

Abstract
We have investigated the ability of Sf-caspase-1 and two mammalian caspases, caspase-1 and caspase-3, to induce apoptosis in Spodoptera frugiperda Sf-21 insect cells. While the transient expression of the pro-Sf-caspase-1 did not induce apoptosis, expression of the pro-domain deleted form, p31, or coexpression of the two subunits of mature Sf-caspase-1, p19 and p12, induced apoptosis in Sf-21 cells. The behavior of Sf-caspase-1 resembled that of the closely related mammalian caspase, caspase-3, and contrasted with that of the mammalian caspase-1, the pro-form of which was active in inducing apoptosis in Sf-21 cells. The baculovirus caspase inhibitor P35 blocked apoptosis induced by active forms of all three caspases. In contrast, members of the baculovirus inhibitor of apoptosis (IAP) family failed to block active caspase-induced apoptosis. However, during viral infection, expression of OpIAP or CpIAP blocked the activation of pro-Sf-caspase-1 and the associated induction of apoptosis. Thus, the mechanism by which baculovirus IAPs inhibit apoptosis is distinct from the mechanism by which P35 blocks apoptosis and involves inhibition of the activation of pro-caspases like Sf-caspase-1.
Apoptosis or programmed cell death is a genetically regulated cell suicide program that plays an important role during normal development, tissue homeostasis, and disease processes of metazoans (1–3). Apoptosis also serves as an important defense strategy employed by host cells against viral invasion (4). In turn, viruses have evolved mechanisms to block the defense-by-death response of host cells. The baculovirus Autographa californica nuclear polyhedrosis virus (AcMNPV) was one of the first viruses known to induce and inhibit apoptosis (5). Infection of Spodoptera frugiperda Sf-21 cells with the wild-type (wt) AcMNPV results in the production of both budded virus and occluded virus (6). In contrast, infection with an AcMNPV mutant lacking the P35 gene results in induction of apoptosis with limited budded virus production and no occluded virus production (7). Virus-induced apoptosis involves the activation of caspases, an aspartate-specific cysteine protease family of pro-apoptotic proteins that play a central role in the evolutionarily conserved apoptotic pathway (8). The caspases are synthesized as proenzymes that can be activated either by autoprocessing or by other family members or by other proteases (e.g., granzyme B) involved in induction of apoptosis. Recently, Sf-caspase-1, a caspase from S. frugiperda cells implicated in the virus-induced apoptosis, has been described (9). Mammals possess at least 10 different caspases, caspase-1–10, that can be subdivided into three subfamilies based on their sequence similarities. Caspase-1, also known as interleukin-1β converting enzyme (ICE), is the founding member of the family and belongs to a different subfamily than caspase-3. Caspase-3 is the mammalian caspase most closely related to CED-3, a nematode caspase that is a central component of the cell death pathway in developing nematodes, and genetic evidence substantiates a role for caspase-3 in apoptosis (10).
P35, a general caspase inhibitor, serves to inhibit virus-induced caspase activity (11–13) and thereby blocks apoptosis (5, 7, 11, 14, 15). In addition, ectopic expression of P35 is known to block apoptosis induced by diverse signals in both vertebrates and other invertebrates (5, 7, 15).
The inhibitors of apoptosis (IAPs) were first identified in a genetic screen for baculovirus genes that can complement the loss of the p35 gene and block apoptosis induced by p35 mutants of AcMNPV (7). Op-IAP (16) and Cp-IAP (17) also block apoptosis in Sf-21 cells induced with actinomycin D (14, 16, 17), Reaper (RPR) expression (18, 19), or UV treatment (20). These baculovirus IAPs also exhibit some anti-apoptotic activity in other types of cells; Op-IAP partially blocks apoptosis induced by overexpression of caspase-1 or FADD (Fas-associating protein with a death domain) in HeLa cells (21, 22) and Cp-IAP is partially effective in blocking apoptosis induced by overexpression of RPR in the developing eye of Drosophila (18). The baculovirus IAPs, OpIAP and CpIAP, physically interact with and block the pro-apoptotic activity of Drosophila RPR (19) and Doom (23) in Sf-21 cells. Cellular homologs of IAPs that show anti-apoptotic activity have also been described in mammals and Drosophila (24). Like the baculovirus IAPs, the mammalian IAPs block apoptosis in response to different stimuli (25). The mammalian xIAP inhibits the activity of caspase-3 and caspase-7 in vitro (26). Although, recent studies place the baculovirus IAPs at or upstream of P35 (20) in the apoptotic pathway, the mechanism by which the baculovirus IAPs block virus-induced apoptosis is currently not known.
In this study we show that baculovirus infection activates the processing of Sf-caspase-1, and that the IAPs, but not P35, are able to effectively block the activation of pro-Sf-caspase-1. This caspase, like mammalian caspase-3, is inactive when expressed in its pro-form in Sf-21 cells. P35 but not IAPs were able to block apoptosis induced by the active form of these proteases. Thus, the mechanism by which IAPs block apoptosis is distinct from P35 and involves the inhibition of caspase processing.
MATERIALS AND METHODS
Cell Line and Viruses.
Sf-21 cells [S. frugiperda (Lepidoptera: noctuidae) IPLB-SF-21] were cultured at 27°C in TC-100 medium (GIBCO/BRL) supplemented with 10% fetal bovine serum (Intergen, Purchase, NY) and 0.26% tryptose broth (6, 27).
Wild-type (wt) AcMNPV strain L-1 (28) and the previously described recombinant viruses vOPIAPR-11 (16) and vASB6–1 (17), expressing OpIAP and CpIAP, respectively, were propagated in Sf-21 cells. Virus vP35del containing a deletion in the P35 gene was propagated in TN-368 cells (14).
Plasmid Constructs.
Plasmid pHSP70PLVI+CAT, contains the cat gene encoding chloramphenicol acetyltransferase (CAT) under the control of Drosophila 70-kDa heat-shock protein (hsp70) promoter (14). All plasmids used in transient expression assays were derived by replacing cat in pHSP70PLVI+CAT with other coding sequences. Plasmids pHSc-Epi-pro-Sf-Casp-1, pHSc-Epi-Sf-Casp-1p31, pHSSf-Casp-1p19, and pHSc-Epi-Sf-casp-1p12 contain a BglII-EcoRI fragment encoding the Sf-caspase-1 proenzyme (35-kDa form) (9), the prodomain deleted form p31 (residues 29–299), the larger subunit p19 (residues 29–184), and the smaller subunit p12 (residues 185–299), respectively. The Sf-caspase-1 pro-enzyme, the p31 form, and the p12 coding sequences contain HA.11 epitope (YPYDVPDYA) and His6 tags as C-terminal fusions. In other pHSP70PLVI+CAT-based plasmids, cat was replaced with the following: in pHSPCR-ICE45VI+, a PCR-derived BglII-EcoRI fragment coding for the mammalian pro-caspase-1 (p45) (29); in pHSP70PLVI+ICE32 a 0.8 kb sequence coding for the caspase-1 p32 (residues 120–404); in pHSICE20VI+ a SpeI-NotI fragment encoding the caspase-1 p20 subunit (residues 120–297); in pHSICE10VI+ a BglII-EcoRI fragment coding for the caspase-1 p10 subunit (residues 317–404); in pHSP70CPP32βVI+ a SpeI-NotI fragment coding for the mammalian pro-caspase-3 p32 form (30); in pHSCPP29βVI+ a BglII-EcoRI fragment coding for the caspase-3 p29 (residues 29–277); in pHSCPP17βVI+ a BglII-EcoRI fragment encoding the caspase-3 p17 subunit (residues 29–175); and in pHSCPP12βVI+ a BglII-EcoRI fragment encoding the caspase-3 p12 subunit (residues 176–277). Plasmids pHSP35VI+, pHSOpIAPVI+, pHSCpIAPVI+, pHSAcIAPVI+, pHSAd19KVI+, pHSBcl-2VI+, pHSP70CrmAVI+, pHSP70Bcl-XLVI+, and pHSced-9VI+ were described (14, 19). Plasmid pKVEpihisCPP32β contains N-terminal HA.11- and His6-tagged pro-caspase-3 under the control of the T7 promoter in a previously described vector pKV (11).
DNA Fragmentation and Viability Assay.
Sf-21 cells (0.5 × 106 per 35-mm diameter tissue culture dish) were transfected with 0.5 μg of indicated plasmids using Lipofectin (GIBCO/BRL). Cells were heat-shocked in a 42°C water bath for 30 min beginning at 16 h posttransfection. Viable cells excluding trypan blue were counted at 10–12 h after heat-shock administration as described (14). For DNA fragmentation, cells were collected at 10 h after heat-shock and processed as described (14).
In Vitro Transcription/Translation of Pro-Caspase-3.
Coupled transcription/translation was performed with the T7 polymerase and rabbit reticulocyte lysate kit (Promega), with pKVEpihisCPP32β as template, according to the manufacturer’s instructions. The in vitro translated 35S-labeled pro-caspase-3 was purified sequentially on DEAE-Sepharose and Ni2+-resin columns as described (31).
Cell Extract Preparation.
Sf-21 cells (2 × 106 per 60-mm diameter tissue culture dish) were treated either with actinomycin D (1 μg/ml) for 12 h or infected with wt AcMNPV or vP35del, a p35 mutant of AcMNPV, at a multiplicity of infection of 10 plaque-forming units for 24 h. The cells were resuspended in lysis buffer (10 mM Hepes, pH 7.0/2 mM EDTA/0.1% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate/5 mM DTT) and subjected to three freeze-thaw cycles. Cell lysates were then clarified by centrifugation and aliquots of the supernatant were stored at −80°C.
In Vitro Activation Assay.
Purified pro-caspase-3 (5 μl) was incubated with the cell extracts at 37°C for 8 h in reaction buffer (50 mM Hepes, pH 7.4/0.1M NaCl/0.1% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate/10% sucrose) supplemented with 10 mM DTT. The samples were then mixed with SDS-sample buffer (2% SDS/62.5 mM Tris⋅HCl, pH 6.7/15% glycerol/1% 2-mercaptoethanol/0.001% Bromophenol blue) and were resolved on a SDS/12% polyacrylamide gel and visualized using a PhosphorImager (Molecular Dynamics).
Sf-Capase-1 Activation and Western Blot Analysis.
Sf-21 cells (0.5 × 106) were transfected with 1.5 μg of plasmid, pHSc-Epi-pro-Sf-Casp-1 as described above. At 14 h after transfection the cells were heat shocked for 30 min at 42°C. At 4 h after heat shock administration the cells were infected with different viruses at a total multiplicity of infection of 20. In treatments involving coinfections, a multiplicity of infection of 10 for each virus was used maintaining a total multiplicity of infection of 20. Samples were collected at various times indicated. The cells were then lysed in SDS sample buffer and equal volume representing the same cell number were resolved on a SDS/10% polyacrylamide gel. The resolved proteins were then blotted on to a polyvinylidene difluoride membranes (Millipore). The membranes were blocked and incubated with anti-HA.11 mouse IgG (Babco, Richmond, CA), followed by anti-mouse IgG conjugated to horse radish peroxidase. Immunoreactive proteins were visualized with the Enhanced Chemiluminescence Western Blotting System (Amersham).
RESULTS AND DISCUSSION
Pro-Sf-Caspase-1 and the Mammalian Pro-Caspase-3 Do Not Induce Apoptosis in Sf-21 Cells.
Infection of Sf-21 cells with a P35 mutant of the baculovirus AcMNPV results in apoptosis (5, 11), which is mediated through activation of a caspase activity that is inhibitable by P35 (13). Sf-caspase-1 may be responsible for induction of apoptosis during AcMNPV infection since over expression of the pro-enzyme form during AcMNPV infection results in the induction of apoptosis (9). However, since Sf-21 cells do not require additional protein or RNA synthesis to undergo apoptosis (14), one would predict that the caspase that mediates the apoptotic response probably exists as an inactive zymogen that is activated upon viral infection. To determine if pro-Sf-caspase-1 (35-kDa form) initiates apoptosis in Sf-21 cells in the absence of virus infection, we transiently expressed pro-Sf-caspase-1 under the control of the Drosophila hsp70 promoter (see Materials and Methods). Expression from the plasmid was induced by heat-shock, a procedure that does not induce apoptosis in Sf-21 cells (e.g., see Fig. 1A, lane 1). Under these conditions, pro-Sf-caspase-1 failed to induce apoptosis as evidenced by the absence of DNA laddering (Fig. 1A, lane 2), the absence of membrane blebbing, and the absence of apoptotic bodies (data not shown), all of which are characteristic of apoptosis in Sf-21 cells (5). However, transient expression of the prodomain-deleted form of Sf-caspase-1, p31, induced membrane blebbing, apoptotic body formation (data not shown), and DNA laddering (Fig. 1A, lane 3). While expression of either one of the active subunits of Sf-caspase-1, p19 or p12, failed to induce apoptosis (Fig. 1A lanes 5 and 6), the combined expression of both p19 and p12, induced apoptosis with characteristics similar to those observed for p31 (Fig. 1A, lane 4).
Figure 1.
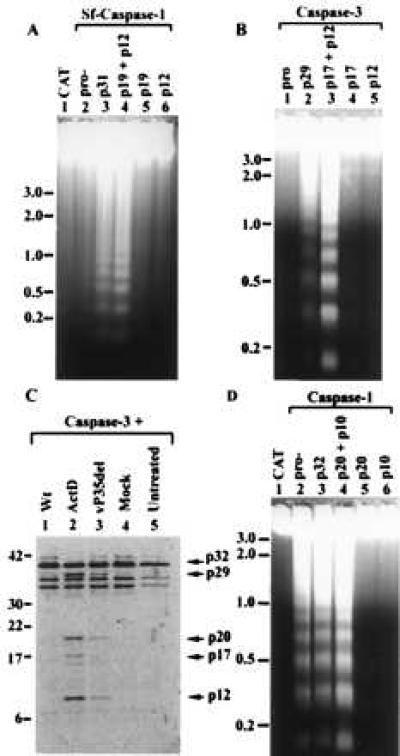
Induction of apoptosis and caspase processing activity in Sf-21 cells. (A, B, D) DNA isolated from Sf-21 cells 10 h after transfection with plasmids expressing various caspases and their forms, as indicated in the figure, was electrophoresed through a 1.2% (A) or 1.5% (B, D) agarose gel and visualized by ethidium bromide staining. CAT-transfected cells (lane 1 of A and D) served as negative control for induction of apoptosis. (C) Purified, in vitro translated, and 35S-labeled pro-caspase-3 was incubated for 12 h with cell extracts prepared from Sf-21 cells treated as follows: wt baculovirus-infected (Wt, lane 1), actinomycin D-treated cells (Act D, lane 2), vP35del-infected (lane 3), mock-infected cells (lane 4). Untreated, purified in vitro-translated pro-caspase-3 is shown in lane 5. Samples were resolved on a SDS/12% polyacrylamide gel and visualized using a PhosphorImager. Size standards are indicated to the left of the gel. Arrows on the right show the position of pro-caspase-3 and known processed forms of caspase-3. Extra bands in the untreated lanes are other products of the in vitro translation reaction including products possibly initiated from internal methionine codons.
Transient expression of the different forms of the closely related mammalian homolog caspase-3 resulted in a similar pattern of apoptosis in Sf-21 cells. Expression of pro-caspase-3 (p32) failed to induce apoptosis (Fig. 1B, lane 1) while expression of caspase-3 lacking the pro-domain (p29) induced apoptosis (Fig. 1B, lane 2). Coexpression of the two subunits of the active caspase-3, p17 and p12, induced higher levels of apoptosis compared with p29 (Fig. 1B, lane 3) while expression of p17 or p12 individually did not induce apoptosis (Fig. 1B, lanes 4, 5). The ability of the pro-domain deleted forms of Sf-caspase-1 and mammalian caspase-3 to induce apoptosis suggests that the prodomain is a negative regulator of enzyme activity under these conditions.
Although our data demonstrate that pro-caspase-3 did not induce apoptosis in uninfected cells (Fig. 1B), previous studies showed that over-expression of pro-caspase-3 in the context of a baculovirus expression vector induces apoptosis in Sf-9 cells (a clonal derivative of Sf-21) (32). To explore the basis for this difference in cellular response to pro-caspase-3 expression, we tested whether extracts from infected and uninfected Sf-21 cells differ in their ability to proteolytically process pro-caspase-3 to its subunit forms. When incubated with cell extracts derived from Sf-21 cells infected with a p35 mutant baculovirus (vP35del) that induces apoptosis during viral infection, the 32 kDa pro-caspase-3 was processed into fragments corresponding to p29, p20 (a previously described pro-caspase-3-derived product; ref. 33), and p12 subunit of the active enzyme (Fig. 1C, lane 3) (30). Extracts derived from Sf-21 cells induced to undergo apoptosis by treatment with actinomycin D also processed pro-caspase-3 to p29, p20, p17, and p12 products (Fig. 1C, lane 2). This processing activity was not detected in extracts derived from mock-infected (Fig. 1C, lane 4) or wt virus-infected cells (Fig. 1, lane 1). The inability of the extracts from wt virus-infected Sf-21 cells to process pro-caspase-3 can be attributed to the expression of the caspase inhibitor P35 during infection.
In contrast to Sf-caspase-1 and mammalian caspase-3, expression of the proenzyme form of the mammalian caspase-1 (p45) induced apoptosis in Sf-21 cells (Fig. 1D, lane 2) suggesting an unregulated autocatalytic mechanism of activation of this zymogen. Sf-21 cells transfected with a plasmid expressing a form of caspase-1 lacking the pro-domain (p32), or cotransfected with plasmids expressing the two active caspase-1 subunits (p20 and p10), also induced apoptosis (Fig. 1D, lanes 3, 4). Transient expression of either the p20 or p10 alone did not induce apoptosis (Fig. 1D, lanes 5 and 6) as expected.
Baculovirus IAPs Do Not Block Active Caspase-Induced Apoptosis.
Although P35 is known to be a stoichiometric inhibitor of a variety of caspases (11), the ability of baculovirus anti-apoptotic genes Op-iap and Cp-iap to block caspase activity in Sf-21 cells has not been examined. We therefore tested whether these anti-apoptotic genes could block apoptosis induced by active caspases in vivo. P35, OpIAP, CpIAP, and AcIAP, an AcMNPV IAP homolog with no known function (14, 16), were coexpressed with the active caspases and the transfected cells were observed for signs of apoptosis. P35 served as a positive control for inhibition of caspase-induced apoptosis, while the cat gene served as a negative control.
Coexpression of the two subunits, p19 and p12, of the active Sf-caspase-1 with cat induced apoptosis in ≈40% of Sf-21 cells (Fig. 2A). As expected, coexpression of P35 with the active Sf-caspase-1 subunits resulted in a 4- to 5-fold reduction in levels of apoptosis compared with the cat control (Fig. 2A). However, the expression of OpIAP or CpIAP failed to block the active Sf-caspase-1-induced apoptosis (Fig. 2A). The activity of OpIAP and CpIAP from the same expression construct used in these experiments was confirmed by their ability to block actinomycin D- and RPR-induced apoptosis as reported previously (14, 19) (data not shown). The AcMNPV homolog AcIAP was also unable to block active Sf-caspase-1 induced apoptosis consistent with its inability to block apoptosis induced by virus infection or actinomycin D treatment. We additionally tested the cowpox virus CrmA, a selective caspase inhibitor, and several anti-apoptotic Bcl-2 family of death regulators for their ability to block active Sf-caspase-1 induced apoptosis. We found that the coexpression of CrmA or Bcl-2 family members failed to block apoptosis induced by active Sf-caspase-1-induced apoptosis (Fig. 2A). The expression of Bcl-2, Bcl-xL, CED-9, and E1B19K from these plasmids in Sf-21 cells has been verified elsewhere in similar experiments (14, 19, 34).
Figure 2.
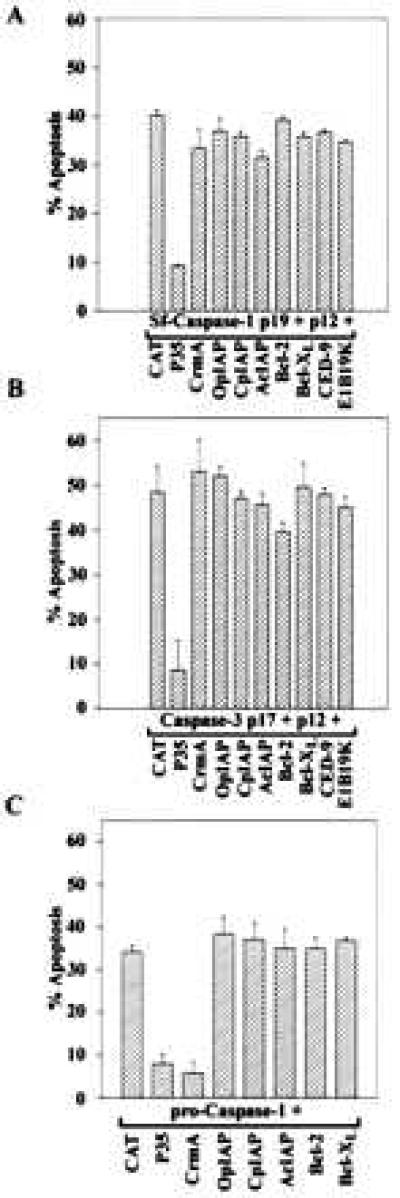
Baculovirus IAPs do not block active caspase-induced apoptosis. Sf-21 cells were cotransfected with plasmids expressing the active subunits of Sf-caspase-1 (p19 + p12) (A), the active subunits of mammalian caspase-3 (p17 + p12) (B), or the zymogen form of mammalian pro-caspase-1 (p45) (C), and a plasmid expressing one of the following as indicated in the figure: CAT, P35, CrmA, OpIAP, CpIAP, AcIAP, Bcl-2, Bcl-xL, E1B19K, or CED-9. Apoptosis was quantitated as described in Material and Methods. The results presented are relative to viability of cells transfected with cat-expressing plasmid alone (not shown) set at 100%. Bars = SD of at least two independent experiments, each with two replications.
We also tested the ability of the P35, IAPs, CrmA, and Bcl-2-family members to block apoptosis induced by coexpression of p17 and p12 subunits of caspase-3 in Sf-21 cells. Like the closely related Sf-casapse-1, apoptosis induced by the active form of caspase-3 was inhibited only by coexpression with P35 (Fig. 2B). The IAPs, CrmA, and Bcl-2 family members were not effective in blocking the active caspase-3 induced apoptosis (Fig. 2B).
Additionally, we tested various anti-apoptotic genes for activity against pro-caspase-1-induced apoptosis. P35 was an effective inhibitor of pro-caspase-1 induced apoptosis. Consistent with its strong inhibitory activity against caspase-1 in vitro, CrmA was also effective in blocking pro-caspase-1-induced apoptosis in Sf-21 cells (Fig. 2C). However, expression of IAPs did not block pro-caspase-1-induced apoptosis suggesting that the IAPs were unable to block the autocatalytic activation of the unregulated pro-caspase-1 in Sf-21 cells. As observed with active Sf-caspase-1 and mammalian caspase-3, all the Bcl-2 family members tested were ineffective against pro-caspase-1-induced apoptosis (Fig. 2C).
Although Cp-IAP and human IAPs have been reported to inhibit apoptosis induced by pro-caspase-1 and caspase-1p32 in some mammalian cells (21, 22), we found that the baculovirus-derived IAPs—OpIAP, CpIAP, and AcIAP—were unable to block any of the active caspases in Sf-21 cells. The ability of IAPs to block apoptosis in a given cell type is possibly influenced by the types of caspases present in these cells and the presence or nature of their regulators. Because Sf-21 cells are poised to undergo apoptosis in the absence of additional protein or RNA synthesis, their behavior may be more easily interpreted than many other cell lines. The inactive nature of pro-Sf-caspase-1, the ability of the active enzyme to induce apoptosis and the ability of P35 to inhibit Sf-caspase-1-induced apoptosis support the function of this caspase in the virus-induced apoptotic pathway in Sf cells. The inability Op-iap and Cp-iap to block active Sf-caspase-1 induced apoptosis, but their ability to block apoptosis induced by other inducers like actinomyin D (14), RPR (19), Doom (23), or viral infection (7, 14) suggests that these viral IAPs may function by blocking the activation of Sf-caspase-1.
OpIAP and CpIAP Block Activation of Sf-Caspase-1 during Baculovirus Infection.
To test if viral IAPs can block the activation of Sf-caspase-1, we transfected Sf-21 cells with pro-Sf-caspase-1 and then studied the activation of the caspase following mock infection or infection with wt, P35 mutant, or IAP-expressing recombinants of AcMNPV. Mock infection of Sf-21 cells transiently expressing pro-Sf-caspase-1 did not induce apoptosis at any of the times observed (Fig. 3A, data not shown). Infection with wt virus in the absence of transiently expressed pro-Sf-caspase-1 did not induce apoptosis as expected (Fig. 3B). In contrast, infection of Sf-21 cells transiently expressing pro-Sf-caspase-1 with wt resulted in membrane blebbing and apoptotic body formation beginning at 12 h and continuing through 24 h after infection (Fig. 3D and data not shown). Although, wt AcMNPV expresses the caspase inhibitor P35, apoptosis observed in infections with the wt virus in Sf-21 cells transiently expressing Sf-capase-1 is probably due to limiting levels of P35 that are normally sufficient to block endogenous Sf-caspase-1. In Sf-21 cells transiently expressing an active site mutant (Cys-178 changed to Ala) of Sf-caspase-1, infection with wt virus did not induce apoptosis (Fig. 3C). This indicates that the apoptosis induced by wt virus was due to the activation of the pro-Sf-caspase-1 and was dependent on the proteolytic activity of the Sf-caspase-1.
Figure 3.
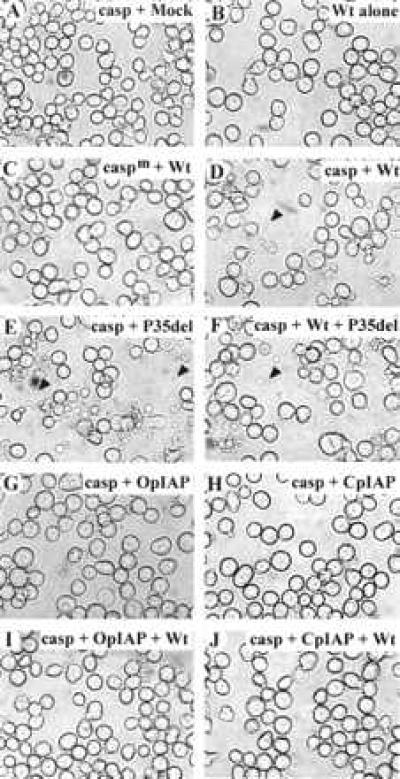
IAP-expressing viruses block virus-initiated apoptosis in Sf-21 cells expressing Sf-caspase-1. (A, D–J) Sf-21 cells transiently expressing pro-Sf-caspase-1 (casp) or (B) mock transfected, or (C) transiently expressing a mutant of pro-Sf-caspase-1 (caspm) were infected with (A) mock, (B–D) Wt, (E) vP35del, (F) Wt + vP35del, (G) vOpIAPR-11 (virus expressing OpIAP), (H) vASB6–11 (virus expressing CpIAP), (I) Wt + vOpIAPR-11, and (J) Wt + vASB6–11. Cells at 18 h after infection were photographed using an Olympus IX50 microscope. The arrowheads in D–F point to apoptotic bodies.
Infection of Sf-21 cells with a P35 mutant AcMNPV, vP35del, induces apoptosis (5). In Sf-21 cells, transiently expressing pro-Sf-caspase-1, infection with vP35del results in widespread apoptosis from 12 h after infection through 24 h after infection (Fig. 3E). As observed in infections involving either vP35del or wt virus alone, coinfection with vP35del and wt virus-induced apoptosis in Sf-21 cells expressing Sf-caspase-1 (Fig. 3F).
In contrast, infection of Sf-21 cells expressing Sf-caspase-1 with viruses expressing OpIAP or CpIAP did not induce apoptosis at any of the time points observed (Fig. 3 G and H and data not shown). Similarly, coinfection of wt virus with OpIAP or CpIAP did not induce apoptosis indicating that the IAPs could act in trans to block apoptosis induced by wt virus infection. These results are consistent with the possibilty that the viral IAPs prevent apoptosis by blocking the activation of Sf-caspase-1 during viral infection.
To determine whether pro-Sf-caspase-1 was being processed to its active subunits, we examined lysates of infected cells transiently expressing an epitope-tagged version of pro-Sf-caspase-1. The presence of an epitope tag on the C terminus of the pro-Sf-caspase-1 allowed the detection of the zymogen (pro-form) and the smaller subunit (p12) of the active enzyme. As expected, the pro-Sf-caspase-1 was not processed in mock-infected cells (Fig. 4A). However, infection with wt virus resulted in processing of the pro-Sf-caspase into its p12 subunit by 12 h after infection and caused a significant decline in the levels of the pro-form by 30 h postinfection (Fig. 4B). This is consistent with the observation that wt virus induces apoptosis under these conditions (Fig. 3D).
Figure 4.
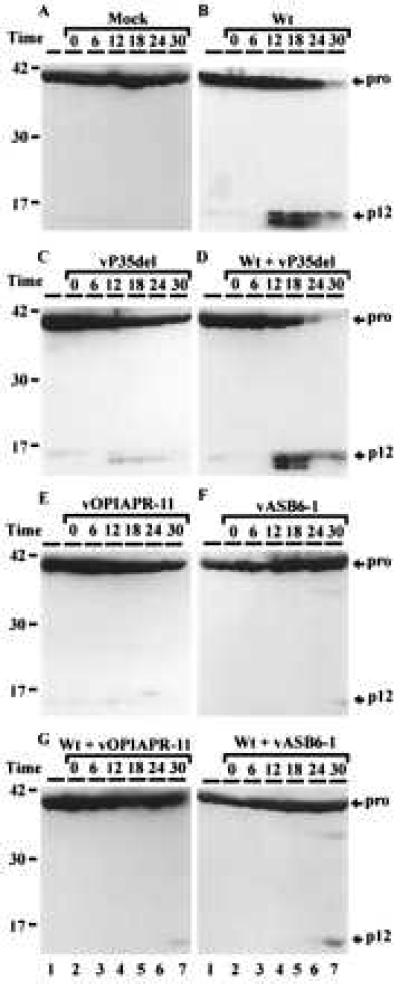
OpIAP and CpIAP block the processing of pro-Sf-caspase-1. Sf-21 cells transiently expressing pro-Sf-caspase-1 were infected with various viruses as indicated in each panel. Samples were collected at the times indicated and were subjected to immunoblot analysis. The presence of the C-terminal epitope tag allowed the detection of pro-Sf-caspase-1 and its processed p12 subunit. The identity of the p12 subunit was established based on its comigration with the product derived independently from plasmid expressing tagged p12 in Sf-21 cells (data not shown). The position of the size standards are shown on the left. The product seen below p12 (∗) is probably derived from cleavage after Asp-209 instead of Asp-195, the previously described (9) cleavage site from which the p12 subunit is derived. Lane 1 in A–H represents samples collected before virus infection.
Infection of Sf-21 cells expressing the pro-Sf-caspase-1 with vP35del induced wide-spread apoptosis, as expected (Fig. 3E). However, only low levels of the p12 subunit were observed under these conditions (longer exposures of the immunoblot revealed a significant presence of the p12 subunit; Fig. 4C and data not shown). This we attribute to the absence of P35 that forms stable complexes with caspases (11) and could stabilize the caspase subunits. We tested this by coinfecting Sf-21 cells with wt and vP35del viruses. As predicted, the coinfection with both the wt and vP35del viruses resulted in the stabilization of the p12 subunit of the active Sf-caspase-1 beginning at 12 h after infection (Fig. 4D).
We then determined whether infection of Sf-21 cells transiently expressing pro-Sf-caspase-1 with recombinant viruses expressing anti-apoptotic IAPs could block the activation of the Sf-caspase-1. Infection with either vOPIAPR-11 or vASB6–1, which express OpIAP and CpIAP, respectively, did not result in processing of the caspase; only very low levels of the p12 subunit of the pro-Sf-caspase-1 were observed. (Fig. 4 E and F). This correlated with the absence of apoptosis in these cells (Fig. 3 G and H). However, these viruses lack P35 so we tested if the IAP expressing viruses could block the pro-Sf-caspase-1 processing observed in the presence of P35 during wt infection. Coinfection of the Sf-21 cells expressing pro-Sf-caspase-1 with the wt virus and either vOpIAPR-11 or vASB6–1 prevented the processing of the caspase and the appearance of the p12 subunit (Fig. 4 compare G and H with B or D). Although a small amount of processed p12 subunit was observed at 30 h in coinfections with wt and vOPIAPR-11 or vASB6–1, no apoptosis was observed in these cells even at 30 h and >95% of the cells contained occluded virus indicating the normal progression of infection (data not shown).
While these viral IAPs do not block the active form of Sf-caspase-1 (Fig. 2A) there are several possible mechanisms by which IAPs block apoptosis. Human xIAP can inhibit the activity of activated mammalian caspase-3 and caspase-7 in vitro (26). It is possible that OpIAP and CpIAP block the activity of another, yet to be defined, Sf-caspase that acts upstream of Sf-caspase-1 in the apoptotic pathway. However, viral IAPs have also been found to physically interact with cellular inducers of apoptosis such as Drosophila RPR (19) and Doom (23) and block their ability to induce apoptosis. The viral IAPs could interact with a Sf-homolog of Drosophila RPR or Doom and prevent the apoptosis-inducing signals from transducing to the downstream effectors (e.g., caspases). Our results show that, unlike P35, OpIAP and CpIAP do not block active Sf-caspase-1 but function by blocking the activation of pro-Sf-caspase-1.
Acknowledgments
We thank Jeanne R. McLachlin, Julie Olszewski, Domagoj Vucic, and Alex Harvey for their helpful discussions during the course of the project; Wen-teh Chang for constructing Sf-caspase-1 clones; and Elena Prikhodka for preparing the cells. We also thank Winne W. Wong (BASF Bioresearch Corp., Worcester, MA) for cDNAs encoding caspase-1, caspase-3, and crmA and for the pKV vector; Emad S. Alnemri (Jefferson Medical College, Philadelphia) for Sf-caspase-1; Craig B. Thompson (University of Chicago) for bcl-xL cDNA; and H. Robert Horvitz (Massachusetts Institute of Technology) for ced-9 cDNA. This work was supported in part by U.S. Public Health Services Grant AI38262 from the National Institute of Allergy and Infectious Disease.
ABBREVIATIONS
- IAP
inhibitor of apoptosis
- ICE
interleukin-1β converting enzyme
- AcMNPV
Autographa californica nuclear polyhedrosis virus
- CAT
chloroamphenicol acetyltransferase, wt, wild type
- RPR
Reaper
- hsp70
70-kDa heat shock protein
References
- 1.Kerr J F, Wyllie A H, Currie A R. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wyllie A H, Kerr J F, Currie A R. Int Rev Cytol. 1980;68:251–306. doi: 10.1016/s0074-7696(08)62312-8. [DOI] [PubMed] [Google Scholar]
- 3.Thompson C B. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 4.Shen Y, Shenk T E. Curr Opin Genet Dev. 1995;5:105–111. doi: 10.1016/s0959-437x(95)90061-6. [DOI] [PubMed] [Google Scholar]
- 5.Clem R J, Fechheimer M, Miller L K. Science. 1991;254:1388–1390. doi: 10.1126/science.1962198. [DOI] [PubMed] [Google Scholar]
- 6.O’Reilly D A, Miller L K, Luckow V A. Baculovirus Expression Vectors: A Laboratory Manual. New York: Freeman; 1992. [Google Scholar]
- 7.Clem R J. In: The Baculoviruses. Miller L K, editor. New York: Plenum; 1997. pp. 237–266. [Google Scholar]
- 8.Alnemri E S, Livingston D J, Nicholson D W, Salvesen G, Thornberry N A, Wong W W, Yuan J. Cell. 1996;87:171. doi: 10.1016/s0092-8674(00)81334-3. [DOI] [PubMed] [Google Scholar]
- 9.Ahmad M, Srinivasula S M, Wang L, Litwack G, Fernandes-Alnemri T, Alnemri E S. J Biol Chem. 1997;272:1421–1424. doi: 10.1074/jbc.272.3.1421. [DOI] [PubMed] [Google Scholar]
- 10.Kuida K, Zheng T S, Na S, Kuan C, Yang D, Karasuyama H, Rakic P, Flavell R A. Nature (London) 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- 11.Bump N J, Hackett M, Hugunin M, Seshagiri S, Brady K, Chen P, Ferenz C, Franklin S, Ghayur T, Li P, Mancovich J, Shi L, Greenberg A H, Miller L K, Wong W W. Science. 1995;269:1885–1888. doi: 10.1126/science.7569933. [DOI] [PubMed] [Google Scholar]
- 12.Xue D, Horvitz H R. Nature (London) 1995;377:248–251. doi: 10.1038/377248a0. [DOI] [PubMed] [Google Scholar]
- 13.Bertin J, Mendrysa S M, LaCount D J, Gaur S, Krebs J F, Armstrong R C, Tomaselli K J, Friesen P D. J Virol. 1996;70:6251–6259. doi: 10.1128/jvi.70.9.6251-6259.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clem R J, Miller L K. Mol Cell Biol. 1994;14:5212–5222. doi: 10.1128/mcb.14.8.5212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clem R J, Hardwick J M, Miller L K. Cell Death Differ. 1996;3:9–16. [PubMed] [Google Scholar]
- 16.Birnbaum M J, Clem R J, Miller L K. J Virol. 1994;68:2521–2528. doi: 10.1128/jvi.68.4.2521-2528.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crook N E, Clem R J, Miller L K. J Virol. 1993;67:2168–2174. doi: 10.1128/jvi.67.4.2168-2174.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hay B A, Wassarman D A, Rubin G M. Cell. 1995;83:1253–1262. doi: 10.1016/0092-8674(95)90150-7. [DOI] [PubMed] [Google Scholar]
- 19.Vucic D, Seshagiri S, Miller L K. Mol Cell Biol. 1997;17:667–676. doi: 10.1128/mcb.17.2.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Manji G A, Hozak R R, LaCount D J, Friesen P D. J Virol. 1997;71:4509–4516. doi: 10.1128/jvi.71.6.4509-4516.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duckett C S, Nava V E, Gedrich R W, Clem R J, Van Dongen J L, Gilfillan M C, Shiels H, Hardwick J M, Thompson C B. EMBO J. 1996;15:2685–2694. [PMC free article] [PubMed] [Google Scholar]
- 22.Uren A G, Pakusch M, Hawkins C J, Puls K L, Vaux D L. Proc Natl Acad Sci USA. 1996;93:4974–4978. doi: 10.1073/pnas.93.10.4974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harvey A J, Bidwai A P, Miller L K. Mol Cell Biol. 1997;17:2835–2843. doi: 10.1128/mcb.17.5.2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clem R, Duckett C. Trends Cell Biol. 1997;7:337–339. doi: 10.1016/S0962-8924(97)01088-X. [DOI] [PubMed] [Google Scholar]
- 25.Liston P, Roy N, Tamai K, Lefebvre C, Baird S, Cherton-Horvat G, Farahani R, McLean M, Ikeda J E, MacKenzie A, Korneluk R G. Nature (London) 1996;379:349–353. doi: 10.1038/379349a0. [DOI] [PubMed] [Google Scholar]
- 26.Devereaux Q L, Takahashi R, Salvesen G S, Reed J C. Nature (London) 1997;388:300–304. doi: 10.1038/40901. [DOI] [PubMed] [Google Scholar]
- 27.Vaughn J L, Goodwin R H, Tompkin G J, McCawley P. In Vitro. 1977;13:213–217. doi: 10.1007/BF02615077. [DOI] [PubMed] [Google Scholar]
- 28.Lee H H, Miller L K. J Virol. 1978;27:754–767. doi: 10.1128/jvi.27.3.754-767.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Walker N P, Talanian R V, Brady K D, Dang L C, Bump N J, et al. Cell. 1994;78:343–352. doi: 10.1016/0092-8674(94)90303-4. [DOI] [PubMed] [Google Scholar]
- 30.Nicholson D W, Ali A, Thornberry N A, Vaillancourt J P, Ding C K, Gallant M, Gareau Y, Griffin P R, Labelle M, Lazebnik Y A, Munday N A, Raju S M, Smulson M E, Yamin T, Yu V L, Miller D K. Nature (London) 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- 31.Quan L T, Tewari M, K, O R, Dixit V, Snipas S J, Poirier G G, Ray C, Pickup D J, Salvesen G S. Proc Natl Acad Sci USA. 1996;93:1972–1976. doi: 10.1073/pnas.93.5.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fernandes-Alnemri T, Litwack G, Alnemri E S. J Biol Chem. 1994;269:30761–30764. [PubMed] [Google Scholar]
- 33.Schlegel J, Peters I, Orrenius S, Miller D K, Thornberry N A, Yamin T T, Nicholson D W. J Biol Chem. 1996;271:1841–1844. doi: 10.1074/jbc.271.4.1841. [DOI] [PubMed] [Google Scholar]
- 34.Seshagiri S, Miller L K. Curr Biol. 1997;7:455–460. doi: 10.1016/s0960-9822(06)00216-8. [DOI] [PubMed] [Google Scholar]