract, assumes a highly hydrated “A-tract” conformation, and exhibits anomalously slow migration in a polyacrylamide gel. The other duplex, d(GT4A4C)2, contains two 4-bp A⋅T tracts separated by a TpA dinucleotide step, assumes a less hydrated “B-like” conformation, and, on the basis of its molecular weight, migrates normally in a polyacrylamide gel. In short, we characterize 5PTB binding to the d(GA4T4C)2 and d(GT4A4C)2 duplexes, which have identical base compositions but differ by the sequence of the central core of eight A⋅T base pairs, a sequence alteration that induces differences in duplex conformation and degree of hydration. As noted above, we selected these two duplexes because electrophoretic (22), thermodynamic (21), and dynamic (20) evidence suggests that the d(GA4T4C)2 duplex is conformationally distinct from the d(GT4A4C)2 duplex. At least two different interpretations have been proposed to define the nature of this structural distinction. According to one interpretation, the d(GA4T4C)2 duplex adopts a “bent” conformation, whereas the d(GT4A4C)2 duplex assumes a “normal” conformation (21). According to a second interpretation, the d(GA4T4C)2 duplex adopts a poly(dA)⋅poly(dT)-like structure (hereafter referred to as an “A-tract” structural motif), while the d(GT4A4C)2 duplex assumes a more normal “B-like” conformation because, as emphasized by the Dickerson group, the central TpA step disrupts the A-tract structural motif (23, 24). Independent of the veracity of either interpretation, the differential structures of these two duplexes make them well suited for studies designed to assess if 5PTB binding is sensitive to such sequence-directed conformational/hydration differences in the ligand-free states of the targeted duplex domains. The results presented here reveal that, relative to the d(GT4A4C)2 duplex, 5PTB preferentially binds to and structurally alters the d(GA4T4C)2 duplex. This binding preference, which contrasts with the lack of binding discrimination exhibited by netropsin and DAPI, suggests that TOP1 poisons such as 5PTB may preferentially target highly hydrated duplex domains with altered A-tract conformations which are characterized by reduced electrophoretic mobilities.
Figure 1.

Structure and atomic numbering of the terbenzimidazole derivative 5PTB.
MATERIALS AND METHODS
Oligonucleotide and Ligand Molecules.
Oligomers were synthesized on a BioSearch 8600 Synthesizer using standard cyanoethyl phosphoramidite chemistry, followed by purification with reverse-phase HPLC. Molar extinction coefficients (ɛ) for the single-stranded oligomers were determined in 10 mM sodium cacodylate (pH 7.0) by enzymatic digestion and subsequent phosphate analysis (25). The following ɛ values [in units of (mol of strand)/liter)−1⋅cm−1] at 260 nm and 95°C were so obtained: 93,300 for d(GA4T4C) and 94,400 for d(GT4A4C).
5PTB was synthesized as previously described (16). The extinction coefficient of 5PTB at 329 nm and 25°C was determined by dry weight to be 22,600 M−1⋅cm−1 in the buffer described below. Netropsin was obtained from Boehringer Mannheim, and DAPI was obtained from Sigma. Both ligands were used without further purification.
All spectroscopic and calorimetric experiments were conducted in 10 mM sodium cacodylate, pH 6.8/10 mM MgCl2/100 mM KCl/1 mM Na2EDTA.
UV Absorption Spectrophotometry.
Absorbance vs. temperature profiles were measured at 260 nm on an AVIV model 14DS spectrophotometer (AVIV Associates; Lakewood, NJ) equipped with a thermoelectrically controlled cell holder and a cell pathlength of 1 cm. The temperature was raised in 0.5°C increments and the samples were allowed to equilibrate for 1 min at each temperature setting. The DNA concentration was 2 μM in duplex, while the ligand concentration was 0–4 μM.
Circular Dichroism (CD) Spectropolarimetry.
All CD measurements were performed at 20°C on an AVIV Model 60DS Spectropolarimeter equipped with a thermoelectrically controlled cell holder and a cell pathlength of 1 cm. Individual 1-ml samples containing 5 μM 5PTB and DNA at concentrations ranging from 0 to 35 μM in duplex were prepared and allowed to equilibrate at room temperature for 9–12 hr prior to their use in the CD experiments. The CD spectrum of each sample then was recorded from 220 to 420 nm, with an averaging time of 3 sec.
Isothermal Stopped-Flow Mixing Microcalorimetry.
Isothermal calorimetric measurements were performed at 20°C in an all-tantalum, differential, stopped-flow, heat conduction microcalorimeter (model DSFC-100, Commonwealth Technology, Alexandria, VA), developed by Mudd and Berger (26, 27). The calorimeter was calibrated chemically by measuring the heat associated with a 1:2 dilution of 10 mM NaCl (26–29). The enthalpies of ligand binding to both the d(GA4T4C)2 and d(GT4A4C)2 duplexes were determined by dividing the measured heat for each reaction by the concentration of bound ligand. When necessary, the appropriate correction for the concentration of bound ligand was deduced from CD equilibrium binding data.
Polyacrylamide Gel Electrophoresis (PAGE).
Nondenaturing PAGE experiments were conducted at room temperature. Stacking gels measured 13 cm × 16 cm × 0.2 cm, with the upper stack containing 10% acrylamide (1 bisacrylamide/19 acrylamide) and the lower stack containing 28% acrylamide (1 bisacrylamide/19 acrylamide). Both the gel and running buffer contained 89 mM Tris⋅borate (pH 8.0), 10 mM MgCl2, 100 mM KCl, and 1 mM Na2EDTA (TBMgKE buffer). DNA samples in TBMgKE buffer were 80 μM in duplex and contained ligand at concentrations ranging from 0 to 400 μM. Glycerol was added to a final concentration of 5% (vol/vol) and a final sample volume of 18 μl. The samples then were loaded on to the gel and electrophoresed at 9 V/cm (≈45 mA) for 14 hr. The gels were stained for 8 hr in a solution of 1:1 formamide/distilled water containing 0.01% Stains-all dye (Acrōs Organics, Fisher Scientific), destained for 45 min in distilled water, and then photographed.
RESULTS AND DISCUSSION
5PTB Binds to and Enhances the Thermal Stabilities of Both Decameric Duplexes, with the Extent of This Binding-Induced Enhancement Being Greater When the d(GA4T4C)2 Duplex Serves as the Host.
UV melting experiments were conducted in the absence and presence of 5PTB to assess the impact, if any, of the ligand on the thermal stabilities of the d(GA4T4C)2 and d(GT4A4C)2 duplexes. The resulting melting profiles are shown in Fig. 2. Note that as one increases the ratio of the total ligand concentration to duplex concentration (rDup), the thermal stabilities of both the d(GA4T4C)2 host duplex (Fig. 2A) and the d(GT4A4C)2 host duplex (Fig. 2B) increase, with rDup ratios higher than those shown having little or no effect on the melting temperature (Tm) of either duplex. These ligand-induced changes in duplex thermal stability are consistent with 5PTB binding to each duplex, with a preference for the duplex state vs. the single-stranded state (30). In fact, optical titrations (not shown) suggest that 5PTB does not bind to the single-stranded states at the temperatures at which they are formed. Consequently, for this system, the ΔTm data reflect differences in the duplex binding properties of 5PTB.
Figure 2.

UV melting profiles at 260 nm for the d(GA4T4C)2 duplex (A) and the d(GT4A4C)2 duplex (B) and their 5PTB complexes at the indicated values of [total ligand]/[duplex], rDup. For clarity of presentation, the melting curves for a given duplex and its 5PTB complexes are normalized by subtraction of both upper and lower baselines.
Further inspection of Fig. 2 reveals that the maximal extent of 5PTB-induced enhancement in duplex thermal stability (ΔTm) is greater for the d(GA4T4C)2 duplex (ΔTm = 19.7°C) than for the d(GT4A4C)2 duplex (ΔTm = 13.2°C). Thus, as measured by differences in ΔTm, 5PTB is able to distinguish between a duplex with an A4T4 central core and the corresponding sequence-isomeric duplex with a T4A4 central core. These melting experiments, however, do not allow us to assess if this differential affinity of 5PTB results from an intrinsic sequence preference for an A4T4 vs. a T4A4 binding site, or if the drug is selecting for a difference in overall duplex structure/hydration (e.g., A-tract/highly hydrated DNA vs. B-like/less hydrated DNA, or “bent” vs. “normal” DNA). Distinguishing between sequence and structural recognition is difficult because the sequence difference between the two duplexes is what induces the difference in structure/hydration; in other words, the properties are coupled.
CD Reveals That 5PTB Binds to Both Duplexes by a Single Motif and with a Common Stoichiometry of One Ligand Molecule per Duplex.
In addition to Tm shifts in the UV melting studies described above, CD spectropolarimetry provides a second means for detecting and characterizing the DNA binding of 5PTB. In this approach, the drug rather than the duplex optical properties are monitored. Fig. 3 shows the CD spectra from 220 to 420 nm obtained by addition of either d(GA4T4C)2 (A) or d(GT4A4C)2 (B) to a solution of 5PTB. Neither the free 5PTB ligand nor the two ligand-free duplexes (spectra not shown) exhibit CD signals between 300 and 390 nm. However, upon addition of either duplex to a solution of 5PTB, substantial CD signals arise near 355 nm (Fig. 3). These induced CD signals, which are indicative of interactions between 5PTB and each host DNA duplex, can be used to detect and to monitor any CD-active DNA binding mode(s).
Figure 3.

CD titrations at 20°C of 5PTB with either d(GA4T4C)2 (A) or d(GT4A4C)2 (B). From bottom to top at 350 nm, the CD spectra in A correspond to 1/rDup ratios ranging from 0 to 3.5, while the CD spectra in B correspond to 1/rDup ratios ranging from 0 to 7.0. (Insets) Normalized molar (mol of total 5PTB/liter) ellipticities at 340 nm vs. 1/rDup for titration. The apparent binding stoichiometries are indicated and correspond to the apparent inflection points.
Inspection of Fig. 3 A and B reveals a single discrete isoelliptic point for either host duplex in the wavelength range between 300 and 390 nm. For the d(GA4T4C)2 duplex, an isoelliptic point is observed at 317 nm, whereas, for the d(GT4A4C)2 duplex, an isoelliptic point is observed at 311 nm. The presence of a single isoelliptic point for both duplexes is consistent with a single optically detectable 5PTB binding event when either duplex serves as the host.
Single-wavelength titration curves extracted from the CD spectra shown in Fig. 3 A and B are presented in the Insets. In these curves, the points represent the experimental molar ellipticities of 5PTB at 340 nm, and the solid lines reflect linear least-squares fits of each apparent linear domain before and after the apparent inflection points. Note that a single inflection point is observed for 5PTB binding to both DNA host duplexes. The presence of one inflection point is consistent with 5PTB binding to each duplex by a single motif. We previously have shown that 5PTB exhibits linear dichroism properties characteristic of binding to the minor groove of an all-A⋅T polymeric DNA duplex (19). By extension, it is reasonable to propose that the single motif by which 5PTB interacts with the d(GA4T4C)2 and d(GT4A4C)2 duplexes also is through binding to the minor groove. The 1/rDup value that corresponds to the inflection point observed for 5PTB binding to each host duplex provides an estimate of the ligand/duplex stoichiometry. Inspection of the Fig. 3 Insets reveals the inflection points for 5PTB binding to the d(GA4T4C)2 and d(GT4A4C)2 duplexes to correspond to 1/rDup values of 0.9 and 1.1, respectively. Thus, 5PTB binds to both duplexes with a stoichiometry of approximately one ligand molecule per duplex.
Further inspection of the Fig. 3 Insets reveals the curvature of the single-wavelength titration curve for the d(GA4T4C)2 duplex to be substantially sharper than the curvature of the corresponding curve for the d(GT4A4C)2 duplex. This difference in curvature is consistent with 5PTB binding to the d(GA4T4C)2 duplex with a greater affinity than it binds to the d(GT4A4C)2 duplex, a correlation that is substantiated by the ligand binding constants at 20°C (K20), which we present in a later section. Despite this correlation, it should be noted that the K20 of 1.7 × 108 M−1 we report below (see Table 1) for the 1:1 binding of 5PTB to the d(GT4A4C)2 duplex should yield a much sharper curve than the one shown in the Fig. 3B Inset. We found this apparent incongruity to be due to the competing equilibrium of ligand self-association. Under the solution conditions employed in this study, 5PTB aggregates at concentrations between 0.1 and 10 μM (data not shown), with a self-association constant of ≈2 × 107 M−1. The affinity of 5PTB for the d(GA4T4C)2 duplex is sufficiently high so that ligand self-association does not significantly compete with DNA binding. By contrast, the affinity of 5PTB for the d(GT4A4C)2 duplex is not high enough to overcome the competing effects of ligand self-association. Consequently, a larger concentration of d(GT4A4C)2 [compared with the concentration of d(GA4T4C)2] is required to saturate an identical concentration of 5PTB (5 μM), thereby resulting in the broader CD titration curve for d(GT4A4C)2 (Fig. 3B Inset) relative to d(GA4T4C)2 (Fig. 3A Inset).
Table 1.
ΔTm-derived binding affinities of 5PTB for the d(GA4T4C)2 and d(GT4A4C)2 duplexes at 20°C
| Duplex | Tm°,* °C | Tm,* °C | K20,† M−1 |
|---|---|---|---|
| d(GA4T4C)2 | 34.1 ± 0.1 | 53.3 ± 0.2 | (8.9 ± 2.4) × 109 |
| d(GT4A4C)2 | 29.3 ± 0.1 | 42.5 ± 0.3 | (1.7 ± 0.6) × 108 |
Tm values were derived from UV melting profiles at 2 μM duplex in the absence (Tm°) and presence of 5PTB at an rDup ratio of either 1.1 for d(GA4T4C)2 or 2.0 for d(GT4A4C)2. Each Tm value is an average derived from at least two independent experiments, with the indicated errors corresponding to the standard deviation from the mean.
Binding constants at 20°C (K20) were determined by using Eq. 1 and the following calorimetrically determined duplex-to-single strand transition enthalpies (ΔHdup) for the two host duplexes: 86.4 kcal/mol for d(GA4T4C)2 and 79.1 kcal/mol for d(GT4A4C)2. The indicated uncertainties reflect the maximal errors in K20 that result from the corresponding uncertainties noted above in Tm, Tm°, and ΔHb, as propagated through Eq. 1 and the standard thermodynamic relationship: ∂(lnK)/∂(1/T) = −ΔHb/R.
In light of the ligand self-association properties noted above, we considered the possibility that 5PTB binds to the host duplex as a preformed aggregate (e.g., a dimer). However, if 5PTB complexed with duplex DNA as a dimer or higher aggregate, then the well defined inflection point of the CD titration curve for 5PTB binding to the d(GA4T4C)2 duplex (Fig. 3A Inset) would occur at a [duplex]/[total ligand] ratio of ≤0.5 rather than the observed value of 0.9. Hence, our CD titration data are consistent with monomeric 5PTB binding to its host duplex.
5PTB Exhibits Similar Binding Enthalpies When It Complexes with the d(GA4T4C)2 and d(GT4A4C)2 Duplexes.
Isothermal, stopped-flow mixing calorimetry was used to measure the binding enthalpies (ΔHb) for 5PTB complexation with the d(GA4T4C)2 and d(GT4A4C)2 duplexes. These studies revealed enthalpies for 5PTB binding to d(GA4T4C)2 and d(GT4A4C)2 duplexes of −4.9 ± 0.6 and −5.9 ± 0.6 kcal/mol, respectively. Thus, 5PTB binding to the d(GA4T4C)2 and d(GT4A4C)2 duplexes is enthalpically similar. In other words, 5PTB does not enthalpically discriminate significantly between these two duplexes, which have identical base compositions but isomeric core sequences that cause them to exhibit different structures (e.g., A-tract vs. B-like or bent vs. normal).
Despite Exhibiting Similar Binding Enthalpies, 5PTB Binds with Greater Affinity to the d(GA4T4C)2 Duplex Than to the d(GT4A4C)2 Duplex.
To assess, by a single method, the relative strength of 5PTB binding to the d(GA4T4C)2 and d(GT4A4C)2 duplexes, we used a ΔTm approach previously described in detail (30) and briefly summarized below. It should be noted that the magnitude of 5PTB binding to both decameric duplexes, as well as the competing self-association equilibrium of the ligand noted above, precludes a Scatchard analysis of the optical data. Consequently, to calculate ligand binding affinities, we used the Crothers ΔTm approach (30) described below, which considers only differences between the ligand-free and ligand-saturated duplexes and therefore is not compromised by the nature of the aggregation state of the free ligand.
We used our measured ligand-induced changes in the thermal stabilities (ΔTm) of the d(GA4T4C)2 and d(GT4A4C)2 duplexes (see Fig. 2) to estimate apparent drug binding constants at Tm (KTm) by using the expression (30):
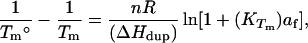 |
1 |
where Tm° and Tm are the melting temperatures of the ligand-free and ligand-saturated duplexes, respectively; n is the number of ligand binding sites on the duplex; R is the molar gas constant; ΔHdup is the enthalpy change for the melting of the DNA duplex in the absence of bound ligand (values we determined independently for each duplex by using differential scanning calorimetry); and af is the free ligand activity at Tm. For the tightly bound 5PTB–d(GA4T4C)2 complex, we estimated af as one-half the total ligand concentration (Ltot), whereas, for the more weakly bound 5PTB–d(GT4A4C)2 complex, we estimated af as Ltot minus one-half the total duplex concentration. We then extrapolated the KTm values calculated in this manner to a common reference temperature of 20°C, using the binding enthalpies (ΔHb) noted above. The resulting K20 values are listed in Table 1. Inspection of these data reveals that 5PTB binds to the d(GA4T4C)2 duplex with an approximately 52-fold greater affinity than it binds to the d(GT4A4C)2 duplex, which corresponds to a 2.3 kcal/mol binding free energy preference at 20°C. Note the agreement between this hierarchy of binding affinity and that noted above for binding-induced enhancement in duplex thermal stability (ΔTm). In other words, for this system, the relative extent to which 5PTB thermally stabilizes the target duplex correlates with its relative binding affinity.
The 2.3-kcal/mol Binding Preference of 5PTB for the d(GA4T4C)2 Duplex Relative to the d(GT4A4C)2 Duplex Is Entropic in Origin.
Armed with the binding constants listed in Table 1, we calculated the corresponding binding free energies (ΔGb) by using the standard relationship ΔGb = −RTlnK. These binding free energies, coupled with our calorimetrically determined binding enthalpies, allowed us to calculate the corresponding binding entropies (ΔSb) by using the relationship ΔSb = (ΔHb − ΔGb)/T. These calculations enabled us to generate complete thermodynamic profiles for the binding of 5PTB to both duplexes. These binding profiles are summarized in Table 2. Inspection of these data reveals that, at 20°C, 5PTB exhibits a 2.3 kcal/mol binding preference for d(GA4T4C)2 relative to d(GT4A4C)2. Further, note that the preferential binding of 5PTB to the d(GA4T4C)2 duplex is entropic in origin. This favorable differential entropic contribution [Δ(TΔSb)] of +3.3 kcal/mol may, in part, reflect differential minor groove desolvation of the d(GA4T4C)2 relative to the d(GT4A4C)2 duplex (31–35). In this regard, crystallographic studies from the Dickerson group (23, 31–33, 36) and examination of the nucleic acid database (37) reveal that A-tract DNA (whose formation is facilitated by ApA, ApT, and TpT dinucleotide steps, but disrupted by TpA steps) exhibits a narrow minor groove that contains a well ordered “spine” of water molecules. Nadeau and Crothers (38) have shown that full A-tract structural behavior requires ≥7 A⋅T base pairs (i.e., at least six ApA, ApT, and/or TpT dinucleotide steps), with only partial A-tract behavior being exhibited by duplexes that contain only 4–6 A⋅T base pairs, and no A-tract behavior being exhibited by duplexes containing ≤3 A⋅T base pairs. Because the d(GA4T4C)2 duplex contains an 8-bp A-tract, it is reasonable to propose that it exhibits full A-tract structural behavior, including “complete” narrowing of the minor groove with a well ordered spine of hydration. By contrast, the d(GT4A4C)2 duplex contains two A-tracts of only 4 base pairs separated by a disruptive TpA step, with neither A-tract 5′ or 3′ to the TpA step being of sufficient length to exhibit full A-tract structural behavior. In other words, the water molecules in the all-AT minor groove of the d(GT4A4C)2 duplex should be less ordered than those in the all-AT minor groove of the d(GA4T4C)2 duplex. On the basis of this picture, 5PTB binding to the minor groove of the d(GA4T4C)2 duplex should exhibit a greater entropic driving force due to ligand-induced desolvation than 5PTB minor groove binding to the less hydrated d(GT4A4C)2 duplex. Inspection of Table 3 reveals that this expectation is realized in our data, in that we find a +3.3 kcal/mol TΔSb advantage for 5PTB binding to the d(GA4T4C)2 duplex.
Table 2.
Thermodynamic parameters for the binding of 5PTB, netropsin, and DAPI to the d(GA4T4C)2 and d(GT4A4C)2 duplexes
| Duplex | Ligand | ΔHb, kcal/mol* | TΔSb,† kcal/mol | ΔGb-20‡ kcal/mol | K20,* M−1 |
|---|---|---|---|---|---|
| d(GA4T4C)2 | 5PTB | −4.9 ± 0.6 | +8.4 ± 0.8 | −13.3 ± 0.2 | (8.9 ± 2.4) × 109 |
| Netropsin | −3.4 ± 0.7 | +9.0 ± 0.9 | −12.4 ± 0.2 | (1.7 ± 0.5) × 109 | |
| DAPI | −3.6 ± 0.6 | +7.4 ± 0.8 | −11.0 ± 0.2 | (1.5 ± 0.4) × 108 | |
| d(GT4A4C)2 | 5PTB | −5.9 ± 0.6 | +5.1 ± 1.2 | −11.0 ± 0.2 | (1.7 ± 0.6) × 108 |
| Netropsin | −9.2 ± 0.9 | +2.7 ± 1.1 | −11.9 ± 0.2 | (7.9 ± 2.1) × 108 | |
| DAPI | −5.5 ± 0.7 | +5.4 ± 0.9 | −10.9 ± 0.2 | (1.4 ± 0.3) × 108 |
ΔHb values were determined at rDup ratios of 1.0, with the indicated uncertainties corresponding to the sum of the standard deviations from two separate mixing experiments (DNA–ligand and buffer–buffer) of at least 14 independent injections each. The indicated errors in K20 are as described in the footnote to Table 1.
ΔSb is the binding entropy, as determined using the corresponding values of ΔHb and ΔGb-20. The indicated uncertainties reflect the maximal possible errors in TΔSb that result from the corresponding errors noted above in ΔHb and ΔGb-20, as propagated through ΔSb = (ΔHb − ΔGb)/T.
ΔGb-20 is the binding free energy at 20°C, as determined using the corresponding value of K20. The indicated uncertainties reflect the errors in ΔGb-20 that result from the corresponding uncertainties noted above in K20, as propagated through ΔGb = −RTln K.
Comparison Between the Duplex Binding Profiles of 5PTB, a TOP1 Poison, and Two Other AT-Specific, Minor Groove-Directed Ligands That Are Not TOP1 Poisons.
In addition to listing the thermodynamic profiles for 5PTB binding to the d(GA4T4C)2 and d(GT4A4C)2 duplexes, Table 2 also summarizes the corresponding binding profiles for netropsin and DAPI, two AT-specific, minor groove-directed ligands that, in contrast to 5PTB, are inactive as TOP1 poisons (8). Inspection of these data reveals that netropsin and DAPI exhibit similar binding affinities (K20, ΔGb-20) for the two host duplexes, in contrast to the 52-fold (2.3 kcal/mol) preferential binding affinity exhibited by 5PTB for the d(GA4T4C)2 duplex. Note that this preferential binding affinity of 5PTB for the d(GA4T4C)2 duplex relative to the d(GT4A4C)2 duplex is entirely entropic in origin. Further note that, while netropsin and DAPI binding also exhibit entropic preferences for the d(GA4T4C)2 duplex, these preferences are compensated by losses in binding enthalpy, with these enthalpy–entropy compensations causing netropsin and DAPI to exhibit similar affinities for the two host duplexes. This behavior contrasts with that of 5PTB, which fully expresses its entropically driven binding preference for d(GA4T4C)2, because its affinity is not dampened by a compensating loss in binding enthalpy. In short, 5PTB, a TOP1 poison, exhibits differential thermodynamic DNA binding properties relative to netropsin and DAPI, two other minor groove-directed ligands that do not poison TOP1, perhaps reflecting a correlation between biophysical binding properties and TOP1 biochemical poisoning activity. Clearly, additional studies are required to assess the generality of this observation.
The above discussion focused on the differential binding properties for two host duplexes of several minor groove-directed ligands. It also is of interest to compare the binding properties of these ligands to a single, common host duplex, particularly one for which some structural data are available on the corresponding or related drug–DNA complexes. The d(GA4T4C)2 duplex provides such an opportunity. Inspection and comparison of the information in the first three rows of Table 2 provides the relevant data. Note that 5PTB exhibits a 5- and 59-fold greater binding affinity for the d(GA4T4C)2 duplex relative to netropsin and DAPI, respectively. Interestingly, this enhanced binding affinity for 5PTB is significantly enthalpic in origin. In the section that follows, we use available structural data to propose a possible microscopic basis for the enhanced DNA binding enthalpy of 5PTB.
Structural Interpretation of the Enhanced Binding Enthalpy of 5PTB for the d(GA4T4C)2 Duplex Relative to That Exhibited by Netropsin and DAPI.
A recent crystallographic study by Neidle and co-workers (39) on a terbenzimidazole–oligonucleotide complex suggests a possible molecular basis for the enhanced d(GA4T4C)2 binding enthalpy exhibited by 5PTB relative to the corresponding DNA binding enthalpies of netropsin and DAPI. Three structural features of the complex between a terbenzimidazole analog of Hoechst 33258 (TRIBIZ) and the d(CGCA3T3GCG)2 duplex distinguish it from complexes of other minor groove-binding ligands, such as netropsin (31, 32) and DAPI (33), with DNA duplexes that possess similar A-tract domains. These three structural features are as follows: (i) an extremely narrow minor groove width in the ligand-bound A-tract region; (ii) a highly twisted ligand structure, with an overall twist of 50° between aromatic rings; and (iii) an unwound A-tract region, with a 10–13° reduction in overall helical twist. These three features reflect the ability of TRIBIZ to alter both the conformation of the DNA and its own inherent twist to achieve the proper phasing of its aromatic subunits with the edges of the base pairs. This “induced fit” binding behavior appears to enhance both hydrogen bonding interactions and van der Waals contacts between the ligand and atoms lining the walls of the minor groove. Such hydrogen bonds and van der Waals contacts, generally speaking, are energetically favorable, and, if maximized through the structural accommodations just noted, should provide the 5PTB–d(GA4T4C)2 complex with a source of enhanced enthalpic stability. By contrast, crystal structures from the Dickerson group of netropsin (31, 32) and DAPI (33) complexes with duplex DNA reveal significantly less binding-induced distortions of either the DNA host structures or the ligands relative to those observed when TRIBIZ binds to DNA (39). Such a “lock and key” binding behavior may mean that the unbound rest states of the ligand and the host duplex are fortuitously poised so as to minimize the need for any structural alterations required to maximize favorable ligand–DNA interactions in the final state. Alternatively, the complex simply may tolerate suboptimal ligand–DNA interactions because the energetic cost(s) of significant alterations in the ligand and host duplex exceeds the energetic gain achieved in the final complex. Independent of the veracity of these speculations, it is of interest to note that the strongest DNA binding ligand (5PTB) forms a complex with its host duplex that exhibits the greatest degree of structural accommodation, a feature that is reflected in a more favorable binding enthalpy. This observation, in part, may reflect the fact that the larger terbenzimidazole ligand is able to more fully take advantage of the inherent, sequence-directed propensity of the A-tract domain of the unbound duplex to form a narrow minor groove with an underwound helical twist, features that previously have been emphasized by Dickerson and co-workers. Clearly, additional parallel macroscopic and microscopic studies are needed to evaluate the generality of these interpretations, which are offered here purely as a basis for further discussions.
Comparison Between the Effect of 5PTB and Netropsin Binding on the Electrophoretic Mobilities of Both Decameric Duplexes.
PAGE experiments were conducted to assess the impact, if any, of 5PTB and netropsin binding on the global conformations, as assessed by electrophoretic mobility, of the d(GA4T4C)2 and d(GT4A4C)2 duplexes. The resulting gels are shown in A (5PTB) and B (netropsin) of Fig. 4. A comparison of lanes 1–3 in A (as well as lanes 1, 5, 6, and 10 in B) reveals that, in the absence of ligand, d(GA4T4C)2 migrates more slowly than d(GT4A4C)2. This observation is consistent with a previous electrophoretic study (22) on the same two decameric duplexes. A reduction in electrophoretic mobility relative to that expected of a DNA molecule on the basis of its molecular weight has been employed as the “gold standard” for scoring DNA bending (40–45). Hence, the anomalously slow migration of d(GA4T4C)2 is consistent with this duplex adopting a “bent” conformation relative to the “normal” conformation assumed by d(GT4A4C)2. Alternatively, the retarded electrophoretic mobility of d(GA4T4C)2 relative to d(GT4A4C)2 may reflect differences in structure/hydration (e.g., minor groove width and base propeller twist) and/or stiffness between the 8-bp A-tract of d(GA4T4C)2 and the two 4-bp A-tracts of d(GT4A4C)2, which are separated by a disruptive TpA step. Cognizant of these two possible explanations for the retarded electrophoretic mobility of the d(GA4T4C)2 duplex relative to the d(GT4A4C)2 duplex, we discuss below the impact of 5PTB and netropsin binding on the global conformations of both decameric duplexes.
Figure 4.

Nondenaturing 28% polyacrylamide gels containing the d(GA4T4C)2 duplex (lanes 1 and 3–7 of A; lanes 1–5 and 10 of B), the d(GT4A4C)2 duplex (lanes 2, 3, and 8–12 of A; lanes 5–10 of B), and their complexes with either 5PTB (A) or netropsin (B) at the indicated ligand concentrations.
Inspection of Fig. 4A reveals that the addition of 5PTB retards the migration of both the d(GA4T4C)2 duplex (lanes 4–7) and the d(GT4A4C)2 duplex (lanes 8–12), with the extent of this retardation being greater for the d(GA4T4C)2 duplex. By contrast, electrophoretic “control” experiments on these two duplexes and their complexes with netropsin (Fig. 4B) and DAPI (not shown) reveal little or no binding-induced retardation in duplex migration. Thus, it is unlikely that the observed 5PTB-induced retardation is due to differences in size, charge, or stiffness between the ligand-free duplex and the ligand-bound complex. Instead, this retardation of mobility may reflect a binding-induced “bending” of both target duplexes, with d(GA4T4C)2 being “bent” to a greater extent than d(GT4A4C)2. Alternatively, this retardation of mobility may reflect a binding-induced unwinding of the host DNA as has been observed by the Neidle group in their crystallographic studies on the TRIBIZ–DNA complex (39). Thus, the sequence-directed structure of the d(GA4T4C)2 duplex (whether bent or fully A-tract) poises it for further alteration (e.g., bending and/or unwinding) through complexation with 5PTB. Our thermodynamic studies described here reveal the basis for this ligand-induced effect to be entropic in origin. Independent of the veracity of either a DNA bending or a DNA unwinding model, our electrophoretic data are consistent with 5PTB binding being associated with an alteration in DNA structure/conformation, a feature that may give rise to its TOP1 poisoning properties.
Concluding Remarks.
We have used a combination of spectroscopic and calorimetric techniques to demonstrate that the minor groove-directed terbenzimidazole 5PTB preferentially binds to the d(GA4T4C)2 duplex relative to the sequence-isomeric d(GT4A4C)2 duplex, with this preferential binding being entropic in origin. In addition, we have shown that 5PTB binding to both target duplexes induces changes in their electrophoretic mobilities that are consistent with ligand-induced bending and/or unwinding of the host duplexes, with the sequence-directed structure of the ligand-free d(GA4T4C)2 duplex (whether bent or fully A-tract) poising it for further structural alteration upon complexation with 5PTB. We also have shown that, in contrast to 5PTB, the minor groove-directed, non-TOP1 poisoning ligands netropsin and DAPI do not exhibit a greater affinity for d(GA4T4C)2 relative to d(GT4A4C)2, nor do they significantly alter the electrophoretic mobilities of the two host decameric duplexes. In the aggregate, these observations suggest a potential mechanism by which 5PTB and related bi- and terbenzimidazoles may poison TOP1. In this connection, Liu and co-workers have shown that these families of compounds, but not netropsin or DAPI, stimulate enzyme-mediated DNA cleavage, ultimately leading to cell death (5, 6). Our studies suggest that the poisoning of TOP1 may correlate with ligand-induced bending and/or unwinding of the host DNA, which, in turn, may stabilize the covalent intermediate of the TOP1 reaction—namely, the reversible cleavable complex. Such knowledge of the mechanism(s) and molecular forces that govern ligand-induced TOP1 poisoning is required for the ultimate development of more rational approaches to the design and screening of new TOP1-directed chemotherapeutic agents.
Acknowledgments
We are indebted to Ms. Yong Mao for her assistance with the gel photography. We also thank Drs. Eric H. Rubin and Craig A. Gelfand for their helpful discussions and Prof. Richard E. Dickerson for critical evaluation of and commentary on the manuscript, as well as for sharing with us some of his recent data in advance of publication. This work was supported by National Institutes of Health Grants GM23509, GM34469, CA47995, and CA39962.
ABBREVIATIONS
- 5PTB
5-phenyl-2-[2′-(benzimidazol-5"-yl)benzimidazol-5′-yl]benzimidazole
- TOP
topoisomerase
- DAPI
4′,6-diamidino-2-phenylindole
- Tm
melting temperature
- rDup
[total ligand]/[duplex]
References
- 1.Wang J C. Annu Rev Biochem. 1985;54:665–697. doi: 10.1146/annurev.bi.54.070185.003313. [DOI] [PubMed] [Google Scholar]
- 2.D’Arpa P, Liu L F. Biochim Biophys Acta. 1989;989:163–177. doi: 10.1016/0304-419x(89)90041-3. [DOI] [PubMed] [Google Scholar]
- 3.Liu L F. Annu Rev Biochem. 1989;58:351–375. doi: 10.1146/annurev.bi.58.070189.002031. [DOI] [PubMed] [Google Scholar]
- 4.Schneider E, Hsiang Y-H, Liu L F. Adv Pharmacol. 1990;21:149–183. doi: 10.1016/s1054-3589(08)60342-7. [DOI] [PubMed] [Google Scholar]
- 5.Chen A Y, Liu L F. Annu Rev Pharmacol Toxicol. 1994;34:191–218. doi: 10.1146/annurev.pa.34.040194.001203. [DOI] [PubMed] [Google Scholar]
- 6.Liu L F, editor. DNA Topoisomerases: Topoisomerase-Targeting Drugs, Advances in Pharmacology. 29B. San Diego: Academic; 1994. [Google Scholar]
- 7.Chen A Y, Yu C, Bodley A, Peng L F, Liu L F. Cancer Res. 1993;53:1332–1337. [PubMed] [Google Scholar]
- 8.Chen A Y, Yu C, Gatto B, Liu L F. Proc Natl Acad Sci USA. 1993;90:8131–8135. doi: 10.1073/pnas.90.17.8131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pjura P E, Grzeskowiak K, Dickerson R E. J Mol Biol. 1987;197:257–271. doi: 10.1016/0022-2836(87)90123-9. [DOI] [PubMed] [Google Scholar]
- 10.Teng M-K, Usman N, Frederick C A, Wang A H-J. Nucleic Acids Res. 1988;16:2671–2690. doi: 10.1093/nar/16.6.2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spink N, Brown D G, Skelly J V, Neidle S. Nucleic Acids Res. 1994;22:1607–1612. doi: 10.1093/nar/22.9.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parkinson J A, Barber J, Douglas K T, Rosamond J, Sharples D. Biochemistry. 1990;29:10181–10190. doi: 10.1021/bi00496a005. [DOI] [PubMed] [Google Scholar]
- 13.Fede A, Labhardt A, Bannwarth W, Leupin W. Biochemistry. 1991;30:11377–11388. doi: 10.1021/bi00112a004. [DOI] [PubMed] [Google Scholar]
- 14.Fede A, Billeter M, Leupin W, Wüthrich K. Structure. 1993;1:177–186. doi: 10.1016/0969-2126(93)90019-d. [DOI] [PubMed] [Google Scholar]
- 15.Sun Q, Gatto B, Yu C, Liu A, Liu L F, LaVoie E J. Bioorg Med Chem Lett. 1994;4:2871–2876. [Google Scholar]
- 16.Sun Q, Gatto B, Yu C, Liu A, Liu L F, LaVoie E J. J Med Chem. 1995;38:3638–3644. doi: 10.1021/jm00018a024. [DOI] [PubMed] [Google Scholar]
- 17.Kim J S, Gatto B, Yu C, Liu A, Liu L F, LaVoie E J. J Med Chem. 1996;39:992–998. doi: 10.1021/jm950412w. [DOI] [PubMed] [Google Scholar]
- 18.Kim J S, Sun Q, Gatto B, Yu C, Liu A, Liu L F, LaVoie E J. Bioorg Med Chem. 1996;4:621–630. doi: 10.1016/0968-0896(96)00047-8. [DOI] [PubMed] [Google Scholar]
- 19.Pilch D S, Xu Z, Sun Q, LaVoie E J, Liu L F, Geacintov N E, Breslauer K J. Drug Des Discovery. 1996;13(3–4):115–133. [PubMed] [Google Scholar]
- 20.Park Y-W, Breslauer K J. Proc Natl Acad Sci USA. 1991;88:1551–1555. doi: 10.1073/pnas.88.4.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leroy J-L, Charretier E, Kochoyan M, Guéron M. Biochemistry. 1988;27:8894–8898. doi: 10.1021/bi00425a004. [DOI] [PubMed] [Google Scholar]
- 22.Chen J-H, Seeman N C, Kallenbach N R. Nucleic Acids Res. 1988;16:6803–6812. doi: 10.1093/nar/16.14.6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goodsell D S, Kaczor-Grzeskowiak M, Dickerson R E. J Mol Biol. 1994;239:79–96. doi: 10.1006/jmbi.1994.1352. [DOI] [PubMed] [Google Scholar]
- 24.Dickerson R E, Goodsell D, Kopka M L. J Mol Biol. 1996;256:108–125. doi: 10.1006/jmbi.1996.0071. [DOI] [PubMed] [Google Scholar]
- 25.Griswold B L, Humoller F L, McIntyre A R. Anal Chem. 1951;23:192–194. [Google Scholar]
- 26.Mudd C P, Berger R L. J Biochem Biophys Methods. 1988;17:171–192. doi: 10.1016/0165-022x(88)90002-4. [DOI] [PubMed] [Google Scholar]
- 27.Remeta D P, Mudd C P, Berger R L, Breslauer K J. Biochemistry. 1991;30:9799–9809. doi: 10.1021/bi00104a032. [DOI] [PubMed] [Google Scholar]
- 28.Robinson A L. J Am Chem Soc. 1932;54:1311–1318. [Google Scholar]
- 29.Gulbransen E A, Robinson A L. J Am Chem Soc. 1934;56:2637–2641. [Google Scholar]
- 30.Crothers D M. Biopolymers. 1971;10:2147–2160. doi: 10.1002/bip.360101110. [DOI] [PubMed] [Google Scholar]
- 31.Kopka M L, Yoon C, Goodsell D, Pjura P, Dickerson R E. Proc Natl Acad Sci USA. 1985;82:1376–1380. doi: 10.1073/pnas.82.5.1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kopka M L, Yoon C, Goodsell D, Pjura P, Dickerson R E. J Mol Biol. 1985;183:553–563. doi: 10.1016/0022-2836(85)90171-8. [DOI] [PubMed] [Google Scholar]
- 33.Larsen T A, Goodsell D S, Cascio D, Grzeskowiak K, Dickerson R E. J Biomol Struct Dyn. 1989;7:477–491. doi: 10.1080/07391102.1989.10508505. [DOI] [PubMed] [Google Scholar]
- 34.Marky L A, Breslauer K J. Proc Natl Acad Sci USA. 1987;84:4359–4363. doi: 10.1073/pnas.84.13.4359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chalikian T V, Plum G E, Sarvazyan A P, Breslauer K J. Biochemistry. 1994;33:8629–8640. doi: 10.1021/bi00195a003. [DOI] [PubMed] [Google Scholar]
- 36.Drew H R, Dickerson R E. J Mol Biol. 1981;151:535–556. doi: 10.1016/0022-2836(81)90009-7. [DOI] [PubMed] [Google Scholar]
- 37.Berman H M, Olson W K, Beveridge D L, Westbrook J, Gelbin A, Demeny T, Hsieh S-H, Srinivasan A R, Schneider B. Biophys J. 1992;63:751–759. doi: 10.1016/S0006-3495(92)81649-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nadeau J G, Crothers D M. Proc Natl Acad Sci USA. 1989;86:2622–2626. doi: 10.1073/pnas.86.8.2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clark G R, Gray E J, Neidle S, Li Y-H, Leupin W. Biochemistry. 1996;35:13745–13752. doi: 10.1021/bi960421m. [DOI] [PubMed] [Google Scholar]
- 40.Marini J C, Levene S D, Crothers D M, Englund P T. Proc Natl Acad Sci USA. 1982;79:7664–7668. doi: 10.1073/pnas.79.24.7664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu H-M, Crothers D M. Nature (London) 1984;308:509–513. doi: 10.1038/308509a0. [DOI] [PubMed] [Google Scholar]
- 42.Hagerman P J. Biochemistry. 1985;24:7033–7037. doi: 10.1021/bi00346a001. [DOI] [PubMed] [Google Scholar]
- 43.Hagerman P J. Nature (London) 1986;321:449–450. doi: 10.1038/321449a0. [DOI] [PubMed] [Google Scholar]
- 44.Koo H-S, Wu H-M, Crothers D M. Nature (London) 1986;320:501–506. doi: 10.1038/320501a0. [DOI] [PubMed] [Google Scholar]
- 45.Koo H-S, Drak J, Rice J A, Crothers D M. Biochemistry. 1990;29:4227–4234. doi: 10.1021/bi00469a027. [DOI] [PubMed] [Google Scholar]