


Abstract
A SELEX approach has been developed in order to select oligonucleotides that bind double-stranded DNA in the presence of a triplex-stabilizing agent, and was applied to a target sequence containing an oligopurine–oligopyrimidine stretch. After only seven rounds of selection, the process led to the identification of oligonucleotides that were able to form triple helices within the antiparallel motif. Inspection of the selected sequences revealed that, contrary to GC base pair which were always recognized by guanines, recognition of AT base pair could be achieved by either adenine or thymine, depending on the sequence context. While thymines are strongly preferred for several positions, some others can accommodate the presence of adenines. These results contribute to set the rules for designing oligonucleotides that form stable triple helices in the presence of triplex-stabilizing agents at physiological pH. They set the basis for further experiments regarding extension of potential target sequences for triple-helix formation or recognition of ligand–DNA complexes.
INTRODUCTION
Sequence-specific DNA-binding agents have been regarded as a great promise toward the development of new therapeutic strategies based on gene regulation or modification. Synthetic molecules that can recognize specific DNA sequences include triplex-forming oligonucleotides (TFOs), peptide nucleic acids and minor-groove binding agents. TFOs were developed in order to modulate the transcription of specific genes, in the so-called antigene strategy, but their affinity and specificity have been exploited for many other purposes, for example to target cleaving or cross-linking agents, transcription factors, or nucleases to a specific site [see refs (1,2) for review]. Moreover, they have been used as tools to induce DNA sequence modifications in live cells (3,4), as well as in various assays aimed at purifying or labelling DNA (5,6) or at investigating DNA–protein interactions (7).
Triple-helix formation is based on sequence-specific recognition of oligopyrimidine–oligopurine sequence by a third nucleic-acid strand. This third-strand binds in the major groove and recognizes the oligopurine strand by establishing a pair of hydrogen bonds with purine bases that remain involved in Watson–Crick base pairing. Different types of triple helices, which contain different types of base triplets, can be formed. The first discovered triple helices, which were formed with third strands containing only pyrimidines, relied on the formation of T.AxT and C.GxC+ base triplets. In this so-called pyrimidine or parallel motif, the third-strand binds in a parallel orientation with respect to the oligopurine strand. Purine-containing oligonucleotides can also bind double-helical DNA, thanks to the formation of C.GxG and T.AxA base triplets. In this case, the third-strand binds in an antiparallel orientation. Recognition of T.A base pairs can be achieved by adenines, but also by thymines. Therefore this recognition scheme has been named the ‘antiparallel’ motif. The stability of triple helices depends on the recognition scheme, on the sequences and on experimental conditions. Whereas the parallel motif is favoured by an acidic pH and a low GC content within the target sequence, the antiparallel motif can be formed at neutral pH but preferentially in the presence of divalent cations and on target sequences with a very high GC content. The design and experimental study of TFOs binding in the antiparallel motif can be complicated by the fact that purine-rich oligonucleotides can self-associate into G-quadruplex containing structures or GA duplexes that can compete with triple-helix formation (8,9). A systematic comparison of the use of A or T to recognize AT base pairs in the antiparallel motif has never been undertaken. Alternative binding patterns have been observed in some instances. For example, GT oligonucleotides have been reported to bind in a parallel orientation when the target sequence contains long runs of AT base pairs (10). The use of TFOs is also mostly limited to oligopyrimidine–oligopurine target sequences, although strategies have been proposed for the recognition of single or double inversions within the target or for recognizing alternated stretches of purines and pyrimidines (11). There is no convenient method for finding the best TFO for a given sequence. There are some instances where both types of TFO can bind the same DNA target, but this is not typical. TFO are generally chosen mostly on an empirical basis, using known rules for various triple helix motifs for the design followed by in vitro binding assays for validation (12).
Stability of triple helices formed by non-modified DNA oligonucleotides can be low at physiological pH and temperature. This stability can be enhanced by using chemically modified oligonucleotides [see ref. (2) for review]. Another very efficient way of stabilizing triple helices is by using triplex-specific ligands (13). The most efficient triplex-stabilizing agents are polyaromatic compounds which bind duplex and triplex DNA by intercalation [see ref. (14) for review]. Although there is no structural data regarding triplex-intercalator complexes, it has been demonstrated that increased stacking interactions obtained by additional cycles lead to more efficient triplex stabilizers (15,16). Most studies performed with such compounds have regarded the stabilization of triple helices with ‘canonical’ base triplets. Some of these intercalators have been shown to bind to both parallel (with TC TFOs) and antiparallel (with GT TFOs) triple helices (17). However, there is no reason to exclude that the presence of some of these compounds intercalated between DNA bases may alter the recognition code and enhance the formation of structures that do not involve canonical base triplets. In addition, it has been shown that triplex-specific intercalators can promote the formation of triple helices on target sequences that were not perfect oligopurine–oligopyrimidine stretches (18,19).
SELEX is an in vitro selection method for oligonucleotides (DNA or RNA) that bind very tightly to a chosen target. It is based on the iterative repetition of cycles that include a binding step, an elution step and an amplification by PCR. Selected oligonucleotide sequences are called aptamers. The process was first described in 1990 using proteins (20) and small molecules (21) as targets. Since then, numerous variants of the original process have been described, and the SELEX technology has been successfully applied for numerous applications on a large variety of targets (22–25), including whole cells and nucleic acids. For example, it has been demonstrated that DNA oligonucleotides selected against DNA secondary structures were able to recognize their target through base pair formation and additional interactions (26). SELEX experiments have also helped in the design of antisense oligonucleotides with the ability to bind specific structures (27–29).
Because of the limitations regarding the potential target sequences and as there is no systematic rule for designing the best TFO for a defined target sequence, the SELEX method represents an interesting tool for identifying nucleic acids sequences that bind double-stranded DNA. Two studies have tried to identify RNA molecules binding to double-stranded DNA using a SELEX approach (30,31). The selection process was carried out at acidic pH in the first study and at different pH values from 5.5 to 7 in the second one, using a target containing an oligopurine–oligopyrimidine sequence. The selected RNAs consisted mostly in uraciles and cytosines that were able to form canonical T.AxU and C.GxC+ base triplets.
In the present study, we have selected DNA oligonucleotides for binding to a DNA target containing an oligopurine.oligopyrimidine sequence with a low GC content (35%) under neutral pH in the presence of a triplex-stabilizing agent. These selections were performed in order to determine what would be the best TFO for such a sequence in the presence of a triplex-binding agent and to investigate if the presence of intercalators may modify the ‘code’ for triple-helix formation by enhancing the formation of non-canonical base triplets. Our idea was also to implement a new approach for identifying new recognition schemes and maybe extend the range of sequences that can be recognized by oligonucleotides in a sequence-specific manner.
MATERIALS AND METHODS
Nucleic acids and chemicals
All DNA oligonucleotides were purchased from Eurogentec (Seraing, Belgium). The DNA library used in our experiments consisted of a pool of oligonucleotides made of a continuous stretch of 40 randomized nucleotides flanked on both sides by fixed sequences which function as primer binding sites in the PCR. The sequence of this library was 5′-CGTACGGTCGACGCTAGC–N40–CACGTGGAGCTCGGATCC-3′. The following primers that anneal to the 5′- and 3′-end of the library fragments were used in the aptamer selection process: primer fw18F (5′-CGTACGGTCGACGCTAGC-3′) is connected at its 5′-end to a 6-carboxyfluorescein (6-FAM) moiety, while primer rv18P (5′-GGATCCGAGCTCCACGTG-3′) is phosphorylated on its 5′-end.
Plasmid pY11, which was constructed by insertion of synthetic oligonucleotides into the pBluescriptSK+ plasmid, has already been described (32). Two primers were used for the preparation of double-stranded DNA from those plasmids by PCR: fw120 (5′-CGAGGTCGACGGTATCGAT-3′) and rv120b (5′-CGCTCTAGAACTAGTGGATC-3′) which is biotinylated at its 5′-end. Polymerase chain reactions were carried out in a total volume of 50 µl of standard taq buffer (10 mM Tris–HCl, pH 8.3, 1.5 mM MgCl2, 50 mM KCl) containing 0.8 µM of each primer, 200 µM each of dNTPs and 2.5 U of Taq polymerase (NEB), using 1.6 ng/µl of plasmid as template. After 30 cycles of 30 s at 94°C, 30 s at 60°C and 1 min at 72°C, the final extension was carried out for 5 min at 72°C. Primers and unincorporated dNTP were removed using PCR purification kits (Qiagen). Plasmids pY11 and pBluescript SK+ led to fragments of 133 and 70 bp, respectively. The resulting sequences are explicited in Figure 1A. For gel shift experiments, the DNA fragments made with these plasmids were radiolabelled with 32P on the 5′ free-end according to standard procedures.
Figure 1.

(A) Sequences of the DNA fragments used for counterselection (top) and for selection (sequence above where the central EcoRI–HindIII sequence is replaced by the sequence below, the oligopurine–oligopyrimidine stretch is shown in darker blue). (B) Schematic representation of the selection protocol.
Two oligonucleotides were synthesized for control experiments. The first one, called GT (5′-CGTACGGTCGACGCTAGCTTATCTATTGTTTTGGTGGTTTGTGTTTTTTATCTATTCACGTGGAGCTCGGATCC-3′), is a 74-mer containing a GT sequence (underlined) that can form a 20 base triplets antiparallel triple helix with the target; the second one, called NS (5′-CGTACGGTCGACGCTAGCTTTTTTTT TTTTGATCCTCTA GAGTCGAC CTGCAGGCATGCTTTTT TTTTTTTCACGTG GAGCTCGGATCC-3′), was a 89-mer oligonucleotide that did not contain any sequence appropriate for triple-helix formation.
The triplex-stabilizing agent used in this study is a 6-[3-aminopropyl]amino-9-methoxy-13H-benzo[4,5]-indolo[3,2-c]quinoline derivative (BIQ) (see structure on Supplementary Figure S1) which synthesis has previously been described (16).
In vitro selection
Fresh aliquots of 6 × 106 streptavidin-coated magnetic beads (Dynabeads M-280 Streptavidin, Invitrogen) were washed twice with washing buffer (20 mM Tris–HCl, pH 7.2) before each selection round. The binding and washing procedures were facilitated by using a magnetic separation stand.
Hybridization of the oligonucleotide pool with the target double-stranded DNA (5 pmol, final concentration 100 nM) was performed in 50 µl binding buffer (washing buffer + 10 mM MgCl2 + 0.03% Triton X-100) in the presence of 6 µM of the BIQ compound. Triton X-100 was introduced in order to reduce non-specific adsorption of nucleic acids to the beads. We used 250 pmol of oligonucleotides from the pool for the first selection (final concentration 5 µM), then 25 pmol (final concentration 500 nM) for the successive rounds. Samples are heated to 80°C for 5 min, then cooled down to room temperature and incubated for another 5 min before they are applied onto the washed beads. After magnetic capture, beads are resuspended in 50 µl of washing buffer and incubated at room temperature for 30 min. Then the beads are captured again and elution was obtained by heating the beads resuspended in 50 µl of washing buffer at 95°C for 10 min, then placing the tube on the magnetic stand and removing the liquid phase as quickly as possible. The supernatant is either directly used for PCR amplification (rounds 1–3) or submitted to counterselection. In this case, the supernatant is incubated with 5 pmol of the 70 bp fragment in 50 µl binding buffer with 6 µM BIQ. The sample is heated to 80°C for 5 min, then incubated at room temperature for another 5 min, before it is applied onto new washed beads for 15 min at room temperature. After magnetic capture, the supernatant was recovered for further PCR amplification. In all these steps, incubation of beads solution is always performed by pipetting up and down every 5 min in order to avoid sedimentation of the beads.
Starting from round 2, it was possible to estimate the quantity of oligonucleotides that were recovered after selection by measuring the fluorescence of 6-FAM after release from the beads. This measurement was performed with a VictorII microplate reader (Wallac).
Mock experiments were conducted using control oligonucleotides in order to optimize the protocol. In those experiments, the GT oligonucleotide is mixed with a non-specific oligonucleotide (NS) in a 1:99 ratio. The sample is submitted to the selection protocol and the relative amounts of GT and NS are measured after oligonucleotide recovery and PCR amplification, thanks to their different sizes which made possible their separation by non denaturing gel electrophoresis (see Supplementary Figure S3).
Amplification and conversion of oligonucleotides into single-stranded DNA
The recovered oligonucleotide pools were amplified by PCR: the reaction was carried out in a total volume of 50 µl of standard taq buffer (10 mM Tris–HCl, pH 8.3, 1.5 mM MgCl2, 50 mM KCl) containing 0.8 µM of each primer (fw18F and rv18P), 200 µM each of dNTPs, and 2.5U of Taq polymerase (NEB). An approximate amount of 0.05 pmol of recovered oligonucleotides was amplified in four tubes. We performed 30 PCR cycles, consisting each of 30 s at 94°C, 30 s at 60°C and 60 s at 72°C. The quantity and quality of PCR products were estimated by agarose gel-electrophoresis, using the 6-FAM fluorescence of PCR amplicons. After elimination of the non-incorporated primers using the Montage centrifuge filter device (Millipore), the quantity of oligonucleotides was estimated by UV absorbance using a nanodrop apparatus (Thermo Scientific). Then, ∼1500 ng of the PCR products were converted into single-stranded by incubation in a total volume of 30 µl of λ exonuclease buffer with 10 U of λ exonuclease (NEB) for 30 min at 37°C, followed by 20 min at 65°C to inactivate the enzyme. Conversion into single-strand was checked by polyacrylamide gel electrophoresis under native conditions. The same protocol was used for the production of fluorescently labelled oligonucleotides corresponding to the random oligonucleotide pool and to the GT and NS oligonucleotides.
Cloning, sequencing and analysis of oligonucleotides binding double-stranded DNA
After several rounds of selection, the oligonucleotide pool was characterized by cloning individual sequences. The chosen pool of oligonucleotides was amplified using the fw29E (5′-CTAATCGAATTCGTACGGTCGACGCTAGC-3′) and the rv18P primers, in order to introduce restriction sites for the Sac I and EcoR I enzymes. The digested fragment was introduced between the corresponding sites in the pBluescript SK+ plasmid. Escherichia coli DH5a (Invitrogen) were transformed by up-taking this vector construct. Clones were randomly picked and sequenced with dye terminators, using the standard T7 primer (Cogenix, Grenoble). Very few sequences displayed insertions or deletions within the 40 nucleotides between the two sequences used for amplification. The sequences were manually analysed and aligned. Some oligonucleotides were selected several times being strictly identical, in which case the number of occurrences is indicated. In nearly all cases, the oligonucleotides share several motifs, and can therefore tentatively be grouped in three different families. The secondary-structure analysis of several aptamers was performed by means of the free-energy minimization algorithm using the internet tool mfold (33), with conditions set up at 0.01 M MgCl2 and 23°C.
Electrophoretic mobility shift assay (EMSA)
Binding of aptamers to the their target double-stranded DNA was investigated by EMSA. The radiolabelled DNA fragment (5 nM) was mixed with increasing concentrations of aptamers in a 50 mM HEPES pH 7.2 buffer containing 10 mM MgCl2, 5 µg tRNA competitor (Roche) and 6 µM of BIQ (total volume 10 µl). Samples were heated to 80°C for 5 min, then incubated at room temperature for 2 h. Then 2 µl of sucrose used as a loading buffer are added to the sample which is analysed in a native 10% (19:1 acrylamide/bisacrylamide) gel made in 50 mM HEPES and migrated for 1 h 30 min at a constant low power of 4 W. Gels were autoradiographed after drying using a Typhoon apparatus (GE Healthcare). A quantitative analysis of the relative amounts of radioactivity present in the different bands was obtained using the ImageQuant software. The percentage of triple helices, y, was determined for each gel lane by measuring the storage phosphor signal. Values of the apparent dissociation constant, Kd, for each oligonucleotides were obtained by least-squares fitting of the data to the binding isotherm: 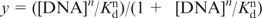 ) where [DNA] is the concentration of the DNA third strand, and n is the Hill coefficient.
) where [DNA] is the concentration of the DNA third strand, and n is the Hill coefficient.
RESULTS
Implementation of a selection process
Our aim was to design an experimental system that enabled the selection of oligonucleotides binding to any double-stranded DNA sequence of interest in the presence of triplex-binding agents. As a model, we chose a target sequence containing a 20 bp oligopurine–oligopyrimidine stretch of which a single copy is present on yeast chromosome XI (32). We chose as a triplex-binding agent a benzoindoloquinoline derivative (BIQ) containing five aromatic rings and an alkylamine side chain [see Supplementary Figure S1 and ref. (16)]. We prepared a biotinylated 133 bp target by PCR using a plasmid containing this sequence. In order to avoid the selection of artifactual sequences that would bind to the magnetic beads or to the ends of the double-stranded DNA, we devised a counterselection step involving a double-stranded DNA sequence that was made of both extremities of the sequence used for the selection. The fragment used for counterselection contained only plasmidic DNA sequences and was shorter (70 bp) than the one used for the selection (133 bp) (see Figure 1A). The basic steps of our process are presented in Figure 1B, and the scheme for counterselection is displayed in Supplementary Figure S2. The recovered oligonucleotides are PCR-amplified using 6-FAM-labelled forward and 5′-phosphorylated reverse primers, complementary to constant sequence segments of the oligonucleotide library. The presence of phosphorylated reverse primer allows lambda exonuclease-induced digestion of the PCR product, to produce single-stranded probe sequences capable of interacting with the target in the next round of hybridizations (34).
Various protocols were tested regarding oligonucleotide capture and elution. We chose to form the triplex in solution at neutral pH and in the presence of magnesium, then proceed to capture of the oligonucleotides by the beads, in order to avoid problems linked with the steric hindrance of the magnetic beads, and to introduce a washing step in the absence of magnesium before elution. In order to validate the selection protocol, we performed preliminary experiments using the GT oligonucleotide that was known to form a triple helix by binding to the 20 bp oligopurine.oligopyrimidine stretch present in the target in the presence of BIQ. Mock selection experiments were performed using as starting material a solution containing 1% of the 74-mer GT oligonucleotide and 99% of the 89-mer NS oligonucleotide, and measuring the composition of the sample after one selection cycle by visualization of the two amplified DNA fragments on native gel. The enrichment was measured by comparing the intensity of the bands (see Supplementary Figure S3). These experiments permitted the determination of the best experimental conditions that would ensure efficient capture of the TFOs and a minimal recovery of non specific oligonucleotides. They showed that including a short heating step (5 min at 80°C) before capture enhanced the enrichment, which may be due to a better hybridization of the binding oligonucleotides. This heating step does not result in denaturation of the long target under our experimental conditions. The influence of ligand concentration could also be investigated by these experiments. Enrichment rate was ∼80% in the presence of 6 µM of BIQ, and only 30% in the absence of the compound. Increasing the concentration of ligand led to lower enrichment rates. Experiments performed with a pyrimidine rich third strand (data not shown) led ∼20%.
The presence of the 6-FAM fluorophore facilitates the monitoring of selection steps and allows quantification of the single-stranded oligonucleotides that are released after each binding step (35). Using preparation of fluorescently labelled NS and GT control oligonucleotides, the amounts of recovered oligonucleotides were ∼0.1 pmol and between 0.65 and 0.8 pmol, respectively, which was sufficient to detect any significant improvement in the binding affinity of the oligonucleotide pools.
Selection of DNA-binding oligonucleotides in the presence of a triplex intercalator
Using the best conditions from the experiments described above, we decided to proceed further and initiate in vitro selection experiments to screen a 250 pmol oligonucleotide pool for its ability to bind to our chosen target sequence.
The detailed protocol is documented in the ‘Materials and methods’ section. Briefly, biotinylated target DNA was mixed with the synthetic random oligonucleotide mixture consisting of a 40-nt random sequence flanked by two 18-nt-long primer sequences. Samples were heated to 80°C for 5 min and then incubated at room temperature for 5 min. Following this hybridization, bound oligonucleotides were captured onto streptavidin-coated magnetic beads while unbound oligonucleotides were washed away. Bound oligonucleotides were eluted by resuspending the beads in 50 µl of washing buffer, heating the sample for 10 min at 95°C and quickly recovering the supernatant. Starting from round 4, a counterselection was included in order to eliminate oligonucleotide binding to the DNA sequences that are adjacent to the sequence of interest. The recovered pool of oligonucleotides was amplified by PCR and converted into single-stranded DNA in order to perform successive selection cycles.
The amount of fluorescently labelled oligonucleotides that was recovered after each binding step was quantified (Figure 2). It increased steadily from round 3 to 7, then remained stable or decreased slightly, which suggests that hybridization cycles enriched the mixture of oligonucleotides in sequences that were efficiently binding the target.
Figure 2.

Enrichment of aptamers during the selection. All probes were labelled with 6-FAM at their 5′ terminus to allow quantification of the recovered oligonucleotide. The bar graph shows the amount of oligonucleotide eluted from the target in each selection round. The quantity is expressed in pmol. Serial dilutions of a 6-FAM oligonucleotide were used to calibrate the quantification assay. Oligonucleotides from round 1 were not labelled and therefore could not be quantified. We prepared a small amount of labelled random oligonucleotides in order to quantitate their affinity for the target (marked here as « ran »).
Sequences of cloned DNAs
As no increase in affinity was observed after round 7, we decided to analyse the sequences. Thirty-eight individual members of the oligonucleotide pool from round 7 were cloned and sequenced for analysis (Table 1). These sequences clearly reveal three different families of oligonucleotide sequences. Alignment of sequences from family I showed the presence of sequences that could form a canonical triple helix within the antiparallel motif (Figure 3A). The target sequence was recognized by formation of C.GxG, T.AxT and T.AxA base-triplets. Only the base pairs located at both ends of this target sequence were not always recognized by the expected nucleotide (four sequences, i.e. 15% in each case). We did not observe very conserved nucleotides outside of the 20 nucleotide triplex forming region, although there may be some biases for the closest positions (Figure 3A). Sequences within family I were all different. Nevertheless, we noticed that several sequences shared the same triplex forming region. Figure 3B reports the triplex-forming regions corresponding to the 19 sequences that could form base triplets all along the target sequence. One sequence was present four times, two sequences were present three times and four sequences were present twice. We observed a very strong (100%) conservation of guanines in front of internal GC base pairs. Recognition of AT base pairs seems to be accommodated by both adenines and thymines, although there was a bias toward one or the other base depending on the position. Four positions (3, 11, 12, 18) were recognized exclusively by thymines, and we observed a very high frequency of thymines at four other positions (2, 4, 17, 19). Note that for the two sequences for which an adenine is observed at position 2, the terminal C.GxG base triplet is not present. On the contrary, adenine was found in 85% of the sequences for recognition of the AT base pair at position 15. Position 13 did not show any preference between A and T. The search for potential common secondary structures was undertaken using the free energy minimization method of the mfold program (33), to see if a common structural pattern would emerge. Structures were computed for all the sequences with or without the flanking primer-binding sites. We did not notice any recurrent folding pattern, and our analysis suggests that selected sequences do not fold into complex structures. Usually only very few base pairs could be formed, which are likely to be much less stable than the triple helical structures.
Table 1.
Alignment of the core regions of 38 aptamer sequences
 |
Figure 3.

Consensus sequence and binding affinity of individual aptamer sequences. (A) Frequency of each nucleobase in the third-strand aligned to form a triple helix with the target. The consensus motif is indicated. The nucleotides that are conserved in all selected sequences are underlined. (B) Sequences of the triplex-forming regions covering the entire target. The number of the corresponding clones are indicated. Adenines are displayed in red. (C) EMSA. Increased concentration of oligonucleotides were incubated in the absence (lanes 1, 7, 13) or in the presence of 20 nM (lanes 2, 8, 14), 50 nM (lanes 3, 9, 15), 100 nM (lanes 4, 10, 16), 200 nM (lanes 5, 11, 17) or 500 nM (lanes 6, 12, 18) of third strand. (D) Quantification of oligonucleotides binding affinity.
Sequences from family II contain a conserved 13 nucleotide region that was able to hybridize to one extremity of the double-stranded DNA target (Supplementary Figure S4). This was somewhat unexpected due to the use of a counterselection process. Interestingly, the complementary sequence is located on the biotinylated side of the double-stranded target, and not on its free end. One possible explanation for the presence of those sequences is that binding of the oligonucleotide precludes attachment of the target to the magnetic beads during the counterselection step.
We also observed two additional sequences (family III) that were containing pyrimidine stretches and therefore might be expected to form a triple helix according to the ‘pyrimidine’ motif. Alignment of these two sequences with respect to the target was possible in the parallel or antiparallel orientation, and involved in both cases two mispairings for clone 04 and three for clone 01 (see Supplementary Figure S5).
Dissociation constant of cloned DNA
Gel mobility shift assays were performed under conditions similar to those of selection at pH 7.2. Triple-helix formation resulted in a slower migration of the radiolabelled target. The fraction of triplex DNA was quantified and plotted as a function of total oligonucleotide concentration in order to generate binding curves. We decided to compare the affinity of clone 19, corresponding to the consensus sequence, to that of clone 20, which appeared to be the further from the consensus, and that of the GT control oligonucleotide (Figure 3C). Dissociation constants were generally in the few tens of nanomolar range (Figure 3D). The sequence that exhibited the highest affinity was clone 19 (43.9 nM). The dissociation constants were 70.9 nM for the GT control sequence and 79.7 nM for clone 20. We were not able to detect any binding in the absence of the triplex-binding agent.
We wondered if sequences that were located outside of the triple-helix motif provide a contribution to the binding affinity. We resynthesized several sequences and compared the affinity of the full-length selected molecule (76-mer) and the sequence derived from the random region (40-mer). We did not observe any difference in the binding affinities. The affinities of 20-mers corresponding to the triplex-forming regions were in the 20 nM range, with no significant difference between the various selected sequences (not shown). We were not able to detect any binding of the TC-containing sequences to the target.
DISCUSSION
In vitro selection experiments have been applied to the recognition of double-stranded nucleic acid targets by only two different groups (30,31). These studies did not result in the discovery of new binding motifs, but identified pyrimidine-rich RNA oligonucleotides that were able to bind their target upon formation of canonical base-triplets within the parallel motif. Two additional studies used a combinatorial approach in order to investigate the specificity of triple-helix formation. In the first one (36), van Dyke and coworkers have selected the best double-stranded DNA substrate for a GT-rich TFO. In the second one (37), both the third strand and the target were randomized, which led to the selection of triple helices containing mostly C.GxG base triplets. None of these studies were able to identify TFOs that could bind a given target double-stranded DNA with an antiparallel configuration. The absence of further combinatorial investigations for the recognition of double-stranded DNA is likely due to the experimental challenge of forming stable triple helical structures at neutral pH, except for sequences that are rich in GC base pairs.
In the present study, we have implemented an in vitro selection scheme for the identification of oligonucleotides that bind duplex DNA in the presence of a triplex intercalator. We chose as triplex intercalator the BIQ compound, which has been shown to increase the melting temperature of a pyrimidine triple-helix by up to 40°C (16), and had also been shown to stabilize triple helices with purine containing third strands in our laboratory. Selection rounds were performed at neutral pH using as a target sequence a DNA fragment containing a 20 bp oligopurine–oligopyrimidine stretch, with 65% AT and 35% GC. The selection process was very convenient as each selection round could be performed in less than a day, and introduction of a fluorescent moiety at the end of a primer enabled the quantification of recovered DNA at each step in a convenient way. After only seven rounds of selection, a strong affinity of the oligonucleotide pool for the target sequence was evident from the amount of oligonucleotide recovered after elution. Sequencing of the oligonucleotides revealed that most selected sequences were able to form antiparallel triple helices with the DNA target sequence through recognition of the oligopurine–oligopyrimidine stretch. Importantly, GC base pairs were always recognized by guanines, whereas recognition of AT base pairs seemed to depend on the sequence context. For four positions, thymines were found exclusively, while others seemed to accommodate both adenine and thymine. Depending on the sequence context, adenine or thymine were found more or less frequently. No clear rule can be established from the observation of this single target sequence. Nevertheless, some trends can be mentioned. Positions located on the 5′ side of a G in the third strand were more likely to accommodate an adenine than those located on the 3′ side. AT base pairs that were surrounded by only AT base pairs were more likely to be recognized by thymines. Only one AT base pair (position 15) seemed to prefer an adenine (85%). Preference for A or T may be related to the existence of distortions in the third strand at TpG and GpT steps within antiparallel triple helices with GT third strands (11), and/or to preferential binding sites for intercalation of BIQ. It is also possible that the selection process favours sequences that do not fold into competing structures. We were not able to explain the presence of adenine at specific positions by such a mechanism. We measured the dissociation constant for clone 20, which corresponded to the consensus sequence involving G, T and a single A in the triplex forming region. The affinity of this sequence for the target was slightly higher than that of clone 19, which has the highest number of adenines. This difference is probably not very significant, and may be explained by the effects of adjacent sequences. Experiments performed with 20-mer oligonucleotides did not show significant differences between the binding affinities of the various selected sequences. Although two pyrimidine rich oligonucleotides were present among the selected sequences, binding of these oligonucleotides could not be detected. The binding scheme and the reason for the presence of these sequences remains to be investigated.
Interesting questions are raised by the issue of our selections. One may wonder why only TFOs were selected. Given the tremendous variety of structures that are adopted by aptamers for the recognition of small molecules [see ref. (38) for review], one may expect the possibility for recognition of a DNA–intercalator complex by a selected oligonucleotide within a new structural framework. Aptamers may indeed potentially establish stacking interactions with planar aliphatic rings as well as electrostatic interactions with the side chains. It should be noted that aptamer can recognize chemical moieties as small as ethanolamine (39), which resembles the side chain of our triplex stabilizing agent. Despite the presence of this large intercalating compound in our selection scheme, no DNA binding solutions that differed from triple-helix formation were discovered. Several factors may have biased selection toward recovery of sequences that bound via triple-helix formation: the chosen DNA contained an oligopurine–oligopyrimidine stretch and therefore was typically suited to form conventional triple helices with an appropriate DNA, and conventional triple helix formation may afford the most efficient DNA binding solution. The size of the pool that we used (250 pmol, i.e. ∼1.5 × 1014 molecules) is sufficient to screen all possible sequences with a length of 22 nt (each sequence is present on average 10 times). All the selected sequences seemed to provide maximal recognition with the same length of 20 nucleotides, as there appears to be little nucleotide sequence conservation outside of the canonical recognition sequence. This suggests that additional interactions involving two additional nucleotides did not improve binding sufficiently to make a difference. This does not exclude that better solutions involving more than 23 nucleotides may exist and have been missed in our screening. One may also not exclude that some binding solutions other than the representatives of the triple helix may have been present in the oligonucleotide pool solution but were eliminated due to a lower affinity. Such other solutions would be recovered by using a target with a much shorter or no oligopurine–oligopyrimidine stretch at all.
One may also wonder why we did recover only TFOs within the antiparallel motif, contrary to previous studies. Our target sequence does not contain long stretches of GC base pairs, and the proportion of GC base pairs (35%), should in principle be favourable for the binding of pyrimidine rich TFOs. The issue of our selection may result from a much better stability of antiparallel TFOs in the presence of BIQ. It may also be due to the presence of magnesium, which favours the antiparallel motif. Other parameters, such as binding kinetics, may also be important in such experiments. The formation of pyrimidine motif triple helices is a slow process, which may have been disfavoured by our fast binding step. Further work will be necessary to evaluate how modifications of the selection protocol will affect the repertory of selected sequences. One should also keep in mind that, in addition to slightly acidic conditions, previous studies were aimed at selecting RNA oligonucleotides, which are known to be poorly compatible with the formation of antiparallel triple-helices (40,41).
Our SELEX process was performed in the presence of a low molecular weight compound expected to enhance interactions between the target and the oligonucleotide pool. This is an original strategy which to our knowledge has never been used before. The presence of the ligand resulted in selection of oligonucleotides that bound to the target only in the presence of the small molecule, although no counterselection was introduced to remove sequences that would bind in the absence of the compound. This is likely due to the very high stability of the complexes formed by the target, the ligand and the selected oligonucleotide. A great effort has been devoted to the expansion of the catalytic repertoire of nucleic acids by addition of protein like functionalities to the nucleic acids to be selected [see ref. (42) and references therein]. The number of modifications that can be incorporated into aptamers is still limited. The use of another functional group, in this case an intercalator, in order to provide these additional functionalities represents an interesting solution to this problem.
Although great progress have been made toward the design of new chemically modified oligonucleotides that permit recognition of a wider variety of sequences by TFOs (43,44), there is still no general solution that is easy to apply to any sequence of interest. Our SELEX approach provides an interesting solution for the design of TFOs for applications that can accommodate the presence of an intercalator. The same selection scheme could by applied to other target DNA sequences, with interesting potential applications. First, the repertoire of target sequences for triple-helix formation may be extended in the presence of DNA binding agents. It has already been demonstrated that addition of a triplex-stabilizing agent could enhance the formation of triple helices at sites containing inversions within the oligopurine–oligopyrimidine sequence (19,45) or at (GT)n tracts (46). Triplex-specific intercalators have also been shown to promote the formation of triple helices which switch from one strand to the other strand of DNA (10). In such cases, molecular modelling has suggested a code for designing the best switch oligonucleotide (47). Although this code has received some experimental validation (10,48), our strategy would provide the best oligonucleotide solution for this switch problem. It would also be interesting to perform selections with other DNA binding agents. Large molecules, such as echinomycin, may be more likely to be recognized by folded oligonucleotides. Work is in progress in order to explore all those possibilities.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
INSERM and CNRS. Doctoral fellowship from the French Ministry of Research and the University Pierre et Marie Curie (to E.A.). Funding for open access charge: INSERM.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Thomas Viel for his contribution to the initial selection experiments and Loïc Perrouault for technical help. They are grateful to Dr Chi Hung Nguyen for the gift of the BIQ compound.
REFERENCES
- 1.Thuong NT, Hélène C. Sequence-Specific Recognition and Modification of Double-Helical DNA by Oligonucleotides. Angew. Chem. Int. Ed. Engl. 1993;32:666–690. [Google Scholar]
- 2.Fox KR. Targeting DNA with triplexes. Curr. Med. Chem. 2000;7:17–37. doi: 10.2174/0929867003375506. [DOI] [PubMed] [Google Scholar]
- 3.Vasquez KM, Narayanan L, Glazer PM. Specific mutations induced by triplex-forming oligonucleotides in mice. Science. 2000;290:530–533. doi: 10.1126/science.290.5491.530. [DOI] [PubMed] [Google Scholar]
- 4.Simon P, Cannata F, Concordet JP, Giovannangeli C. Targeting DNA with triplex-forming oligonucleotides to modify gene sequence. Biochimie. 2008;90:1109–1116. doi: 10.1016/j.biochi.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 5.Potaman VN. Applications of triple-stranded nucleic acid structures to DNA purification, detection and analysis. Expert. Rev. Mol. Diagn. 2003;3:481–496. doi: 10.1586/14737159.3.4.481. [DOI] [PubMed] [Google Scholar]
- 6.Ghosh I, Stains CI, Ooi AT, Segal DJ. Direct detection of double-stranded DNA: Molecular methods and applications for DNA diagnostics. Mol. Biosyst. 2006;2:551–560. doi: 10.1039/b611169f. [DOI] [PubMed] [Google Scholar]
- 7.Firman K, Szczelkun MD. Measuring motion on DNA by the type I restriction endonuclease EcoR124I using triplex displacement. Embo J. 2000;19:2094–2102. doi: 10.1093/emboj/19.9.2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Noonberg SB, Francois JC, Garestier T, Hélène C. Effect of competing self-structure on triplex formation with purine-rich oligodeoxynucleotides containing GA repeats. Nucleic Acids Res. 1995;23:1956–1963. doi: 10.1093/nar/23.11.1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olivas WM, Maher LJ., 3rd Competitive triplex/quadruplex equilibria involving guanine-rich oligonucleotides. Biochemistry. 1995;34:278–284. doi: 10.1021/bi00001a034. [DOI] [PubMed] [Google Scholar]
- 10.de Bizemont T, Sun JS, Garestier T, Hélène C. New junction models for alternate-strand triple-helix formation. Chem. Biol. 1998;5:755–762. doi: 10.1016/s1074-5521(98)90667-6. [DOI] [PubMed] [Google Scholar]
- 11.Sun JS, Hélène C. Oligonucleotide directed triple helix formation. Curr. Opin. Struct. Biol. 1993;3:345–356. doi: 10.1016/s0959-440x(96)80051-0. [DOI] [PubMed] [Google Scholar]
- 12.Vekhoff P, Ceccaldi A, Polverari D, Pylouster J, Pisano C, Arimondo PB. Triplex formation on DNA targets: how to choose the oligonucleotide. Biochemistry. 2008;47:12277–12289. doi: 10.1021/bi801087g. [DOI] [PubMed] [Google Scholar]
- 13.Mergny JL, Duval-Valentin G, Nguyen CH, Perrouault L, Faucon B, Rougée M, Montenay-Garestier T, Bisagni E, Hélène C. Triple helix-specific ligands. Science. 1992;256:1681–1684. doi: 10.1126/science.256.5064.1681. [DOI] [PubMed] [Google Scholar]
- 14.Escudé C, Garestier T. In: Triple Helix Forming Oligonucleotides. Malvy C, Harel-Bellan A, Pritchard LL, editors. Kluwer Academic; 1999. pp. 257–271. [Google Scholar]
- 15.Escudé C, Nguyen C, Kukreti S, Janin Y, Sun J, Bisagni E, Garestier T, Hélène C. Rational design of a triple helix-specific intercalating ligand. Proc. Natl Acad. Sci. USA. 1998;95:3591–3596. doi: 10.1073/pnas.95.7.3591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nguyen CH, Marchand C, Delage S, Sun JS, Garestier T, Hélène C, Bisagni E. Synthesis of 13H-Benzo[6,7] and 13H-Benzo[4,5]-Indolo[3,2-c]quinolines: a new series of potent specific ligands for triplex DNA. J. Am. Chem. Soc. 1998;120:2501–2507. [Google Scholar]
- 17.Escudé C, Sun JS, Nguyen CH, Bisagni E, Garestier T, Hélène C. Ligand-induced formation of triple helices with antiparallel third strands containing G and T. Biochemistry. 1996;35:5735–5740. doi: 10.1021/bi960120c. [DOI] [PubMed] [Google Scholar]
- 18.de Bizemont T, Duval-Valentin G, Sun JS, Bisagni E, Garestier T, Hélène C. Alternate strand recognition of double-helical DNA by (T,G)-containing oligonucleotides in the presence of a triple helix-specific ligand. Nucleic Acids Res. 1996;24:1136–1143. doi: 10.1093/nar/24.6.1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kukreti S, Sun J, Loakes D, Brown D, Nguyen C, Bisagni E, Garestier T, Hélène C. Triple helices formed at oligopyrimidine*oligopurine sequences with base pair inversions: effect of a triplex-specific ligand on stability and selectivity. Nucleic Acids Res. 1998;26:2179–2184. doi: 10.1093/nar/26.9.2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- 21.Ellington AD, Szostak JW. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- 22.Famulok M, Mayer G, Blind M. Nucleic acid aptamers-from selection in vitro to applications in vivo. Acc. Chem. Res. 2000;33:591–599. doi: 10.1021/ar960167q. [DOI] [PubMed] [Google Scholar]
- 23.Perrin DM. Nucleic acids for recognition and catalysis: landmarks, limitations, and looking to the future. Comb. Chem. High. Throughput Screen. 2000;3:243–269. doi: 10.2174/1386207003331599. [DOI] [PubMed] [Google Scholar]
- 24.Joyce GF. Forty years of in vitro evolution. Angew. Chem. Int. Ed. Engl. 2007;46:6420–6436. doi: 10.1002/anie.200701369. [DOI] [PubMed] [Google Scholar]
- 25.Stoltenburg R, Reinemann C, Strehlitz B. SELEX—a (r)evolutionary method to generate high-affinity nucleic acid ligands. Biomol. Eng. 2007;24:381–403. doi: 10.1016/j.bioeng.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 26.Mishra RK, Le Tinevez R, Toulmé JJ. Targeting nucleic acid secondary structures by antisense oligonucleotides designed through in vitro selection. Proc. Natl Acad. Sci. USA. 1996;93:10679–10684. doi: 10.1073/pnas.93.20.10679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scarabino D, Crisari A, Lorenzini S, Williams K, Tocchini-Valentini GP. tRNA prefers to kiss. Embo J. 1999;18:4571–4578. doi: 10.1093/emboj/18.16.4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Toulmé JJ, Darfeuille F, Kolb G, Chabas S, Staedel C. Modulating viral gene expression by aptamers to RNA structures. Biol. Cell. 2003;95:229–238. doi: 10.1016/s0248-4900(03)00036-4. [DOI] [PubMed] [Google Scholar]
- 29.Kikuchi K, Umehara T, Fukuda K, Kuno A, Hasegawa T, Nishikawa S. A hepatitis C virus (HCV) internal ribosome entry site (IRES) domain III-IV-targeted aptamer inhibits translation by binding to an apical loop of domain IIId. Nucleic Acids Res. 2005;33:683–692. doi: 10.1093/nar/gki215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pei DH, Ulrich HD, Schultz PG. A combinatorial approach toward DNA recognition. Science. 1991;253:1408–1411. doi: 10.1126/science.1716784. [DOI] [PubMed] [Google Scholar]
- 31.Soukup GA, Ellington AD, Maher LJ., 3rd Selection of RNAs that bind to duplex DNA at neutral pH. J. Mol. Biol. 1996;259:216–228. doi: 10.1006/jmbi.1996.0314. [DOI] [PubMed] [Google Scholar]
- 32.Géron-Landre B, Roulon T, Escudé C. Stem-loop oligonucleotides as tools for labelling double-stranded DNA. FEBS J. 2005;272:5343–5352. doi: 10.1111/j.1742-4658.2005.04932.x. [DOI] [PubMed] [Google Scholar]
- 33.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brukner I, El-Ramahi R, Gorska-Flipot I, Krajinovic M, Labuda D. An in vitro selection scheme for oligonucleotide probes to discriminate between closely related DNA sequences. Nucleic Acids Res. 2007;35:e66. doi: 10.1093/nar/gkm156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stoltenburg R, Reinemann C, Strehlitz B. FluMag-SELEX as an advantageous method for DNA aptamer selection. Anal. Bioanal. Chem. 2005;383:83–91. doi: 10.1007/s00216-005-3388-9. [DOI] [PubMed] [Google Scholar]
- 36.Hardenbol P, Van Dyke MW. Sequence specificity of triplex DNA formation: analysis by a combinatorial approach, restriction endonuclease protection selection and amplification. Proc. Natl Acad. Sci. USA. 1996;93:2811–2816. doi: 10.1073/pnas.93.7.2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Debin A, Laboulais C, Ouali M, Malvy C, Le Bret M, Svinarchuk F. Stability of G,A triple helices. Nucleic Acids Res. 1999;27:2699–2707. doi: 10.1093/nar/27.13.2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hermann T, Patel DJ. Adaptive recognition by nucleic acid aptamers. Science. 2000;287:820–825. doi: 10.1126/science.287.5454.820. [DOI] [PubMed] [Google Scholar]
- 39.Mann D, Reinemann C, Stoltenburg R, Strehlitz B. In vitro selection of DNA aptamers binding ethanolamine. Biochem. Biophys. Res. Commun. 2005;338:1928–1934. doi: 10.1016/j.bbrc.2005.10.172. [DOI] [PubMed] [Google Scholar]
- 40.Escudé C, Francois JC, Sun JS, Ott G, Sprinzl M, Garestier T, Hélène C. Stability of triple helices containing RNA and DNA strands: experimental and molecular modeling studies. Nucleic Acids Res. 1993;21:5547–5553. doi: 10.1093/nar/21.24.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Semerad CL, Maher LJ., 3rd Exclusion of RNA strands from a purine motif triple helix. Nucleic Acids Res. 1994;22:5321–5325. doi: 10.1093/nar/22.24.5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hollenstein M, Hipolito CJ, Lam CH, Perrin DM. A self-cleaving DNA enzyme modified with amines, guanidines and imidazoles operates independently of divalent metal cations (M2+) Nucleic Acids Res. 2009;37:1638–1649. doi: 10.1093/nar/gkn1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Buchini S, Leumann CJ. Stable and selective recognition of three base pairs in the parallel triple-helical DNA binding motif. Angew. Chem. Int. Ed. Engl. 2004;43:3925–3928. doi: 10.1002/anie.200460159. [DOI] [PubMed] [Google Scholar]
- 44.Rusling DA, Powers VE, Ranasinghe RT, Wang Y, Osborne SD, Brown T, Fox KR. Four base recognition by triplex-forming oligonucleotides at physiological pH. Nucleic Acids Res. 2005;33:3025–3032. doi: 10.1093/nar/gki625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gowers DM, Fox KR. DNA triple helix formation at oligopurine sites containing multiple contiguous pyrimidines. Nucleic Acids Res. 1997;25:3787–3794. doi: 10.1093/nar/25.19.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gowers DM, Fox KR. Triple helix formation at (AT)n adjacent to an oligopurine tract. Nucleic Acids Res. 1998;26:3626–3633. doi: 10.1093/nar/26.16.3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sun JS. Rational Design of Switched Triple Helix-Forming Oligonucleotides: Extension of Sequences for Triple Helix Formation. In: Pullman A, Jortner J, editors. Modelling of Biomolecular Structures and Mechanisms. 1995. pp. 267–288. [Google Scholar]
- 48.Brodin P, Sun JS, Mouscadet JF, Auclair C. Optimization of alternate-strand triple helix formation at the 5′-TpA-3′ and 5′-ApT-3′ junctions. Nucleic Acids Res. 1999;27:3029–3034. doi: 10.1093/nar/27.15.3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.