

Abstract
Parathyroid hormone-related protein (PTHrP) is a prohormone that is posttranslationally processed to a family of mature secretory forms, each of which has its own cognate receptor(s) on the cell surface that mediate the actions of PTHrP. In addition to being secreted via the classical secretory pathway and interacting with cell surface receptors in a paracrine/autocrine fashion, PTHrP appears to be able to enter the nucleus directly following translation and influence cellular events in an “intracrine” fashion. In this report, we demonstrate that PTHrP can be targeted to the nucleus in vascular smooth muscle cells, that this nuclear targeting is associated with a striking increase in mitogenesis, that this nuclear effect on proliferation is the diametric opposite of the effects of PTHrP resulting from interaction with cell surface receptors on vascular smooth muscle cells, and that the regions of the PTHrP sequence responsible for this nuclear targeting represent a classical bipartite nuclear localization signal. This report describes the activation of the cell cycle in association with nuclear localization of PTHrP in any cell type. These findings have important implications for the normal physiology of PTHrP in the many tissues which produce it, and suggest that gene delivery of PTHrP or modified variants may be useful in the management of atherosclerotic vascular disease.
Keywords: cellular proliferation, alternative translational start site, nucleolus
Parathyroid hormone-related protein (PTHrP) (Fig. 1), isolated in 1987, is responsible for the common endocrine paraneoplastic syndrome, humoral hypercalcemia of malignancy (1). PTHrP is now known to be produced by most cells and tissues in the body (1–4). Following translation, PTHrP enters the secretory pathway and, in cell types that posses the regulated secretory pathway such as pancreatic islet cells and atrial cardiocytes, it is packaged into secretory granules and is subject to regulated secretion (5, 6). In tissues that lack the regulated secretory pathway, such as squamous carcinoma cells and fibroblasts, it is secreted constitutively (5, 6). During its transit through the secretory pathway, the precursor is endoproteolytically processed at basic residues to yield a family of mature secretory forms of the peptide (Fig. 1) (2, 3, 7). Other secretory forms are certain to exist, including peptides with the approximate compositions of PTHrP(107–139) and PTHrP(141–173). Each of the secretory forms of PTHrP appears to act via cell surface receptors (8–12).
Figure 1.
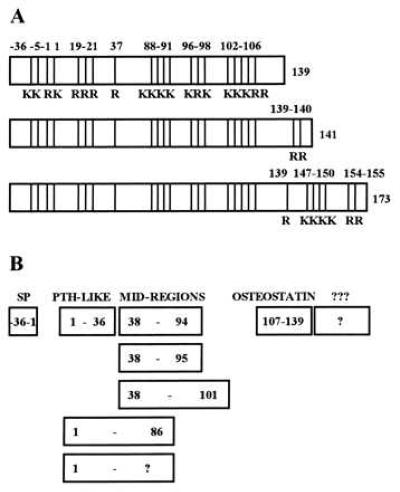
(A) Structural maps of the three PTHrP initial translation products. “K” indicates lysine and “R” arginine. The three translation products are identical in their signal peptide (−36 to −1) region and in their remaining coding regions from amino acids 1 through 139. Alternative splicing gives rise to two C-terminally extended versions of the peptide with lengths of 141 and 173 amino acids. Note the multibasic clusters: the KKXRK at −5 to −1, the single R at +37, and the KR clusters in the 88–106 region are known to be prohormone convertase substrate sites. (B) The mature secretory forms of PTHrP. After cleavage of the signal and pro-peptide in the −36 to −1 region, the secretory forms of PTHrP include PTHrP(1–36) which interacts with the PTH/PTHrP receptor, three mid-region species, and at least two C-terminal species.
In addition to its secretion via the classical secretory pathway, other researchers have provided evidence that PTHrP may signal via translocation to the nucleus or nucleolus (13–15). Recognizing that the multibasic clusters in the 88–106 region are similar to nuclear or nucleolar localization signals in viral and mammalian transcription factors (13–16), these reseachers demonstrated that PTHrP can be observed in the nucleolus of chondrocytes and transfected COS cells, that deletion of the multibasic 87–106 region prevents this targeting, and that expression of this multibasic 87–106 region as a fusion protein with β-galactosidase directs this latter protein to the nucleolus (15). In addition, Henderson et al. (14) have suggested that nuclear targeting of PTHrP inhibits apoptosis in chondrocytes.
PTHrP is expressed in arterial smooth muscle and is up-regulated by mechanical stretch, by hypertension, by vasoconstrictors such as angiotensin II, and by mechanical injury such as angioplasty (17). It is expressed at higher than normal levels in atherosclerotic human coronary arteries (18). PTHrP has been shown to be a potent vasodilator and hypotensive agent (17). Pirola et al. (19), Maeda et al. (20), and Jiang et al. (21) have all demonstrated that PTHrP is an inhibitor of vascular smooth muscle (VSM) cell proliferation, and that PTHrP acts in smooth muscle via the cell surface PTH/PTHrP receptor.
We were interested in studying the posttranslational processing of PTHrP in VSM cells, and therefore stably transfected a commonly used rat smooth muscle cell line, A-10, with a PTHrP-encoding plasmid. To our surprise, and in contrast to the inhibition of proliferation observed by other researchers (19–21), stable transfection with PTHrP dramatically stimulated VSM cell proliferation. These observations have important mechanistic implications for the actions of PTHrP in the several transgenic mouse models of targeted PTHrP overexpression and for the physiologic actions of PTHrP in the myriad tissues that express the peptide under normal conditions.
METHODS
Materials.
Human (h) PTHrP(1–36), hPTHrP(38–94) amide, hPTHrP(107–139), and hPTHrP(109–138) were synthesized by using solid phase methods described in detail previously (7, 22). hPTHrP(1–108) and hPTHrP(1–141) were kindly provided by R. Glenn Hammond (Genentech, South San Francisco, CA). hPTHrP(67–86) amide and hPTHrP(1–86) were purchased from Bachem.
Cell Culture and Stable Transfection.
A-10 cells are a clonal VSM cell line from rat thoracic aorta (23) and were purchased from the American Type Culture Collection. Primary culture of bovine aortic smooth muscle cells were generously provided by Richard Powell (Yale University). A-10 and bovine cells were cultured, unless otherwise specified, at 37°C in humidified 95% 02/5% CO2 atmosphere in RPMI 1640 medium containing 10% fetal bovine serum, antibiotics, and l-glutamine.
A-10 cells were stably transfected as described previously (24) by using lipofectamine (GIBCO/BRL). Two days following transfection, 50 μg/ml G418 (Genticin; GIBCO/BRL) was added to the growth medium and resistant clones were selected. Clones were selected on the basis of PTHrP production as assessed by using the immunoassays described below. Ten to 20 individual clones for each plasmid preparation were picked, placed into individual flasks, and cultured in medium containing 50 μg/ml G418. For each plasmid preparation, two to three clones were used. All clones for all conditions were A-10 cells at the same number of passage.
Plasmids and Mutagenesis.
PTHrP was overexpressed in A-10 cells by using the retroviral vector pLJ, which we have employed previously (24). Constructs that encoded (i) full-length hPTHrP(1–139), (ii) single deletion PTHrP mutants (88–91 or 102–106), and (iii) a double deletion PTHrP mutant (88–91, 102–106) were created by using site-directed mutagenesis as reported (7), and the sequences were confirmed by direct DNA sequencing. The constructs were then directionally subcloned in the pLJ vector and stably transfected in A-10 cells as described above.
Cell Proliferation.
A-10 or bovine aortic smooth muscle cells were plated into 24-well plates at 1 × 104 cells/well and cultured in serum-containing medium until 80% confluent. Cells were then rendered quiescent by culturing them in serum-free medium containing 0.1% BSA for 48 h. Quiescent cells were labeled with 1 μCi/ml [3H]thymidine (Amersham; 1 Ci = 37 GBq) for 24 h in the presence or absence of PTHrP peptides or dibutyryl cAMP (Sigma) as described in Fig. 3A. After 24 h, cells were washed three times in ice-cold PBS. One milliliter per well of ice-cold 10% trichloroacetic acid was then added, and the precipitate was incubated for 20 min on ice, pelleted, air-dried, and dissolved in 0.6 M NaOH. [3H]Thymidine was measured by liquid scintillation.
Figure 3.
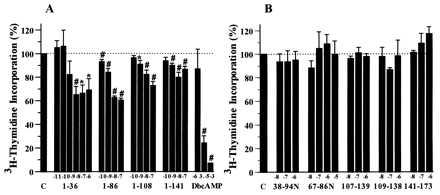
Effects of PTHrP addition to cultured A-10 cells. (A) PTHrP peptides that contain an intact N terminus and dibutyryl cAMP inhibit A-10 cell proliferation. (B) Non-N-terminal PTHrP peptides, comprising all of the known secretory forms of PTHrP (see Fig. 1), have no effect on A-10 cell proliferation. #, P < 0.001; ∗, P < 0.05. “C” indicates control, and the numbers along the x-axis indicate the PTHrP peptide employed [e.g., 1–36 indicates PTHrP(1–36), and 38–94N indicates PTHrP(38–94)amide].
To confirm the results of [3H]thymidine experiments on cell growth, proliferation was confirmed by cell counting. Untransfected and transfected A-10 cell lines were plated into 24-well plates at 1 × 104 cells/well and cultured in serum-containing medium for 10–14 days. Cells were harvested at 48-h intervals by trypsinizing cells and counted by hemocytometer. In all cases, cell viability was greater than 90% as assessed by trypan blue exclusion.
Coculture Experiments.
Vector-transfected or wild-type PTHrP-transfected A-10 cells were grown on 3-μm pore size 24-well culture inserts (Corning) until 80% confluent. Culture inserts containing either vector-transfected or PTHrP-transfected cells were placed in culture wells containing untransfected A-10 cells. Coculture was allowed to continue for 10 days without medium change. Untransfected A-10 cells cocultured either with vector-transfected or PTHrP-transfected A-10 cells were harvested at 48-h intervals by trypsinizing cells and counted with a hemocytometer.
PTHrP Immunoradiometric Assays in Conditioned Medium and Cell Extracts.
Two PTHrP immunoradiometric assays (IRMAs) were performed. The first is a PTHrP(1–36) IRMA, and the second a PTHrP(1–74) IRMA (6, 25).
PTHrP Immunocytochemistry.
Immunocytochemistry was performed as described by using an affinity-purified rabbit polyclonal primary antibody (6, 24) to hPTHrP(37–74) diluted 15–20 μg/ml (final concentration). The number of nuclei staining for PTHrP were quantified by two independent blinded viewers.
Measurement of DNA Synthesis in PTHrP “Knockout” Mouse Aortic Smooth Muscle.
PTHrP knockout mice were generously provided by A. Karaplis (Montreal) and H. Kronenberg, (Boston). Heterozygous males and females were mated and pregnant dams were injected subcutaneously with BrdU (100 mg/kg body weight; Amersham) on day 18.5 of gestation. Embryos were harvested 6 h after labeling, fixed in paraformaldehyde, and paraffin-embedded. Transverse sections including the aorta were stained with hematoxylin/eosin or examined immunohistologically for BrdU incorporation. The proportion of BrdU-positive cells in the aortic media layer of wild-type (+/+) and homozygous (−/−) embryos was quantitated in a blinded fashion.
Statistical Analysis.
All values are expressed as mean ± SEM, and statistical comparison are based on the Student’s t test. P < 0.05 was considered significant.
RESULTS
The growth consequences of stable transfection of A-10 VSM cells with PTHrP(1–139) are shown in Fig. 2. As can be seen from the figure, the rate of proliferation of three PTHrP-expressing VSM clones was four to five times more rapid than that of three vector-transfected clones. The rate of proliferation of untransfected A-10 cells was the same as that of the vector-transfected lines (data not shown). The marked difference in the proliferation rates of PTHrP-expressing as compared with vector-transfected and untransfected A-10 cells was confirmed by using [3H]thymidine incorporation (data not shown).
Figure 2.
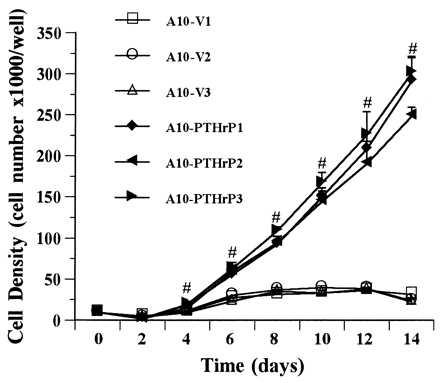
Proliferation of three PTHrP-overexpressing A-10 cells clones (filled symbols) and of three vector-transfected clones (open symbols). Note that the PTHrP-overexpressing clones proliferate at a rate far higher than the vector transfected clones. Untransfected A-10 cells are indistinguishable from the vector-transfected clones. Similar results were observed by using [3H]thymidine incorporation. #, P < 0.01.
To examine the effects of PTHrP and PTH peptides in our system, we examined the rate of proliferation of VSM in culture medium to which PTHrP or PTH peptides had been added. As can be seen in Fig. 3, the addition of PTHrP peptides that contain an intact N terminus inhibited [3H]thymidine incorporation into A-10 cells by ≈30–40%, and displayed an IC50 in the nM range. These results were confirmed in studies in which direct cell counting was performed. Identical results were observed by using primary cultures of bovine aortic VSM (data not shown). Finally, because these studies were performed over 24 h, whereas those in Fig. 2 were performed over 2 weeks, we examined the effects of daily addition of PTHrP(1–36) over 2 weeks to A-10 cells in culture. Again, under these conditions that precisely mimicked those shown in Fig. 2, exogenous PTHrP(1–36) inhibited A-10 VSM proliferation (data not shown).
PTH has been reported to inhibit VSM proliferation (19–21). As with N-terminal PTHrP peptides, hPTH(1–34), rat PTH(1–34), and hPTH(1–84) all inhibited A-10 proliferation both as measured both by [3H]thymidine incorporation, as well as by cell number (data not shown). The magnitude and IC50 of the inhibition were the same as those shown for PTHrP in Fig. 3A. The inhibition was also observed in cells exposed to dibutyryl cAMP (Fig. 3A), suggesting that the inhibition may be cAMP-mediated, because it is known that cAMP inhibits VSM proliferation and because the PTH/PTHrP receptor in VSM is coupled to adenylyl cyclase (20).
PTHrP is processed to a family of mature secretory peptides, most of which do not contain the N terminus (Fig. 1). One explanation for the contrasting effects of exogenous vs. stably transfected PTHrP on VSM proliferation could be that these additional non-N-terminal PTHrP peptides secreted by stably transfected VSM might stimulate VSM proliferation. To examine this hypothesis, as shown in Fig. 3B, proliferation of A-10 cells was studied following the addition of a panel non-N-terminal PTHrP peptides. None of these peptides influenced A-10 proliferation.
To determine whether additional as yet unidentified secretory forms of PTHrP might stimulate VSM proliferation, proliferation of untransfected A-10 cells was studied in a coculture system in which A-10 cells were exposed to the secretory products of PTHrP-transfected A-10 cells grown on filters within culture inserts. Proliferation of untransfected A-10 cells exposed to medium from PTHrP-transfected A-10 cells was 52.5 ± 4.1% (mean ± SE) the rate of control (untransfected A-10 cells exposed to untransfected A-10 cells).
One explanation for the findings described above could be that the multibasic clusters of amino acids in the PTHrP(88–106) region could serve as a nuclear localization signal, as hypothesized (13–15). To test this hypothesis, we prepared a panel of PTHrP deletion mutants (Fig. 4). These mutants were transfected into A-10 cells and clones were selected on the basis of PTHrP secretion as determined by two PTHrP immunoradiometric assays (Fig. 4). Two clones were selected for each of the deletion mutants, and each produced amounts of PTHrP that were equivalent to or greater than the wild-type PTHrP-transfected A-10 cells used in Fig. 2.
Figure 4.
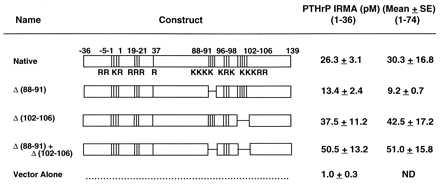
Map of the wild-type and native PTHrP constructs, the three deletion mutants, and vector alone. Values on the right indicate the concentrations of PTHrP in the conditioned medium from the various cell lines. Note that the mutant PTHrP constructs lead to similar levels of PTHrP production as the native construct, and that all constructs lead to the far greater production of PTHrP than the vector-transfected A-10 cells.
The proliferation rates of A-10 cells transfected with the “nuclear targeting” deletion mutants and with A-10 cells transfected with wild-type PTHrP are shown in Fig. 5. These studies demonstrate that A-10 cells expressing the three nuclear targeting deletion mutants proliferate at a rate that is much slower than the rate of the wild-type PTHrP-expressing A-10 cells. Indeed, the rate is even slower than that of the untransfected A-10 cells, indicating that expression of the “nuclear targeting signal deletion” mutants inhibits proliferation in a fashion similar to that observed with the addition of exogenous PTHrP peptides shown in Fig. 3A.
Figure 5.
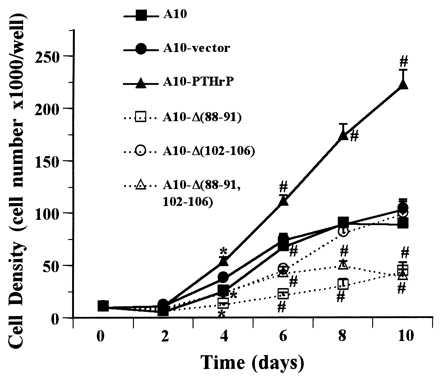
Proliferation of A-10 cells expressing the native PTHrP construct (filled triangles), the three mutant constructs (open symbols), the vector alone (filled circles), or untransfected A-10 cells (filled squares). #, P < 0.01; ∗, P < 0.05. Note that, as seen in the prior experiment in Fig. 2, PTHrP-overexpressing A-10 cells proliferate far more rapidly than their normal or vector-transfected counterparts. Note also that the three mutant-transfected clones proliferate at a rate that is even slower than the untransfected or vector-transfected cells. Each symbol represents the mean of three different clones.
As shown in Fig. 6, A-10 cells overexpressing wild-type PTHrP contained many PTHrP-staining nuclei (≈5–6% of cells), and these nuclei were most often associated with cells in the process of, or completing, cell division. The pattern of staining was a diffuse reticulated pattern. To determine whether PTHrP was present in the nucleus of untransfected VSM under normal circumstances, A-10 cells were stained for PTHrP by using immunofluorescence. As shown in Fig. 6 Bottom, untransfected and vector-transfected A-10 cells contain rare nuclei (≈0.3% of cells), which stain for PTHrP. Importantly, PTHrP-containing nuclei were observed in untransfected cells that were dividing or in the process of completing cell division. The pattern of staining was identical in intensity and pattern to the reticulated diffuse pattern observed in overexpressing cells, but distinct from the nucleolar pattern described by Henderson et al. (15) in chondrocytes and COS cells. Cells stained in the absence of primary antiserum or in the presence of excess synthetic PTHrP showed no nuclear staining (data not shown).
Figure 6.
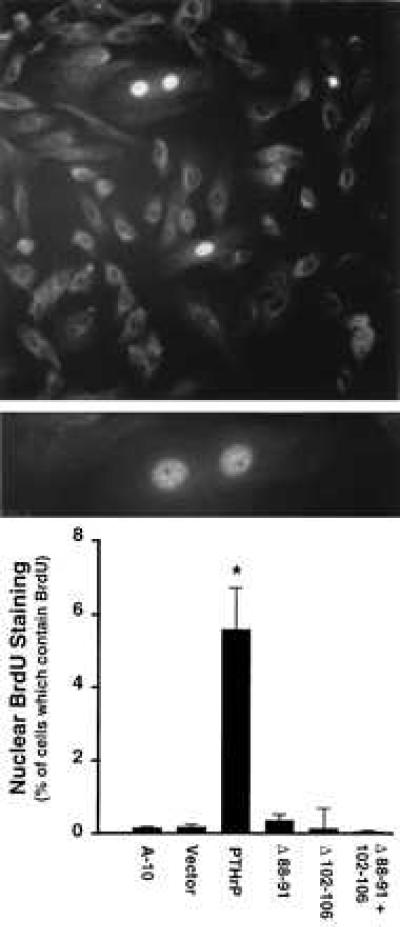
(Top) PTHrP immunocytochemistry showing nuclear staining in PTHrP-overexpressing A-10 cells. (×200.) (Middle) A higher power magnification of the PTHrP-overexpressing cell nuclei. (×400.) Note that the pattern is a diffuse reticulated pattern and that the two most prominent nuclei are in a single dividing cell. For immunocytochemistry, negative controls (no primary antibody and competition with excess PTHrP) showed no staining (data not shown). (Bottom) The percentage of nuclei that contain PTHrP in the various A-10 cell clones. Each bar represents the mean of nine slides, and 300-1000 cells were counted per slide by two blinded observers. The bars indicate standard error; ∗, P < 0.01.
The three PTHrP deletion mutant-expressing A-10 cell types displayed a marked reduction in frequency of nuclear staining (Fig. 6 Bottom), despite levels of PTHrP production which were equivalent to or greater than the wild-type PTHrP overexpressing lines (Fig. 4). These studies demonstrate an association among the rates of A-10 cell proliferation, the frequency of nuclear staining, the presence of the putative nuclear localization signal, and the process of cell division.
Disruption of the PTHrP gene has been accomplished in transgenic mice (4). To the extent that they have been studied, there are no known vascular abnormalities in the PTHrP null mouse. To determine the rate of VSM proliferation in vivo in PTHrP null mouse embryos, heterozygous PTHrP null parents were bred, the pregnant dams labeled with BrdU on day embryonic 18.5, and the rates of nuclear labeling in aortic smooth muscle of homozygous PTHrP null mutants was compared with that of wild-type PTHrP fetuses. As shown in Fig. 7, aortic smooth muscle proliferation in the PTHrP “knockout” mice was dramatically reduced as compared with the rate of proliferation in normal littermates.
Figure 7.
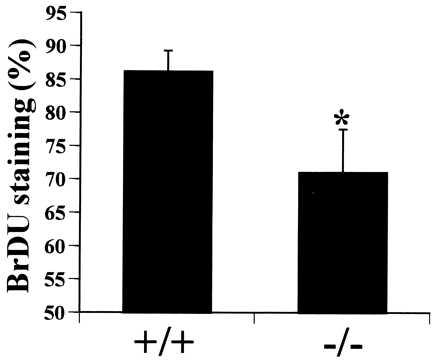
The percent of aortic smooth muscle cell nuclei that contain BrdU in PTHrP knockout mouse fetuses (−/−) or in their normal littermates (+/+). Each bar represents the mean of nine embryos; ∗, P < 0.02.
DISCUSSION
PTHrP, in addition to being a smooth muscle vasodilator (17), is a factor that regulates the growth, development, or differentiation in virtually every tissue in which it has been examined (2). For example, studies in transgenic mice have demonstrated that PTHrP is required for normal chondrocyte maturation and differentiation in the epiphyseal growth plate (4), for normal epidermal and hair follicle development (26), and for normal mammary development (27). In the pancreatic islet, targeted overexpression of PTHrP leads to a dramatic increase in pancreatic β-cell mass (28).
In VSM cells in culture and in vivo, PTHrP is up-regulated by vasoconstrictive agents such as angiotensin II, by mechanical stretching of arterial smooth muscle, and by the induction of hypertension with mineralocorticoids (17). Human coronary artery segments removed during coronary arterial revascularization procedures demonstrate a marked increase in PTHrP content (18). In animal models of vascular damage induced by balloon angioplasty, PTHrP is up-regulated by arterial damage and, as assessed by immunohistochemical and in situ hybridization techniques, is associated with the formation of proliferating vascular cells in the neointima (29).
Because PTHrP has been associated with proliferative, anti-proliferative, and apoptotic responses, the role of PTHrP in neointima could in theory be as a participant in the neointimal proliferation process or as an endogenous inhibitor of the same process. In support of the “VSM anti-proliferative” hypothesis, Pirola et al. (19), Maeda et al. (20), and Jiang et al. (21) have all reported that the addition of PTHrP(1–34) and/or PTHrP(1–141) inhibit VSM proliferation in vitro. Here, we confirm that PTHrP(1–36), PTHrP(1–86), PTHrP(1–108), and PTHrP(1–141) are inhibitors of VSM proliferation. On the other hand, we unexpectedly observed that overexpression of PTHrP by gene transfer dramatically stimulates VSM proliferation. This paradoxical result is not the result of secretion of cell cycle stimulatory PTHrP species because a panel of all of the known non-N-terminal PTHrP secretory forms had no effect on proliferation, and because coculture of normal A-10 VSM cells with PTHrP-overexpressing A-10 cells resulted in inhibition of the proliferation of untransfected A-10 cells.
The biologic relevance of PTHrP to vascular development in vivo is underscored by the observation that the aortic VSM proliferation rate in the PTHrP knockout mouse is markedly lower than that in aortic VSM from normal animals. The long-term consequences of this deceleration of VSM proliferation in vivo are unknown, because PTHrP null mice die immediately after birth (4). Nonetheless, these in vivo findings make the point that PTHrP must be involved in the regulation of the rate of proliferation in vivo under normal circumstances. Moreover, they indicate that the proliferative response to PTHrP is not likely an artifact of transfection and overexpression. Finally, they suggest that in vivo the nuclear targeting effect on proliferation may override the receptor-mediated effects to inhibit proliferation.
Kaiser, Henderson and Karaplis and colleagues (13–15) have suggested that PTHrP, in addition to its well-studied endocrine, paracrine, and autocrine effects, may also act in an “intracrine” fashion. The three PTHrP initial translation products all contain three clusters of basic amino acids in the 88–106 region of the peptide. These clusters resemble the bipartite nuclear localization signals and nucleolar localization signals observed in a number of viral and eukaryotic transcription factors (such as c-jun and c-fos) and in growth factors (such as members of the fibroblast growth factor family) (reviewed in refs. 15 and 16). Indeed, as they had predicted, PTHrP was observed in the nucleolus of mouse chondrocytes and osteoblasts as well as in PTHrP-overexpressing COS cells by immunocytochemistry and immunoelectron microscopy (15). Deletion of the entire basic 87–106 region prevented the nucleolar targeting in COS cells. Importantly, expression of this 88–106 region as a fusion protein with β-galactosidase in COS cells resulted in targeting of β-galactosidase, a cytosolic protein under normal circumstances, to the nucleolar compartment. These observations strongly suggest that PTHrP can enter the nucleus under normal circumstances, and that specific amino acids in the 88–106 region are responsible for this nuclear/nucleolar targeting.
In the current study, we have demonstrated that nuclear targeting of PTHrP occurs in VSM cells as well. It is significant that PTHrP could be observed in the nuclear compartment of untransfected A-10 cells, for this indicates that nuclear targeting is not the artifactual result of overexpression. Moreover, we have pinpointed the nuclear targeting sequences to two clusters of basic amino acids in the 88–91 and 102–106 positions. Deletion of either one of these clusters prevents nuclear targeting, suggesting that the PTHrP nuclear localization signal is indeed a bipartite nuclear localization signal (13–16).
In functional terms, our results contrast rather strikingly from those of Henderson et al. (15). Those investigators found an inhibitory effect of PTHrP on apoptosis, whereas we found a dramatic stimulatory effect on proliferation. We did not examine our A-10 cells for apoptotic events, and it is possible that the rate of cell death in our cells is lower than normal, but this could not explain the dramatic effects of PTHrP on proliferation. The proliferative effects of PTHrP are particularly intriguing because the nuclear PTHrP staining was clearly associated with cells in the process of mitosis. This association with mitotic nuclei was observed in both untransfected and overexpressing A-10 cells. This observation has been independently confirmed: Okano et al. (30) examined the relation of cell cycle phase to PTHrP immunoreactivity in primary cultures of rat aortic VSM. They did not perform immunocytochemistry on their cells, but observed with cell sorting that ≈10% of rat VSM cells contained immunoreactive PTHrP, and that 34% of these were in either G2 or M. In contrast, in PTHrP immunonegative cells, only 4% were in G2 or M.
Importantly in the current study, deletion of the nuclear targeting sequences not only normalized, but dramatically reduced, the rate of proliferation. This finding suggests either that overexpression of the mutant protein interferes with proliferation through a “dominant negative” type of interaction, or, more likely, that the nuclear targeting deletions still permit secretion of large amounts of N-terminally intact PTHrP species which act, like PTHrP(1–36) added to the medium, to inhibit VSM proliferation. Indeed, PTHrP concentrations in the medium of the mutant cell lines were in the 10−10–10−9 M range, far higher than the <10−12 M range in control cultures, and well within the range required to inhibit proliferation.
Given the above observations, one must question the mechanism that allows PTHrP to be directed either into the classical secretory pathway under one set of circumstances, but directed to the nucleus in another set of circumstances. Clearly, the nascent peptide must be directed by its signal peptide into the endoplasmic reticulum if it is to enter the classical secretory pathway. Equally clearly, the precursor must avoid the endoplasmic reticulum if it is to remain in the cytosol prior to its nuclear translocation. The intracellular targeting of fibroblast growth factor-3 is particularly instructive here (31–33). In this example, translation may initiate at a classic AUG translational initiation site, or may initiate at an upstream CUG codon. If the former is used, the resultant peptide is directed into the secretory pathway by the signal peptide, whereas if the latter is used, the resultant peptide is directed to the cytosol and then the nucleus. This finding appears to result from the interference of the extended N-terminal sequence with the interaction between the signal peptide and the signal recognition particle. A potential alternative translational start site (CUG) is present in the PTHrP precursor in the −32 position. If translation initiated at this site, key functional features of the signal peptide would also be bypassed, and the nascent peptide would be free to enter the cytosol, and then the nucleus.
Another question relates to the precise nuclear localization of PTHrP within the nucleus. Henderson et al. (15) described nucleolar accumulation of PTHrP in chondrocytes and osteoblasts, whereas the nuclear staining in A-10 cells appears in a diffuse nuclear pattern. Whether this difference in the nuclear staining pattern is a reflection of the different cells under study, or whether it reflects the different antisera employed remains to be studied. Another important question relates to the mechanism whereby PTHrP activates the cell cycle once it gains entry to the nucleus, and what role the multibasic clusters play within the nucleus. Do they interact with the nuclear pore proteins such as the importins/karyopherins? Do they interact with nucleic acids within the nucleus? Does PTHrP interact with other proteins in the nucleus? The answers to these questions will require further study.
PTHrP is an unusual protein. (i) It can be secreted by either the regulated secretory pathway or the constitutive secretory pathway. Although this type of secretory pathway “promiscuousness” may occur with other peptides expressed in non-native cell types, it would appear to be unusual under normal circumstances (6). (ii) As reported by Ditmer et al. (34), the multibasic clusters within PTHrP appear to be able to control the rate of intracellular PTHrP degradation. (iii) As reported by Henderson et al. (15) and herein, PTHrP, like fibroblast growth factor-3, appears to act via an “intracrine” mechanism. (iv) As described herein, the cellular response to PTHrP nuclear interactions appears to be cell-type specific, in some cell types preventing apoptosis, in others inhibiting proliferation, and in still other cell types, such as VSM, activating the cell cycle. Activation of the cell cycle by PTHrP in association with nuclear targeting has not been reported previously in any cell type. (v) As also described herein, the very same multibasic clusters [e.g., the PTHrP(102–106) cluster] that are used as a substrate for prohormone convertases when the precursor is in the secretory pathway, can also serve as nuclear targeting signals when the peptide is allowed to enter the cytoplasm. This phenomenon has not been described for any other peptide to date.
Acknowledgments
We thank Drs. Andrew Karaplis and Henry M. Kronenberg for the generous donation of the PTHrP gene-disrupted mouse, and Richard Powell for the primary bovine aortic smooth muscle cultures. We thank R. Glenn Hammonds for his generous donation of PTHrP(1–141). This research was supported by the American Physiological Society, the Department of Veterans Affairs, National Institutes of Health Grant DK 47168, and Institut National de la Santé et de la Recherche Médicale Grant CJF 9409 and Ministere de l’education Nationale, de la Enseigement Superieur et de la Recherche Grant EA 1314.
ABBREVIATIONS
- PTH
parathyroid hormone
- PTHrP
PTH-related protein
- h
human
- VSM
vascular smooth muscle
References
- 1.Stewart A F, Insogna K L, Broadus A E. In: Endocrinology. 3rd Ed. DeGroot L, editor. Philadelphia: Saunders; 1995. pp. 1061–1074. [Google Scholar]
- 2.Philbrick W M, Wysolmerski J J, Galbraith S, Holt E, Orloff J J, Yang K H, Vasavada R C, Weir E C, Broadus A E, Stewart A F. Physiol Rev. 1996;76:127–173. doi: 10.1152/physrev.1996.76.1.127. [DOI] [PubMed] [Google Scholar]
- 3.Yang K H, Stewart A F. In: Principles of Bone Biology. Bilezikian J P, Raisz L, Rodan G, editors. San Diego: Academic; 1996. pp. 347–376. [Google Scholar]
- 4.Karaplis A C, Luz A, Glowacki J, Bronson R T, Tybulewicz V L J, Kronenberg H M, Mulligan R C. Genes Dev. 1994;8:277–289. doi: 10.1101/gad.8.3.277. [DOI] [PubMed] [Google Scholar]
- 5.Deftos L J, Burton D W, Brandt D W. J Clin Invest. 1993;92:727–735. doi: 10.1172/JCI116643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Plawner L L, Philbrick W M, Burtis W J, Broadus A E, Stewart A F. J Biol Chem. 1995;270:14078–14084. doi: 10.1074/jbc.270.23.14078. [DOI] [PubMed] [Google Scholar]
- 7.Wu T, Vasavada R, Yang K, Massfelder T, Ganz M, Abbas S K, Care A D, Stewart A F. J Biol Chem. 1996;271:24371–24381. doi: 10.1074/jbc.271.40.24371. [DOI] [PubMed] [Google Scholar]
- 8.Segre G V. In: The Parathyroids. Bilezikian J P, Levine M A, Marcus R, editors. New York: Raven; 1994. pp. 213–229. [Google Scholar]
- 9.Orloff J J, Kats Y, Urena P, Schipani E, Vasavada R C, Philbrick W M, Behal A, Abou-Samra A-B, Segre G V, Jüppner H. Endocrinology. 1995;136:3016–3023. doi: 10.1210/endo.136.7.7789327. [DOI] [PubMed] [Google Scholar]
- 10.Orloff J J, Kats Y, Nathanson M H, Moyer S M, Mitnick M A, et al. Endocrinology. 1996;137:5376–5385. doi: 10.1210/endo.137.12.8940360. [DOI] [PubMed] [Google Scholar]
- 11.Valin A, Garcia-Ocana A, De Miguel F, Sarasa J L, Esbrit P. J Cell Physiol. 1997;170:209–215. doi: 10.1002/(SICI)1097-4652(199702)170:2<209::AID-JCP13>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 12.Cornish J, Callon K E, Nicholson G C, Reid I R. Endocrinology. 1997;138:1299–1304. doi: 10.1210/endo.138.3.4990. [DOI] [PubMed] [Google Scholar]
- 13.Kaiser S M, Laneuville P, Bernier S M, Rhim J S, Kremer R, et al. J Biol Chem. 1992;267:13623–13628. [PubMed] [Google Scholar]
- 14.Kaiser S M, Sebag M, Rhim J S, Kremer R, Goltzman D. Mol Endocrinol. 1994;8:139–147. doi: 10.1210/mend.8.2.8170470. [DOI] [PubMed] [Google Scholar]
- 15.Henderson J E, Amizuka N, Warshawsky H, Diasotto D, Lanske B M K, et al. Mol Cell Biol. 1995;15:4064–4075. doi: 10.1128/mcb.15.8.4064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dingwall C, Laskey R. Trends Biochem Sci. 1991;16:478–481. doi: 10.1016/0968-0004(91)90184-w. [DOI] [PubMed] [Google Scholar]
- 17.Massfelder T, Helwig J-J, Stewart A F. Endocrinology. 1996;137:3151–3153. doi: 10.1210/endo.137.8.8754732. [DOI] [PubMed] [Google Scholar]
- 18.Nakayama T, Ohtsuru A, Enomoto H, Namba H, Ozeki S, Shibata Y, Yokota T, Nobuyoshi M, Ito M, Sekine I, Yamashita S. Biochem Biophys Res Commun. 1994;200:1038–1045. doi: 10.1006/bbrc.1994.1553. [DOI] [PubMed] [Google Scholar]
- 19.Pirola C J, Wang H-M, Kamyar A, Wu S, Enomoto H, Sharifi B, Forrester J S, Clemens T L, Fagin J A. J Biol Chem. 1993;286:1987–1994. [PubMed] [Google Scholar]
- 20.Maeda S, Wu S, Juppner H, Green J, Aragay A M, Simon M I, Fagin J A, Clemens T L. Endocrinology. 1996;137:3154–3162. doi: 10.1210/endo.137.8.8754733. [DOI] [PubMed] [Google Scholar]
- 21.Jiang B, Morimoto S, Fukuo K, Yasuda O, Chen S, Ogihara T. Miner Electrolyte Metab. 1995;21:157–160. [PubMed] [Google Scholar]
- 22.Stewart A, Mangin M, Wu T, Goumas D, Insogna K L, Burtis W J, Broadus A E. J Clin Invest. 1988;81:596–600. doi: 10.1172/JCI113358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kimes B W, Brandt B L. Exp Cell Res. 1976;98:349–366. doi: 10.1016/0014-4827(76)90446-8. [DOI] [PubMed] [Google Scholar]
- 24.Yang K H, de Papp A E, Soifer N S, Wu T L, Porter S E, Bellantoni M, Burtis W J, Broadus A E, Philbrick W M, Stewart A F. Biochemistry. 1994;33:7460–7469. doi: 10.1021/bi00189a054. [DOI] [PubMed] [Google Scholar]
- 25.Burtis W J, Brady T G, Orloff J J, Ersbak J B, Warrell R P, Olson B R, Wu T L, Mitnick M A, Broadus A E, Stewart A F. N Engl J Med. 1990;322:1106–1112. doi: 10.1056/NEJM199004193221603. [DOI] [PubMed] [Google Scholar]
- 26.Wysolmerski J J, Broadus A E, Zhou J, Fuchs E, Milstone L M, Philbrick W M. Proc Natl Acad Sci USA. 1994;91:1133–1137. doi: 10.1073/pnas.91.3.1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wysolmerski J J, McCaughern-Carucci J F, Daifotis A G, Broadus A E, Philbrick W M. Development (Cambridge, UK) 1995;121:3539–3547. doi: 10.1242/dev.121.11.3539. [DOI] [PubMed] [Google Scholar]
- 28.Vasavada R, Cavaliere C, D’Ercole A J, Dann P, Burtis W J, Madlener A L, Zawalich K, Zawalich W, Philbrick W M, Stewart A F. J Biol Chem. 1996;271:1200–1208. doi: 10.1074/jbc.271.2.1200. [DOI] [PubMed] [Google Scholar]
- 29.Ozeki S, Ohtsuru A, Seto S, Takeshita S, Yano H, Nakayama T, Ito M, Yokota T, Nobuyoshi M, Segre G, Yamashita S, Yano K. Arterioscler Thromb Vasc Biol. 1996;16:565–575. doi: 10.1161/01.atv.16.4.565. [DOI] [PubMed] [Google Scholar]
- 30.Okano K, Pirola C, Wang H-M, Forrester J, Fagin J, Clemens T. Endocrinology. 1995;136:1782–1789. doi: 10.1210/endo.136.4.7895691. [DOI] [PubMed] [Google Scholar]
- 31.Hurley M, Florkiewicz R Z. In: Principles of Bone Biology. Raisz L R, Rodan G A, Bilezikian J P, editors. New York: Academic; 1996. pp. 627–745. [Google Scholar]
- 32.Kiefer P, Acland P, Pappin D, Peters G, Dickson C. EMBO J. 1994;13:4126–4136. doi: 10.1002/j.1460-2075.1994.tb06730.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kiefer P, Dickson C. Mol Cell Biol. 1995;15:4364–4374. doi: 10.1128/mcb.15.8.4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ditmer L S, Burton D W, Deftos L J. Endocrinology. 1996;137:1608–1617. doi: 10.1210/endo.137.5.8612492. [DOI] [PubMed] [Google Scholar]