

Abstract
Quantum mechanical calculations have been used to investigate how the incorporation of an amino group to the Cβ- or Cγ-positions of the pyrrolidine ring affects the intrinsic conformational properties of the proline. Specifically, a conformational study of the N-acetyl-N′-methylamide derivatives of four isomers of aminoproline, which differ not only in the β- or γ-position of the substituent but also in its cis or trans relative disposition, has been performed. In order to further understand the role of the intramolecular hydrogen bonds between the backbone carbonyl groups and the amino side group, a conformational study was also performed on the corresponding four analogues of dimethylaminoproline. In addition, the effects of solvation on aminoproline and dimethylaminoproline dipeptides have been evaluated using a Self Consistent Reaction Field model, and considering four different solvents (carbon tetrachloride, chloroform, methanol and water). Results indicate that the incorporation of the amino substituent into the pyrrolidine ring affects the conformational properties, with backbone⋯side chain intramolecular hydrogen bonds detected when it is incorporated in a cis relative disposition. In general, the incorporation of the amino side group tends to stabilize those structures where the peptide bond involving the pyrrolidine nitrogen is arranged in cis. The aminoproline isomer with the substituent attached to the Cγ-position with a cis relative disposition is the most stable in the gas-phase and in chloroform, methanol and water solutions. Replacement of the amino side group by the dimethylamino substituent produces significant changes in the potential energy surfaces of the four investigated dimethylaminoproline-containing dipeptides. Thus, these changes affect not only the number of minima, which increases considerably, but also the backbone and pseudorotational preferences. In spite of these effects, comparison of the conformational preferences, i.e. the more favored conformers, calculated for different isomers of aminoproline and dimethylaminoproline dipeptides showed a high degree of consistency for the two families of compounds.
Introduction
Proline (Pro) is unique among naturally occurring amino acids in that its side chain is bonded to both the α-carbon and its preceding amide nitrogen. As a consequence, rotation about the N—Cα bond is prohibited and the φ torsion angle is confined to values around −60°. Accordingly, Pro is overwhelmingly found in the α-helical (φ,ψ≈ −60°,−30°) and semi-extended (φ,ψ≈ −60°,140°) regions of the conformational map.1 In addition, Pro shows a higher propensity to promote γ-turn conformations (φ,ψ≈ −70°,60°) than other proteogenic amino acids.1d,2 Another effect derived from its cyclic structure is that the peptide bond preceding Pro (that involving the pyrrolidine nitrogen) has a relatively high probability of accommodating a cis arrangement3 as compared to other peptide bonds, for which the cis form is almost inexistent. Recent studies in Pro dipeptides observed that the cis/trans isomerization is an enthalpy driven process that depends on the polarity of the environment.4 Thus, although the electronic effects that stabilize the cis form are enhanced in polar environments, the cis/trans rotational barriers increase with the polarity of the environment. These structural features play a fundamental role in directing the secondary structure of proteins,5 inducing special motifs like reverse turns and bends.6 Furthermore, the cis-trans isomerization of Pro has been speculated to play a role not only in important biological processes7 but also in the rate determining steps for folding and refolding of some proteins.8
4R-Hydroxyproline (Hyp) is a hydroxylated derivative of Pro that shares the same features as its parent amino acid. It is formed by a post-translational modification where a Pro residue is converted to Hyp by an enzyme with a ferrous ion at its active site, called prolyl hydroxylase. Both Hyp and Pro, along with glycine, are found in collagen, the most abundant protein in vertebrates. As a consequence of their importance, the intrinsic conformational preferences of Pro3a,9-12 and Hyp13 have been examined in detail on the corresponding dipeptide analogues using advanced theoretical methods. Interestingly, in spite of the capabilities of the hydroxyl side group to form intramolecular hydrogen bonds able to induce significant structural distortions, the minimum energy conformations found for the N-acetyl-N′-methylamide derivatives of Pro and Hyp (Ac-Pro-NHMe and Ac-Hyp-NHMe dipeptides, respectively) were very similar. Specifically, a strong correlation was observed between the optimized dihedral angles of these dipeptides. Indeed, the largest effect produced by hydroxylation of Pro was detected in the puckering of the pyrrolidine ring. Thus, the down puckering is preferred for Ac-Pro-NHMe, while the up puckering with the hydroxyl group occupying an equatorial position is the most stable for Ac-Hyp-NHMe.
In recent years we have been involved in a broad project devoted to the design and application of synthetic amino acids with restricted conformational mobility in different fields of nanobiology. Non-proteogenic amino acids have been found to be very useful for the re-engineering of physical protein modules and the generation of nanodevices.14 More specifically, we observed that insertion of chemically constrained residues with suitable backbone conformational tendencies enhance the thermodynamic stability of the nanotubular structures constructed by self-assembling protein fragments with a β-helical conformation.15 We have further selectively incorporated synthetic amino acids to impart resistance against proteases not only at the mutated position but also at neighboring amino acids.16 In this work, we investigate the intrinsic conformational preferences of different aminated derivates of Pro. These non-proteogenic amino acids, which have been already used to construct β-peptides with helical secondary structures,17 are expected to be of potential interest in many nanobiological applications. This is because the topological characteristics of the amino and hydroxyl groups are different and, therefore, intramolecular hydrogen bonds in aminoproline (Amp) derivatives are expected to alter significantly the structural properties of Pro.
Theoretical calculations based on Density Functional Theory (DFT) methods have been used to investigate the conformational properties of the N-acetyl-N′-methylamide derivatives of Amp that incorporate an amino group to the Cβ- or Cγ-positions of the pyrrolidine ring, both the cis and trans isomers being considered in each case. Accordingly, calculations were performed on the four compounds displayed in Scheme 1: Ac-βtAmp-NHMe, Ac-βcAmp-NHMe, Ac-γtAmp-NHMe and Ac-γcAmp-NHMe.
Scheme 1.

In order to provide a better understanding of the crucial role of intramolecular hydrogen bonds, the study has been further extended to the four dipeptides constructed by replacing the Amp residue by the corresponding dimethylaminoproline (Dmp) analogue: Ac-βtDmp-NHMe, Ac-βcDmp-NHMe, Ac-γtDmp-NHMe and Ac-γcDmp-NHMe in Scheme 1. In addition we have examined how the incorporation of amino and dimethylamino substituents at the β and γ positions of Pro affects the trans/cis disposition of the peptide bond involving the pyrrolidine nitrogen. Finally, the influence of the environment, in particular of the solvent, on the conformational preferences of the different Amp- and Dmp-containing dipeptides has been evaluated using a Self Consistent Reaction Field (SCRF) method. Results have been compared with those recently reported for Ac-Pro-NHMe,12b which were calculated using the same theoretical procedures.
Methods
All calculations were carried out using the Gaussian 03 computer program.18 DFT calculations were performed using the following combination: the Becke's three-parameter hybrid functional (B3)19 with the local functional developed by Lee, Yang and Parr (LYP),20 which is gradient corrected. Thus, all the calculations presented in this work were performed using the B3LYP method combined with the 6-31+G(d,p) basis set.21 This computational procedure provided a very satisfactory description of the conformational properties of cyclic constrained amino acids, including Pro and its dehydro- and α-substituted derivatives.22,12
The backbone (ω0,φ,ψ,ω) and side chain (χi; endocyclic) dihedral angles of the N-acetyl-N′-methylamide derivatives of conventional Pro, Amp and Dmp are defined in Figure 1. Since φ is fixed by the geometry of the five-membered ring, only three minima may be anticipated for the potential energy surfaces E=E(ψ) of the dipeptides for a given arrangement of the peptide bonds. Thus, the flexible angle ψ is expected to have three minima, i.e. gauche+ (60°), trans (180°) and gauche− (−60°), while each amide bond (ω0, ω) can be arranged in trans or cis. It should be noted that only the peptide bond involving the pyrrolidine nitrogen, which corresponds to the ω0 torsion angle, is likely to adopt a cis configuration. Therefore, both the trans and cis states were considered for ω0, while the amide bond involving the N-methylamide blocking group (given by ω) was arranged in trans only. The cyclic side chains of the compounds under study may adopt two main different conformational states that correspond to the down and up puckering of the five-membered ring. They are defined as those in which the Cγ atom and the carbonyl group of the Pro residue (or analogue) lie on the same and opposite sides, respectively, of the plane defined by the Cδ, N and Cα atoms.
Figure 1.

Dihedral angles used to identify the conformations of the N-acetyl-N′-methylamide derivatives of the Amp and Dmp analogues studied in this work. The dihedral angles ω0, φ, ψ and ω are defined using backbone atoms while the endocyclic dihedral angles χi are given by the atoms of the five-membered ring. In particular, the sequence of atoms used to define φ and χ0 are C(=O)–N–Cα–C(=O) and Cδ–N–Cα–Cβ, respectively.
Accordingly, for each of the eight dipeptides under study (Scheme 1), 3(ψ backbone) × 2(ω0trans-or-cis) × 2(cyclic side chain) = 12 structures were considered as starting points for complete geometry optimizations at the B3LYP/6-31+G(d,p) level. Frequency analyses were carried out to verify the nature of the minimum state of all the stationary points obtained and to calculate the zero-point vibrational energies (ZPVE) as well as both thermal and entropic corrections, these statistical terms being used to compute the conformational Gibbs free energies in the gas phase (ΔGgp) at 298 K.
To obtain an estimation of the solvation effects on the relative stability of the different minima, single point calculations were conducted on the B3LYP/6-31+G(d,p) optimized structures using a Self-Consistent Reaction Field (SCRF) model. SCRF methods treat the solute at the quantum mechanical level, while the solvent is represented as a dielectric continuum. Specifically, the Polarizable Continuum Model (PCM) developed by Tomasi and co-workers was used to describe the bulk solvent.23 This method involves the generation of a solvent cavity from spheres centered at each atom in the molecule and the calculation of virtual point charges on the cavity surface representing the polarization of the solvent. The magnitude of these charges is proportional to the derivative of the solute electrostatic potential at each point calculated from the molecular wave function. The point charges may, then, be included in the one-electron Hamiltonian, thus inducing polarization of the solute. An iterative calculation is carried out until the wave function and the surface charges are self-consistent. PCM calculations were performed using the standard protocol implemented in Gaussian 0318 and considering the dielectric constants of carbon tetrachloride (ε = 2.228), chloroform (ε = 4.9), methanol (ε= 32.6) and water (ε= 78.4). The conformational free energies in solution (ΔG#sol#, where #sol# refers to the solvent) were computed using the classical thermodynamics scheme, i.e. the free energies of solvation provided by the PCM model were added to the ΔGgp values.
Nomenclature and Pseudorotational Parameters
The minimum energy conformations of the eight dipeptides studied in this work have been denoted using a three-label code that specifies the arrangement of the ω0 peptide bond, the (φ,ψ) backbone conformation and the puckering of the five-membered ring. The first letter refers to the trans (t) or cis (c) arrangement of the peptide bond involving the pyrrolidine nitrogen. The second label identifies the backbone conformation using the nomenclature introduced by Perczel et al.24 more than fifteen years ago. Accordingly, nine different backbone conformations can be distinguished in the potential energy surface E=E(φ,ψ) of amino acids: γD, δD, αD, εD, βL, εL, αL, δL and γL. In the case of Pro, only the γL (γ-turn or C7), αL (α-helical), and εL (polyproline II-like) conformations are accessible due to the fact that φ is fixed in the neighborhood of −60°. Next, the up or down puckering of the five-membered ring is indicated using the [u] and [d] labels, respectively. In particular, the down ring puckering was identified when χ1 and χ3 were positive while χ2 and χ4 were negative. Conversely, the up ring puckering is characterized by negative values of χ1 and χ3 and positive values of χ2 and χ4.
The puckering of the five-membered ring was described using the classical pseudorotational algorithm, which uses a very simple model based on only two parameters, as previously applied to proline by Perczel et al.25 The pseudorotational parameters A and P, which describe the puckering amplitude and the state of the pucker in the pseudorotation pathway, respectively, are derived from the endocyclic dihedral angles as follows:
and
Accordingly, parameter A is defined to be positive while P falls between −180° and 180°.
Results and Discussion
Aminoproline (Amp) dipeptides
This section reports the results obtained at the B3LYP/6-31+G(d,p) level for the four Amp-containing dipeptides displayed in Scheme 1, which have been compared to the dipeptide of conventional Pro that was recently reported at the same level of theory.12b Table 1 lists the more relevant structural parameters together with the relative energy (ΔEgp) for the 4, 6, 3 and 7 minimum energy conformations characterized for Ac-βtAmp-NHMe, Ac-βcAmp-NHMe, Ac-γtAmp-NHMe and Ac-γcAmp-NHMe, respectively, selected minima being displayed in Figures 2 and 3. The relative stability of the four dipeptides is indicated in Table 1 through ΔE#gp#, which corresponds to the energy relative to the lowest energy conformation of the most stable isomer. Table 2 compares the relative free energies in the gas-phase (ΔGgp), carbon tetrachloride (ΔGCCl4), chloroform (ΔGCHCl3), methanol (ΔGMeOH) and water (ΔGH2O) solutions for the minima of the four dipeptides mentioned above. Calculations in solution were performed by applying the PCM method to the geometries optimized in the gas phase. Thus, previous studies on simple organic and bio-organic compounds indicated that solute geometry relaxations in solution and single point calculations on the optimized geometries in the gas phase give almost identical free energies of salvation,26 even although nuclear relaxation in solute has been found to be essential in some specific cases.27 Finally, Table 3 compares the relative stability of the four Amp-containing dipeptides by showing the free energies in the different environments calculated in each case with respect to the conformation of lowest free energy of the most stable isomer: ΔG#gp#, ΔG#CCl4#, ΔG#CHCl3#, ΔG#MeOH# and ΔG#H2O#.
Table 1.
Backbone dihedral angles (in degrees), pseudorotational parameters (A and P; in degrees), relative energy (ΔEgp; in kcal/mol) and relative energy with respect to the lowest energy conformation of the most stable dipeptide (ΔE#gp#; in kcal/mol) of the minimum energy conformations characterized for Ac-βtAmp-NHMe, Ac-βcAmp-NHMe, Ac-γtAmp-NHMe and Ac-γcAmp-NHMe at the B3LYP/6-31+G(d,p) level in the gas phase.
| # Conf. | ω0 | φ | ψ | ω | (A, P) | ΔEgp | ΔE#gp# |
|---|---|---|---|---|---|---|---|
| Ac-βtAmp-NHMe | |||||||
| t-γL[d] | -174.0 | -83.1 | 71.9 | -177.6 | (38.4, -117.2)a | 0.0b | 1.4 |
| c-εL[u] | 1.2 | -66.7 | 178.8 | 175.9 | (38.2, 92.2)c | 2.8 | 4.2 |
| c-αL[u] | 6.8 | -69.6 | -35.1 | 179.4 | (37.8, 82.2)d | 5.9 | 7.3 |
| c-εL[d] | -0.6 | -77.5 | 145.8 | 176.0 | (37.5, 75.8)e | 5.9 | 7.3 |
| Ac-βcAmp-NHMe | |||||||
| t-γL[d] | -174.1 | -83.6 | 78.6 | -175.5 | (39.8,118.8)f | 0.0g | 0.5 |
| t-αL[d] | -170.5 | -88.7 | -7.5 | 173.9 | (39.2, -112.5)h | 4.0 | 4.5 |
| c-αL[d] | 10.4 | -84.3 | -17.4 | -177.2 | (36.2, 104.0)i | 4.3 | 4.8 |
| c-εL[d] | -3.0 | -74.5 | 172.9 | 177.8 | (39.3, -126)j | 5.0 | 5.5 |
| c-αL[u] | 8.2 | -96.2 | -0.7 | 178.6 | (42.8, -122.2)k | 5.6 | 6.1 |
| t-αL[u] | -170.8 | -70.5 | -19.3 | 175.2 | (40.2, 78.1)l | 8.6 | 9.1 |
| Ac-γtAmp-NHMe | |||||||
| t-γL[u] | -170.8 | -83.7 | 75.2 | -176.6 | (37.3, 102.6)m | 0.0n | 1.3 |
| c-αL[d] | 10.5 | -91.6 | -3.9 | -179.9 | (37.8, -112.5)o | 2.8 | 4.1 |
| c-εL[u] | -0.8 | -62.5 | 147.7 | 175.7 | (39.3, 93.1)p | 5.9 | 7.2 |
| Ac-γcAmp-NHMe | |||||||
| t-γL[d] | -172.8 | -82.9 | 76.6 | -176.0 | (32.73,-116.5)q | 0.0r | 0.0 |
| t-γL[u] | -174.5 | -81.8 | 77.4 | -176.2 | (37.8, 109.3)s | 1.5 | 1.5 |
| c-αL[d] | 8.5 | -68.5 | -47.6 | -176.4 | (34.1, -93.2)t | 2.7 | 2.7 |
| c-αL[u] | 8.0 | -79.0 | -18.9 | -177.6 | (37.2, 89.4)u | 5.2 | 5.2 |
| t-αL[u] | -170.9 | -79.3 | -9.2 | 175.5 | (37.9, 93)v | 5.7 | 5.7 |
| c-εL[d] | 3.8 | -70.8 | 148.6 | 174.6 | (35.4, -104.1)w | 6.0 | 6.0 |
| c-εL[u] | 0.9 | -62.5 | 148.2 | 176.9 | (39, 89.3)x | 7.9 | 7.9 |
χ0= -17.5°, χ1= 33.5°, χ2= -38.0°, χ3= 27.4° and χ4= -6.0°.
E= -628.667446 a.u.
χ0= -1.4°, χ1= -21.6°, χ2= 35.7°, χ3= -36.2° and χ4= 23.9°.
χ0= 5.1°, χ1= -26.01°, χ2= 37.3°, χ3= -33.9° and χ4= 18.1°.
χ0= -10.3°, χ1= -13.4°, χ2= 31.0°, χ3= -36.6° and χ4= 29.8°.
χ0= -18.7°, χ1= 34.5°, χ2= -38.3°, χ3= 27.0° and χ4= -5.0°.
E= -628.668896 a.u.
χ0= -15.0°, χ1= 32.9°, χ2= -39.2°, χ3= 30.2° and χ4= -9.2°.
χ0= -8.8°, χ1= 27.5°, χ2= -36.3°, χ3= 30.7° and χ4= -13.7°.
χ0= -23.1°, χ1= 36.5°, χ2= -37.4°, χ3= 23.8° and χ4= -0.1°.
χ0= -22.8°, χ1= 39.0°, χ2= -41.6°, χ3= 28.0° and χ4= -3.0°.
χ0= 8.3°, χ1= -29.5°, χ2= 39.9°, χ3= -35.0° and χ4= 16.7°.
χ0= -8.2°, χ1= -15.5°, χ2= 31.9°, χ3= -36.3° and χ4= 28.4°.
E= -628.667515 a.u.
χ0= -14.4°, χ1= 31.0°, χ2= -37.7°, χ3= 28.7° and χ4= -9.1°.
χ0= -2.1°, χ1= -21.9°, χ2= 36.6°, χ3= -37.1° and χ4= 25.1°.
χ0=-14.6°, χ1= 28.9°, χ2= -32.4°, χ3= 23.1° and χ4= -5.6°.
E= -628.669674 a.u.
χ0= -12.5°, χ1= -11.8°, χ2= 29.9°, χ3= -36.8° and χ4= 31.3°.
χ0= -1.9°, χ1= 21.8°, χ2= -33.0°, χ3= 31.1° and χ4= -18.7°.
χ0= 0.4°, χ1= -22.5°, χ2= 35.4°, χ3= -34.6° and χ4= 21.9°.
χ0= -2.0°, χ1= -21.2°, χ2= 35.2°, χ3= -35.9° and χ4= 24.1°.
χ0= -8.6°, χ1= 27.4°, χ2= -35.3°, χ3= 29.6° and χ4= -13.4°.
χ0= 0.5°, χ1= -23.7°, χ2= 37.2°, χ3= -36.3° and χ4= 22.8°.
Figure 2.
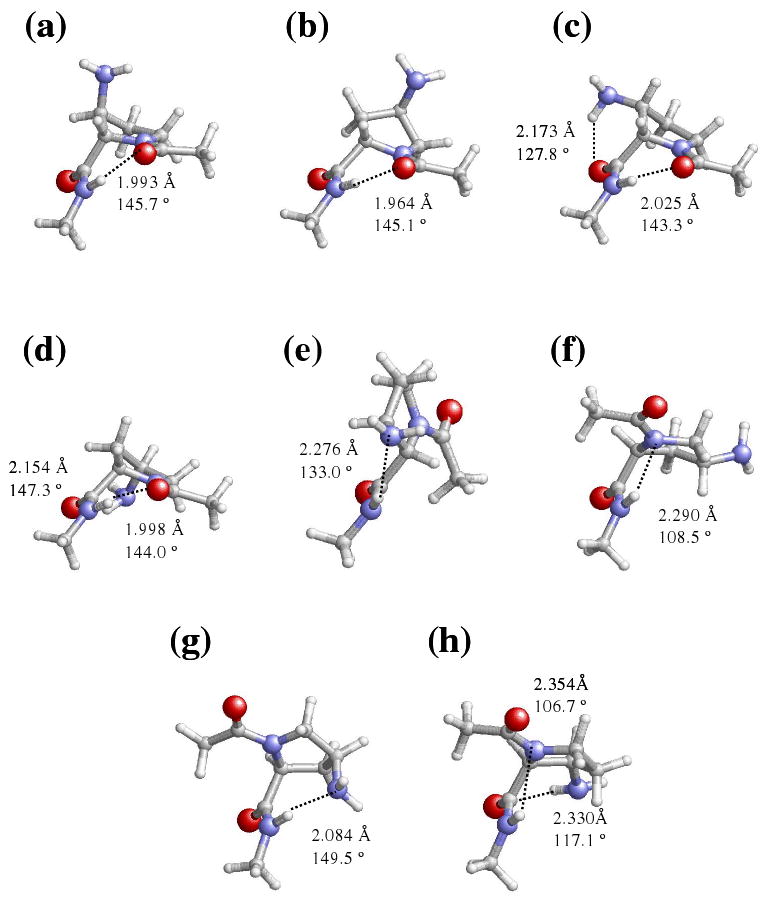
Representation of selected minimum energy conformations characterized in the gas-phase for the Amp-containing dipeptides studied in this work: (a) t-γL[d] for Ac-βtAmp-NHMe; (b) t-γL[u] for Ac-γtAmp-NHMe; (c) t-γL[d] for Ac-βcAmp-NHMe; (d) t-γL[d] for Ac-γcAmp-NHMe; (e) c-εL[u] for Ac-βtAmp-NHMe; (f) c-αL[d] for Ac-γtAmp-NHMe; (g) c-αL[d] for Ac-γcAmp-NHMe; and (h) c-αL[d] for Ac-βcAmp-NHMe.
Figure 3.
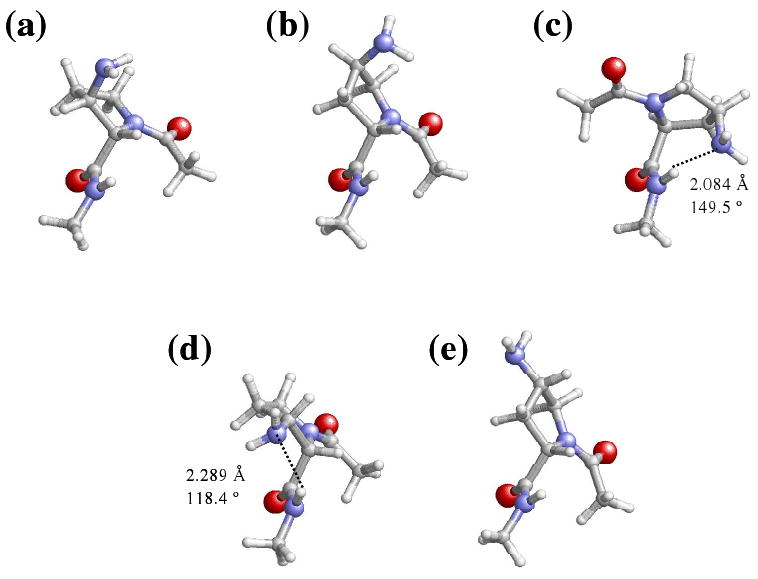
Representation of selected minimum energy conformations characterized for the Amp-containing dipeptides studied in this work: (a) c-εL[d] for Ac-βtAmp-NHMe; (b) c-εL[u] for Ac-γtAmp-NHMe; (c) c-αL[d] for Ac-γcAmp-NHMe; (d) c-εL[d] for Ac-βcAmp-NHMe; (e) c-εL[u] for Ac-γtAmp-NHMe. These minima are especially relevant because they are relatively stable in chloroform, methanol and/or aqueous solution.
Table 2.
Relative free energy in the gas-phase (ΔGgp; in kcal/mol) and in carbon tetrachloride, chloroform, methanol and aqueous solutions (ΔGCCl4, ΔGCHCl3, ΔGCH3OH and ΔGH2O, respectively; in kcal/mol) for the minimum energy conformations of Ac-βtAmp-NHMe, Ac-βcAmp-NHMe, Ac-γtAmp-NHMe and Ac-γcAmp-NHMe at the B3LYP/6-31+G(d,p) level.
| # Conf. | ΔGgp | ΔGCCl4 | ΔGCHCl3 | ΔGCH3OH | ΔGH2O |
|---|---|---|---|---|---|
| Ac-βtAmp-NHMe | |||||
| t-γL[d] | 0.0a | 0.0 | 1.3 | 5.8 | 6.4 |
| c-εL[u] | 2.7 | 1.0 | 0.8 | 3.2 | 3.5 |
| c-αL[u] | 4.7 | 3.0 | 2.5 | 4.1 | 4.2 |
| c-εL[d] | 3.9 | 1.6 | 0.0 | 0.0 | 0.0 |
| Ac-βcAmp-NHMe | |||||
| t-γL[d] | 0.0b | 1.0 | 0.0 | 2.7 | 3.3 |
| t-αL[d] | 2.7 | 0.0 | 0.2 | 1.1 | 1.4 |
| c-αL[d] | 3.3 | 0.6 | 1.3 | 2.5 | 2.3 |
| c-εL[d] | 4.4 | 3.3 | 0.3 | 0.0 | 0.0 |
| c-αL[u] | 4.4 | 1.6 | 2.6 | 3.9 | 3.7 |
| t-αL[u] | 7.6 | 6.4 | 2.9 | 1.5 | 1.2 |
| Ac-γtAmp-NHMe | |||||
| t-γL[u] | 0.0c | 0.0 | 0.5 | 4.4 | 5.1 |
| c-αL[d] | 1.7 | 4.2 | 0.4 | 2.9 | 2.9 |
| c-εL[u] | 4.4 | 5.4 | 0.0 | 0.0 | 0.0 |
| Ac-γcAmp-NHMe | |||||
| t-γL[d] | 0.0d | 0.4 | 2.3 | 7.0 | 7.9 |
| t-γL[u] | 1.3 | 0.8 | 1.7 | 5.4 | 6.3 |
| c-αL[d] | 2.2 | 0.0 | 0.0 | 2.7 | 3.4 |
| c-αL[u] | 3.9 | 2.3 | 2.2 | 4.4 | 4.3 |
| t-αL[u] | 3.7 | 1.2 | 1.5 | 2.9 | 3.2 |
| c-εL[d] | 4.6 | 2.3 | 1.3 | 1.8 | 2.1 |
| c-εL[u] | 6.0 | 2.8 | 0.8 | 0.0 | 0.0 |
G=-628.468989 a.u.
G= -628.469683 a.u.
G=-628.468329 a.u.
G= -628.470592 a.u.
Table 3.
Relative free energy in the gas-phase (ΔG#gp#; in kcal/mol) and in carbon tetrachloride, chloroform, methanol and aqueous solutions (ΔG#CCl4#, ΔG#CHCl3#, ΔG#CH3OH# and ΔG#H2O#, respectively; in kcal/mol) calculated at the B3LYP/6-31+G(d,p) level for the minimum energy conformations of Ac-βtAmp-NHMe, Ac-βcAmp-NHMe, Ac-γtAmp-NHMe and Ac-γcAmp-NHMe with respect to the lowest energy minimum of the most stable isomer.
| # Conf. | ΔG#gp# | ΔG#CCl4# | ΔG#CHCl3# | ΔG#CH3OH# | ΔG#H2O# |
|---|---|---|---|---|---|
| Ac-βtAmp-NHMe | |||||
| t-γL[d] | 1.0 | 2.6 | 2.0 | 5.8 | 6.6 |
| c-εL[u] | 3.7 | 3.7 | 1.5 | 3.2 | 3.7 |
| c-αL[u] | 5. 7 | 5.6 | 3.2 | 4.1 | 4.4 |
| c-εL[d] | 4. 9 | 4.2 | 0.7 | 0.0 | 0.2 |
| Ac-βcAmp-NHMe | |||||
| t-γL[d] | 0.6 | 2.7 | 2.5 | 7.0 | 7.8 |
| t-αL[d] | 3.2 | 1.7 | 2.8 | 5.4 | 6.0 |
| c-αL[d] | 3.8 | 2.3 | 3.8 | 6.8 | 6.9 |
| c-εL[d] | 5.0 | 5.0 | 2.8 | 4.3 | 4.6 |
| c-αL[u] | 5.0 | 3.3 | 5.1 | 8.2 | 8.3 |
| t-αL[u] | 8.2 | 8.1 | 5.4 | 5.6 | 5.9 |
| Ac-γtAmp-NHMe | |||||
| t-γL[u] | 1.4 | 0.0 | 2.1 | 5.9 | 6.8 |
| c-αL[d] | 3.2 | 3.7 | 2.0 | 4.4 | 4.6 |
| c-εL[u] | 5.8 | 4.9 | 1.6 | 1.5 | 1.7 |
| Ac-γcAmp-NHMe | |||||
| t-γL[d] | 0.0 | 2.2 | 2.3 | 7.0 | 7.9 |
| t-γL[u] | 1.3 | 2.6 | 1.7 | 5.4 | 6.3 |
| c-αL[d] | 2.2 | 1.8 | 0.0 | 2.3 | 3.4 |
| c-αL[u] | 3.9 | 4.1 | 2.2 | 4.4 | 4.3 |
| t-αL[u] | 3.7 | 3.0 | 1.5 | 2.9 | 3.2 |
| c-εL[d] | 4.6 | 4.1 | 1.3 | 1.8 | 2.1 |
| c-εL[u] | 6.0 | 4.6 | 0.8 | 0.0 | 0.0 |
Our previous calculations revealed three minimum energy conformations for Ac-Pro-NHMe when the two amide bonds are arranged in trans: t-γL[d], t-γL[u] and t-αL[u], the two latter being 1.0 and 4.9 kcal/mol, respectively, less stable than the former.12b These results, which are in excellent agreement with those reported by other authors,10,3b indicate that the backbone of conventional Pro tends to adopt a turn-like conformation that is compatible with both the down and up puckerings of the pyrrolidine ring, even though the former is the most favored. Table 1 indicates that β- and γ-amination reduce the intrinsically restricted conformational flexibility of conventional Pro, especially when the amino group is introduced in trans position. Thus, only one minimum energy conformation was found for Ac-βtAmp-NHMe and Ac-γtAmp-NHMe when ω0 is arranged in trans, which corresponds to the t-γL[d] (Figure 2a) and t-γL[u] (Figure 2b), respectively. Moreover, although three minima were characterized for Ac-βcAmp-NHMe, the t-γL[d] conformation (Figure 2c) is favored with respect to the t-αL[d] and t-αL[u] ones by 4.0 and 8.6 kcal/mol, respectively. Finally, the t-γL[d] (Figure 2d), t-γL[u] and t-αL[u] structures were identified as minimum energy conformations of Ac-γcAmp-NHMe, the two latter being unfavored with respect to the former by 1.5 and 5.7 kcal/mol, respectively. It is worth noting that these energy differences are larger by 0.5 and 0.8 kcal/mol, respectively, than those calculated for the same structures in Ac-Pro-NHMe.
As a consequence of their γL conformation, the lowest energy minimum of all the Amp-containing dipeptides studied in this work is stabilized by a seven-membered intramolecular hydrogen bond (typically denoted as C7) between the NH and O=C moieties of the NHMe and Ac blocking groups (Figure 2). However, it is worth noting that only the t-γL conformers of the dipeptides with the amino group arranged in cis are able to form simultaneously an intramolecular hydrogen bond between the side amino group and the O=C of the own Amp residue. Thus, the stability of the Ac-γcAmp-NHMe and Ac-βcAmp-NHMe dipeptides, which is higher than that of the corresponding analogues with the amino group in trans, should be attributed to the formation of this intraresidue hydrogen bond (Figures 2c and 2d). This feature is reflected by the ΔE#gp# values displayed in Table 1. On the other hand, it should be mentioned that, independently of both the arrangement of the peptide bond ω0 and the puckering of the five membered ring, the minima with an αL conformation of the four Amp-containing derivatives are typically stabilized by an intramolecular hydrogen bond between the N-H moiety of the NHMe blocking group and the nitrogen atom of the pyrrolidine ring. This interaction is illustrated in Figure 2 for the c-αL[d] conformation of both Ac-γtAmp-NHMe and Ac-βcAmp-NHMe.
Regarding the structures with ω0 arranged in cis, four minimum energy conformations were characterized for the Ac-Pro-NHMe dipeptide: c-αL[d], c-αL[u], c-εL[d] and c-εL[u], which were destabilized by 3.3, 4.2, 6.3 and 6.6 kcal/mol with respect to the global minimum.12b Interestingly, the energy difference between the global minimum and most stable conformations with ω0 arranged in cis is about 0.5 kcal/mol lower for Ac-βtAmp-NHMe, Ac-γtAmp-NHMe and Ac-γcAmp-NHMe than for Ac-Pro-NHMe. Three minima were characterized for Ac-βtAmp-NHMe, the c-εL[u] structure (Figure 2e) being more stable than the c-αL[u] and the c-εL[d] by 3.1 kcal/mol. The c-εL[u] presents an intramolecular hydrogen bond between the amino group, which acts as hydrogen bonding acceptor, and the NHMe blocking group. Only two minimum energy conformations were detected for Ac-γtAmp-NHMe, the c-αL (Figure 2f) being more stable than the c-εL by 3.1 kcal/mol. Regarding Ac-γcAmp-NHMe, four structures with the dihedral angle ω0 arranged in cis were characterized as energy minima. The most favored one is the c-αL[d] (Figure 2g), while the c-αL[u], c-εL[d] and c-εL[u] are higher in energy by 2.5, 3.3 and 5.6 kcal/mol. In this case, the c-αL[u] and c-εL[u] do not show intramolecular hydrogen bonding interactions, while such type of interactions were found in the c-αL[d] and c-εL[d] structures. Specifically, the amino nitrogen acts as hydrogen bonding acceptor with respect to the NHMe blocking group in the former conformation, whereas in the latter one the amino side group interacts with the carboxyl oxygen of the γcAmp residue.
The situation is completely different for Ac-βcAmp-NHMe since, in this case, the minimum of lowest energy with ω0 in cis, c-αL[d] (Figure 2h), is less stable than the global minimum by 4.3 kcal/mol. This result indicates that the cis-amination of the β-carbon atom produces a significant destabilization of the cis disposition of the ω0 amide bond with reference to that observed for Pro. As can be seen in Figure 2, the c-αL[d] structure shows two intramolecular hydrogen bonds. The first is between the amino side group, which acts as donor, and the carboxyl oxygen of the βcAmp residue, and the second corresponds to the interaction between the N-H of NHMe and the nitrogen of the pyrrolidine. The other two minimum energy conformations with ω0 arranged in cis, c-εL[d] and c-αL[u], are unfavored with respect to the c-αL[d] one by 1.0 and 1.6 kcal/mol, respectively. In spite of their high ΔEgp values (Table 1), these two structures show stabilizing intramolecular hydrogen bonds between the amino side group and the NHMe (c-εL[d]) or Ac (c-αL[u]) blocking groups.
Inspection of the free energies listed in Table 2 shows the importance of the ZPVE, thermal and entropic corrections to the relative stability of the minimum energy conformations found for the Amp-containing dipeptides. Consideration of these statistical terms for the transformation of ΔEgp into ΔGgp represents relative variations usually higher than 1 kcal/mol. Specifically, the largest change which was detected in both the c-αL[u] of Ac-βtAmp-NHMe and the t-αL[u] of Ac-γcAmp-NHMe, is 2.0 kcal/mol. Nonetheless, the t-γL is still the most favored conformer for the four dipeptides, the ΔGgp of the other structures being higher than 1.5 kcal/mol. Thus, according to a Bolzmann distribution of minima, the ΔGgp values indicate that the population at room temperature of all the local minimima is negligible for the four dipeptides under study, i.e. the t-γL is the only populated conformation.
Table 2 includes the relative free energies in carbon tetrachloride, chloroform, methanol and water solutions. The solvent introduces significant changes in the relative stability of the different minima characterized for Amp-containing dipeptides. In general, carbon tetrachloride was found to considerably stabilize conformers with the amide bond ω0 arranged in cis. Thus, although the most stable conformations of Ac-βtAmp-NHMe and Ac-βcAmp-NHMe, t-γL[d] and t-αL, respectively show the two amide bonds in trans, yet ω0 presents a cis arrangement in the most favored conformation of Ac-γcAmp-NHMe, c-αL[d] (Figure 2g). As a consequence of the stabilization produced by this solvent in conformers with a cis peptide bond, these structures are within the set of conformations energetically accessible at room temperature. Interestingly, the conformational behavior detected for Ac-γtAmp-NHMe in carbon tetrachloride is completely different from that discussed for the other three dipeptides, in this case the relative stability of the c-αL[d] and c-εL[u] structures being smaller than in the gas-phase.
The higher polarity of chloroform results in a further stabilization of conformers with cis peptide bonds. Thus, the most favored conformation in this environment shows the amide bond ω0 arranged in cis for three of the dipeptides under study, i.e. c-εL[d] (Figure 3a), c-εL[u] (Figure 3b) and c-αL[d] (Figure 3c) for Ac-βtAmp-NHMe, Ac-γtAmp-NHMe and Ac-γcAmp-NHMe, respectively. The only exception to this behaviour was for Ac-βcAmp-NHMe, in which the lowest energy minimum corresponds to the t-γL[d] conformation (Figure 2c). However, it should be noted that in this case the t-αL[d] and c-εL[d] structures are destabilized by only 0.2 and 0.3 kcal/mol, respectively. On the other hand, the ΔGCHCl3 value of the least stable conformer is 2.5, 2.9, 0.5 and 2.3 kcal/mol for Ac-βtAmp-NHMe, Ac-βcAmp-NHMe, Ac-γtAmp-NHMe and Ac-γcAmp-NHMe, respectively, suggesting that chloroform induces a strong stabilizing effect in all the structures.
The c-εL is the most stable conformation in both methanol and aqueous solutions for all the Amp-containing dipeptides, the only difference between them being the puckering of the ring. Thus, the two β-aminated dipeptides prefer a down puckering, while the ring is arranged up when the substituent is introduced at the γ-carbon atom. The conformational characteristics of these structures are displayed in Figure 3. However, the most remarkable result in polar environments is the destabilization of the remaining structures, especially those with ω0 arranged in trans. This feature is fully consistent with theoretical estimations previously reported for the Pro dipeptide.3 Thus, it was found that the electronic effect that stabilize the cis form of the peptide bond become enhanced in polar environments, even though the cis/trans rotational barriers increase with the polarity of the solvent.
Although the stability of the cis conformers in solution was found to be overestimated by PCM for Pro derivatives, especially in protic solvents able to form specific hydrogen bonds with the solute, the general tendencies provided by this theoretical method describe very satisfactorily the experimental observations from a qualitative point of view.12 Thus, in a recent study PCM calculations predicted that ω0 exhibits a considerably smaller probability of adopting a cis disposition in α-methylproline and α-phenylproline than in Pro,12b which was in good agreement with experimental information.29 Comparison of the results provided in Table 2 for Amp-containing dipeptides with those reported for Ac-Pro-NHMe at the same theoretical level suggests that the incorporation of the substituent to the pyrrolidine ring enhances, in general, the stability of the conformers with ω0 arranged in cis. Thus, although the c-εL[u] conformation was predicted as the most favored for Ac-Pro-NHMe in both chloroform and water, the lowest energy structure with ω0 in trans was unfavored by only 0.3 kcal/mol (t-γL[d]) and t-αL[u], respectively).12b Table 2 illustrates that this energy difference is higher for the investigated Amp-containing dipeptides. However, caution is required in the analysis of PCM results, especially when protic solvents able to form specific solute-solvent interactions are considered.
Table 3 shows the free energies relative to the lowest energy minimum of the most stable Amp-containing isomer for each environment. As can be seen the most favored isomer in the gas-phase, chloroform, methanol and water solutions is the Ac-γcAmp-NHMe dipeptide, even though as reflected in Table 2, the preferred conformation depends on the polarity of the environment. Moreover, in the gas-phase the most stable conformation of each isomer shows ΔG#gp# < 1.5 kcal/mol indicating that the relative stability of the other three dipeptides is still significant. However, in chloroform, methanol and aqueous solutions only one isomer, the Ac-βtAmp-NHMe dipeptide, satisfies such condition. These results clearly indicate that the stability of Ac-βcAmp-NHMe and Ac-γtAmp-NHMe decreases with the polarity of the environment. Finally, it should be noted that the Ac-γtAmp-NHMe is the lowest energy isomer in carbon tetrachloride solution. Accordingly, it can be concluded that Ac-βtAmp-NHMe is stabilized by the favorable electrostatic interactions between the solute and the solvent, while Ac-γtAmp-NHMe is preferred in non-polar organic solvent where solute-solvent interactions are dominated by non-electrostatic terms, i.e. van der Waals and cavitation.
Dimetylaminoproline (Dmp) dipeptides
Results provided by B3LYP/6-31+G(d,p) calculations for the four Dmp-containing dipeptides (Scheme 1) are reported in Tables 4, 5 and 6, atomistic pictures of the more relevant minima being displayed in Figures 4 and 5.
Table 4.
Backbone dihedral angles (in degrees), pseudorotational parameters (A and P; in degrees), relative energy (ΔEgp; in kcal/mol) and relative energy with respect to the lowest energy conformation of the most stable dipeptide (ΔE#gp#; in kcal/mol) of the minimum energy conformations characterized for Ac-βtDmp-NHMe, Ac-βcDmp-NHMe, Ac-γtDmp-NHMe and Ac-γcDmp-NHMe at the B3LYP/6-31+G(d,p) level in the gas phase.
| # Conf. | ω0 | φ | ψ | ω | (A, P) | ΔEgp | ΔE#gp# |
|---|---|---|---|---|---|---|---|
| Ac-βtDmp-NHMe | |||||||
| t-γL[d] | -170.6 | -85.0 | 72.0 | -176.9 | (40.5, -110.2)a | 0.0b | 2.3 |
| t-γL[u] | -175.1 | -81.5 | 78.8 | -175.8 | (37.5, 110.2)c | 0.2 | 2.5 |
| c-εL[u] | 0.1 | -67.4 | 179.9 | 176.0 | (38.7, 95.0)d | 2.9 | 5.2 |
| c-αL[d] | 12.0 | -94.1 | -4.9 | 179.3 | (39.4, -111.2)e | 3.2 | 5.5 |
| c-αL[u] | 3.9 | -84.1 | -17.2 | 179.8 | (35.6, 113.7)f | 3.4 | 5.7 |
| c-εL[d] | 3.6 | -79.6 | 141.4 | 177.8 | (39.1, -111.9)g | 5.0 | 7.3 |
| c-εL[u] | -0.2 | -74.8 | 120.7 | -178.5 | (38.4, 106.5)h | 6.3 | 8.6 |
| Ac-βcDmp-NHMe | |||||||
| t-γL[d] | 179.1 | -77.8 | 122.0 | -174.1 | (39.2, -123.2)i | 0.0j | 4.0 |
| c-εL[d] | 0.9 | -87.3 | -137.3 | -177.6 | (43.1, -134.6)k | 1.6 | 5.6 |
| t-αL[d] | -171.8 | -81.7 | -23.9 | 174.4 | (39.6, -106.7)l | 1.7 | 5.7 |
| t-εL[d] | 175.7 | -92.9 | -159.7 | 180.0 | (46.2, -142.2)m | 1.8 | 5.8 |
| c-αL[d] | 7.6 | -83.7 | -27.2 | 178.0 | (38.2, -110.9)n | 2.0 | 6.0 |
| t-εL[u] | 179.5 | -65.1 | 143.8 | -176.5 | (32.5, 99.3)o | 4.2 | 8.2 |
| c-εL[d] | -1.5 | -81.8 | 138.3 | 174.1 | (40.6, -125.2)p | 4.4 | 8.4 |
| t-αL[u] | -172.1 | -65.9 | -26.9 | 175.9 | (42.6, 75.3)q | 5.6 | 9.6 |
| c-αL[u] | 9.8 | -66.0 | -32.0 | -175.4 | (41.8, 76.2)r | 5.6 | 9.6 |
| c-εL[u] | 2.3 | -62.0 | 156.9 | 171.6 | (31.8, 88.3)s | 6.6 | 10.6 |
| Ac-γtDmp-NHMe | |||||||
| t-γL[d] | -171.6 | -83.9 | 69.5 | -178.1 | (37.1, -109.5)t | 0.0u | 0.0 |
| t-γL[u] | -171.0 | -83.1 | 75.7 | -176.5 | (40.3, 98.3)v | 3.1 | 3.1 |
| c-αL[u] | 8.9 | -89.4 | -4.5 | -179.3 | (37.2, -111.4)w | 3.4 | 3.4 |
| c-εL[d] | 1.0 | -73.1 | 148.6 | -175.9 | (36.1, -112.2)x | 6.5 | 6.5 |
| t-αL[u] | -168.1 | -81.0 | -6.9 | 175.3 | (40.8, 87.9)y | 6.7 | 6.7 |
| c-εL[u] | 4.1 | -65.0 | 143.8 | 177.7 | (41.8, 76.2)z | 8.0 | 8.0 |
| Ac-γcDmp-NHMe | |||||||
| t-γL[u] | -173.6 | -81.5 | 74.5 | -176.9 | (38.1, 105.2)aa | 0.0bb | 1.3 |
| t-γL[d] | -171.1 | -79.2 | 47.8 | 175.4 | (36.7, -105.2)cc | 2.0 | 3.3 |
| c-αL[u] | 8.6 | -79.0 | -18.8 | -176.1 | (37.1, 89.1)dd | 2.7 | 4.0 |
| c-αL[d] | 7.8 | -67.5 | -44.5 | -176.3 | (38.1, -92.7)ee | 3.2 | 4.5 |
| t-αL[u] | -170.8 | -77.5 | -11.3 | -176.0 | (37.5, 88.6)ff | 3.5 | 4.8 |
| c-αL[d] | 9.7 | -90.8 | 3.7 | -175.5 | (38.1, -111.7)gg | 3.6 | 4.9 |
| c-εL[u] | 1.1 | -62.1 | 147.5 | 177.0 | (37.4, 87.6)hh | 5.5 | 6.8 |
| c-εL[d] | -0.1 | -76.4 | -178.8 | 178.6 | (40.8, -117.9)ii | 8.9 | 10.2 |
χ0= -14.0°, χ1= 32.9°, χ2= -40.5°, χ3= 32.3° and χ4= -11.3°.
E= -707.281408 a.u.
χ0= -13.0°, χ1= -10.8°, χ2= 29.3°, χ3= -36.7° and χ4= 31.6°.
χ0= -3.4°, χ1= -20.3°, χ2= 35.4°, χ3= -37.0° and χ4= 25.8°.
χ0= -14.3°, χ1= 32.4°, χ2= -39.3°, χ3= 31.1° and χ4= -10.3°.
χ0= -14.3°, χ1= -8.1°, χ2= 26.5°, χ3= -34.7° and χ4= 30.9°.
χ0= -14.6°, χ1= 32.3°, χ2= -39.0°, χ3= 30.6° and χ4= -9.7°.
χ0= -10.9°, χ1= -13.3°, χ2= 31.6°, χ3= -37.7° and χ4= 30.7°.
χ0= -21.4°, χ1= 35.9°, χ2= -37.9°, χ3= 25.1° and χ4= -2.1°.
E= -707.278763 a.u.
χ0= -30.3°, χ1= 41.5°, χ2= -38.6°, χ3= 20.8° and χ4= 6.4°.
χ0= -11.4°, χ1= 31.2°, χ2= -39.8°, χ3= 32.7° and χ4= -13.2°.
χ0= -36.5°, χ1= 44.8°, χ2= -38.0°, χ3= 17.0° and χ4= 12.7°.
χ0= -13.6°, χ1= 31.6°, χ2= -38.3°, χ3= 30.0° and χ4= -10.0°.
χ0= -5.3°, χ1= -14.8°, χ2= 28.8°, χ3= -31.8° and χ4= 23.4°.
χ0= -23.4°, χ1= 37.6°, χ2= -38.8°, χ3= 25.0° and χ4= -0.7°.
χ0= 10.8°, χ1= -32.3°, χ2= 42.4°, χ3= -36.4° and χ4= 15.6°.
χ0= 10.0°, χ1= -31.3°, χ2= 41.7°, χ3= -36.2° and χ4= 15.9°.
χ0= 0.9°, χ1= -19.3°, χ2= 30.4°, χ3= -29.9° and χ4= 18.2°.
χ0= -12.4°, χ1= 30.6°, χ2= -37.2°, χ3= 29.2° and χ4= -10.7°.
E=-707.285136 a.u.
χ0= -5.8°, χ1= -19.6°, χ2= 35.8°, χ3= -38.6° and χ4= 28.6°.
χ0= -13.6°, χ1= 31.2°, χ2= -37.1°, χ3= 28.6° and χ4= -9.6°.
χ0= -13.7°, χ1= 30.6°, χ2= -36.0°, χ3= 27.5° and χ4= -8.8°.
χ0= 1.5°, χ1= -25.8°, χ2= 38.9°, χ3= -37.5° and χ4= 23.2°.
χ0= 10.0°, χ1= -31.3°, χ2= 41.7°, χ3= -36.2° and χ4= 15.9°.
χ0= -10.0°, χ1= -14.3°, χ2= 31.7°, χ3= -37.1° and χ4= 30.0°.
E= -707.283071 a.u.
χ0= -9.6°, χ1= 29.01°, χ2= -36.7°, χ3= 30.1° and χ4= -13.3°.
χ0= 0.6°, χ1= -22.7°, χ2= 35.3°, χ3= -34.5° and χ4= 21.6°.
χ0= -1.8°, χ1= 24.4°, χ2= -36.6°, χ3= 34.7° and χ4= -21.4°.
χ0= 0.9°, χ1= -23.2°, χ2= 35.8°, χ3= -34.8° and χ4= 21.6°.
χ0= -14.1°, χ1= 32.4°, χ2= -38.0°, χ3= 29.0° and χ4= -9.6°.
χ0= 1.6°, χ1= -23.7°, χ2= 36.0°, χ3= -34.4° and χ4= 21.0°.
χ0= -19.0°, χ1= 36.4°, χ2= -40.0°, χ3= 28.5° and χ4 = -6.1°.
Table 5.
Relative free energy in the gas-phase (ΔGgp; in kcal/mol) and in carbon tetrachloride, chloroform. methanol and aqueous solutions (ΔGCCl4, ΔGCHCl3, ΔGCH3OH and ΔGH2O, respectively; in kcal/mol) for the minimum energy conformations of Ac-βtDmp-NHMe, Ac-βcDmp-NHMe, Ac-γtDmp-NHMe and Ac-γcDmp-NHMe at the B3LYP/6-31+G(d,p) level.
| # Conf. | ΔGgp | ΔGCCl4 | ΔGCHCl3 | ΔGCH3OH | ΔGH2O |
|---|---|---|---|---|---|
| Ac-βtDmp-NHMe | |||||
| t-γL[d] | 0.0a | 0.0 | 0.4 | 3.0 | 4.0 |
| t-γL[u] | 0.4 | 0.6 | 0.8 | 3.0 | 4.0 |
| c-εL[u] | 2.6 | 1.0 | 0.0 | 0.7 | 1.4 |
| c-αL[d] | 2.6 | 1.8 | 1.4 | 0.7 | 3.0 |
| c-αL[u] | 2.7 | 1.7 | 1.6 | 3.1 | 3.5 |
| c-εL[d] | 5.3 | 3.4 | 4.3 | 0.5 | 0.6 |
| c-εL[u] | 5.4 | 3.6 | 1.0 | 0.0 | 0.0 |
| Ac-βcDmp-NHMe | |||||
| t-γL[d] | 0.0b | 0.0 | 0.0 | 0.4 | 1.6 |
| c-εL[d] | 0.8 | 0.2 | 0.2 | 0.7 | 1.9 |
| t-αL[d] | 2.6 | 2.4 | 2.0 | 1.5 | 2.2 |
| t-εL[d] | 2.4 | 1.7 | 1.2 | 0.8 | 2.4 |
| c-αL[d] | 0.9 | 1.1 | 1.5 | 2.1 | 2.9 |
| t-εL[u] | 4.8 | 4.0 | 3.4 | 2.7 | 3.8 |
| c-εL[d] | 2.9 | 2.0 | 1. 2 | 0.0 | 0.4 |
| t-αL[u] | 5.9 | 5.0 | 4.6 | 4.2 | 0.0 |
| c-αL[u] | 5.1 | 4.0 | 4.3 | 5.0 | 5.9 |
| c-εL[u] | 5.5 | 4.0 | 2.8 | 1.4 | 2.1 |
| Ac-γtDmp-NHMe | |||||
| t-γL[d] | 0.0c | 0.0 | 0.6 | 4.8 | 5.5 |
| t-γL[u] | 2.9 | 3.1 | 3.8 | 8.2 | 8.9 |
| c-αL[u] | 2.3 | 1.7 | 1.4 | 4.1 | 3.9 |
| c-εL[d] | 4.5 | 2.1 | 0.0 | 0.0 | 0.0 |
| t-αL[u] | 5.6 | 5.2 | 4.5 | 6.9 | 6.7 |
| c-εL[u] | 6.7 | 4.6 | 2.8 | 3.1 | 3.1 |
| Ac-γcDmp-NHMe | |||||
| t-γL[u] | 0.0d | 0.0 | 1.3 | 5.6 | 6.3 |
| t-γL[d] | 2. 6 | 2.8 | 4.5 | 8.9 | 3.7 |
| c-αL[u] | 1.8 | 0.9 | 1.3 | 4.6 | 4.8 |
| c-αL[d] | 2.5 | 0.8 | 1.2 | 5.1 | 5.5 |
| t-αL[u] | 2.4 | 1.5 | 1.6 | 4.2 | 4.6 |
| c-αL[d] | 3.5 | 2.7 | 3.5 | 7.1 | 7.4 |
| c-εL[u] | 4.1 | 1.5 | 0.0 | 0.0 | 0.0 |
| c-εL[d] | 7.7 | 6.8 | 9.1 | 7.6 | 0.5 |
G= -707.030159 a.u.
G= -707.026345 a.u.
G= -707.033869 a.u.
G= -707.031747 a.u.
Table 6.
Relative free energy in the gas-phase (ΔG#gp#; in kcal/mol) and in carbon tetrachloride, chloroform, methanol and aqueous solutions (ΔG#CCl4#, ΔG#CHCl3#, ΔG#CH3OH# and ΔG#H2O#, respectively; in kcal/mol) calculated at the B3LYP/6-31+G(d,p) level for the minimum energy conformations of Ac-βtDmp-NHMe, Ac-βcDmp-NHMe, Ac-γtDmp-NHMe and Ac-γcAmp-NHMe with respect to the lowest energy minimum of the most stable isomer.
| # Conf. | ΔG#gp# | ΔG#CCl4# | ΔG#CHCl3# | ΔG#CH3OH# | ΔG#H2O# |
|---|---|---|---|---|---|
| Ac-βtDmp-NHMe | |||||
| t-γL[d] | 2.3 | 3.8 | 6.9 | 11.8 | 12.7 |
| t-γL[u] | 2.7 | 3.9 | 6.9 | 11.5 | 12.4 |
| c-εL[u] | 4.9 | 2.1 | 3.9 | 6.9 | 7.5 |
| c-αL[d] | 4.9 | 2.9 | 5.3 | 6.5 | 9.2 |
| c-αL[u] | 5.0 | 2.7 | 5.4 | 9.2 | 9.5 |
| c-εL[d] | 7.6 | 1.8 | 5.5 | 4.0 | 4.1 |
| c-εL[u] | 7.7 | 1.9 | 2.1 | 3.5 | 3.4 |
| Ac-βcDmp-NHMe | |||||
| t-γL[d] | 4.7 | 2.2 | 4.0 | 7.0 | 7.5 |
| c-εL[d] | 5.5 | 1.6 | 3.4 | 6.6 | 7.0 |
| t-αL[d] | 7.3 | 1.9 | 3.4 | 5.5 | 5.9 |
| t-εL[d] | 7.1 | 1.5 | 2.8 | 5.0 | 5.8 |
| c-αL[d] | 5.6 | 2.4 | 4.7 | 7.9 | 7.9 |
| t-εL[u] | 9.5 | 1.3 | 2.6 | 4.5 | 4.8 |
| c-εL[d] | 7.6 | 1.3 | 2.4 | 3.7 | 3.4 |
| t-αL[u] | 10.6 | 1.2 | 2.7 | 4.9 | 4.7 |
| c-αL[u] | 9.8 | 1.1 | 3.3 | 6.5 | 6.6 |
| c-εL[u] | 10.2 | 0.6 | 1.3 | 2.4 | 2.4 |
| Ac-γtDmp-NHMe | |||||
| t-γL[d] | 0.0a | 2.8 | 2.6 | 10.5 | 5.1 |
| t-γL[u] | 2.9 | 3.0 | 2.4 | 10.9 | 4.6 |
| c-αL[u] | 2.3 | 2.1 | 4.2 | 7.3 | 7.4 |
| c-εL[d] | 4.5 | 0.4 | 0.7 | 1.1 | 1.4 |
| t-αL[u] | 5.6 | 2.4 | 4.1 | 6.9 | 7.0 |
| c-εL[u] | 6.7 | 0.7 | 1.2 | 2.1 | 2.3 |
| Ac-γcDmp-NHMe | |||||
| t-γL[u] | 1.0 | 2.7 | 5.4 | 9.7 | 10.5 |
| t-γL[d] | 3. 6 | 2.9 | 6.1 | 10.5 | 5.3 |
| c-αL[u] | 2.8 | 1.8 | 3.7 | 7.0 | 7.2 |
| c-αL[d] | 3.5 | 0.9 | 2.9 | 6.6 | 7.1 |
| t-αL[u] | 3.4 | 1.7 | 3.3 | 5.9 | 6.3 |
| c-αL[d] | 4.5 | 1.9 | 4.2 | 7.8 | 8.1 |
| c-εL[u] | 5.1 | 0.0 | 0.0 | 0.0 | 0.0 |
| c-εL[d] | 8.7 | 1.1 | 2.1 | 3.3 | 3.1 |
G= -707.033869 a.u.
Figure 4.
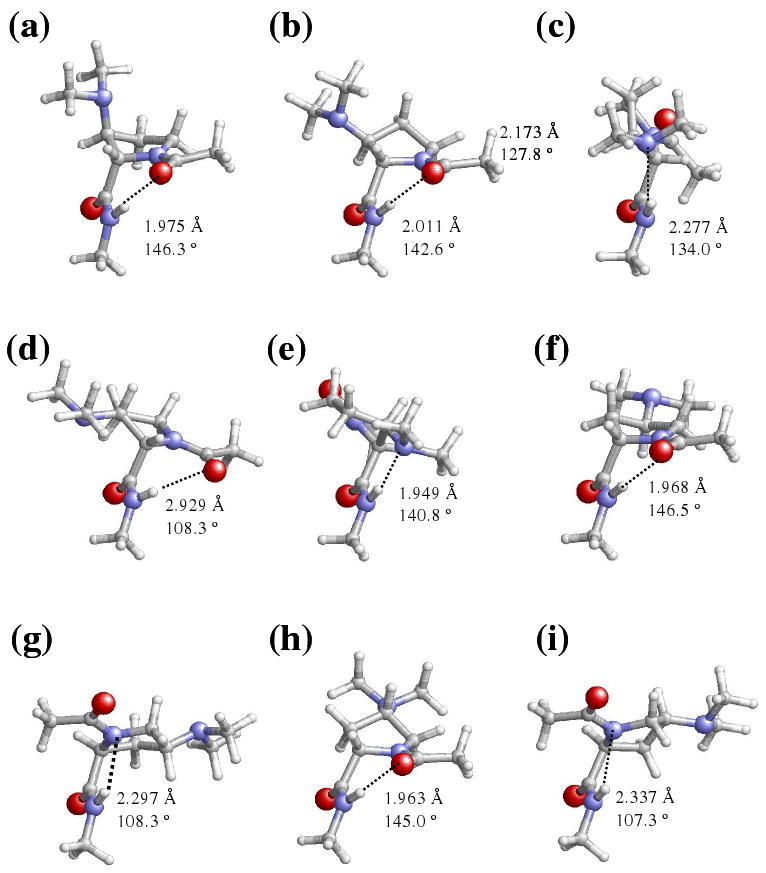
Representation of selected minimum energy conformations characterized in the gas-phase for the Dmp-containing dipeptides studied in this work: (a) t-γL[d], (b) t-γL[u] and (c) c-εL[u] for Ac-βtDmp-NHMe; (d) t-γL[d] and (e) c-εL[d] for Ac-βcDmp-NHMe; (f) t-γL[d] and (g) c-αL[u] for Ac-γtDmp-NHMe; (h) t-γL[u] and (i) c-αL[u] for Ac-γcDmp-NHMe.
Figure 5.
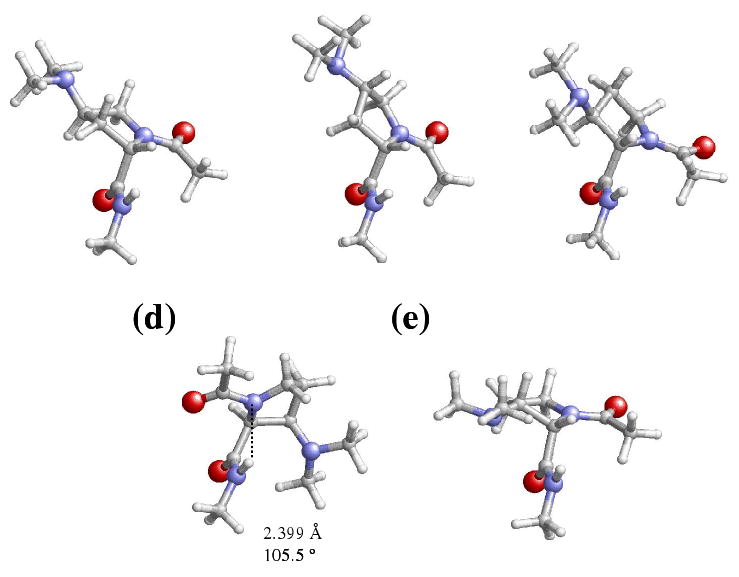
Representation of selected minimum energy conformations characterized for the Dmp-containing dipeptides studied in this work: (a) c-εL[d] for Ac-γtDmp-NHMe; (b) c-εL[u] for Ac-γcDmp-NHMe; (c) c-εL[u] for Ac-βtDmp-NHMe; (d) c-εL[d] and (e) t-αL[u] for Ac-βcDmp-NHMe. These minima are especially relevant because they are relatively stable in chloroform, methanol and/or aqueous solution.
The ΔEgp values displayed in Table 4 indicate that the conformational preferences of the Dmp-containing dipeptides are completely different from those described in the previous section for the Amp-containing ones. Seven minimum energy conformations, including those with the peptide bond ω0 arranged in cis, were obtained for Ac-βtDmp-NHMe, while four were found for Ac-βtAmp-NHMe. The only structures detected for the former dipeptide below a relative energy threshold value of 1.5 kcal/mol were the t-γL[d] and t-γL[u] (Figures 4a and 4b), which are almost isoenergetic and present a seven-membered hydrogen bonded ring. This represents another important difference with respect to Ac-βtAmp-NHMe, since for this compound the only structure with ΔEgp < 1.5 kcal/mol was the t-γL[d]. The relative stability of the c-εL[u] conformation (Figure 4c), which is the most stable structure with ω0 in cis, with respect to the global minimum is similar for both Ac-βtDmp-NHMe and Ac-βtAmp-NHMe. This is a surprising feature since in the former dipeptide, this conformation presents a stabilizing hydrogen bond between the N-H of the NHMe blocking group and the nitrogen of the dimethylamino substituent that was not detected in the latter. Furthermore, remarkable differences appear in the ΔEgp of the minima with c-αL backbone conformation. Thus, these are more stable in Ac-βtDmp-NHMe than in the corresponding βtAmp-containing analogue by about 3 kcal/mol.
A total of 10 minimum energy conformations were characterized for Ac-βcDmp-NHMe, 5 for each arrangement of ω0. Surprisingly, the lowest energy conformation corresponds to a t-γL[d] with the dihedral angles φ and ψ significantly distorted towards those of a conventional t-εL (Figure 4d). As indicated by the corresponding geometric parameters, i.e. dH⋯O= 2.929 Å and ∠N-H⋯O= 108.3°, this structure is stabilized by a very weak intramolecular interaction that defines a seven-membered hydrogen bonded ring between the N-H of NHMe and the C=O of the Ac. Indeed, a standard t-γL conformation with a strong intramolecular hydrogen bond forming a seven-membered ring is not possible for the Ac-βcDmp-NHMe dipeptide. This is because such combination of φ and ψ dihedral angles leads to a strong repulsive interaction between the lone pair of the dimethylamine group and the carbonyl oxygen of the Dmp residue. The ΔEgp of the other four conformations with ω0 arranged in trans ranges from 1.7 (t-αL[d]) to 5.6 (t-αL[u]) kcal/mol, these energy values being significantly lower than those found for Ac-βcAmp-NHMe, i.e. in the latter dipeptide the ΔEgp of the first (t-αL[d]) and the last (t-αL[u]) local minimum were 4.5 and 9.1 kcal/mol, respectively. On the other hand, the c-εL[d] is the lowest energy conformation with ω0 arranged in cis, this structure being destabilized with respect to the global minimum by 1.6 kcal/mol. Figure 4e reveals that this conformation is stabilized by an intramolecular hydrogen bond between the N-H of the NHMe blocking group and the nitrogen of the dimethylamino substituent. Comparison with the results listed in Table 1 for Ac-βcAmp-NHMe indicates that the replacement of the amino substituent by the dimethylamino group also alters the potential energy hypersurface of the dipeptide with the peptide bond arranged in cis. Thus, the least favored cis minimum of Ac-βcDmp-NHMe (c-εL[u]) is destabilized by 5.0 kcal/mol with respect to that of c-εL[d], whereas in Ac-βcAmp-NHMe the most (c-αL[d]) and the least (c-αL[u]) stable conformations with ω0 arranged in cis are separated by only 1.3 kcal/mol.
Six minimum energy conformations, three with ω0 arranged in trans, were found for Ac-γtDmp-NHMe. The lowest energy one corresponds to the t-γL[d] (Figure 4f), the t-γL[u], which was the global minimum of Ac-γtAmp-NHMe, being unfavored by 3.1 kcal/mol. Interestingly, the lowest energy conformation and the t-αL[u], which is destabilized by 6.7 kcal/mol, were not found as energy minima in Ac-γtAmp-NHMe. Regarding the three conformations with ω0 in cis, the most stable, c-αL[u] (Figure 4g), is stabilized by a five-membered intramolecular hydrogen bonded ring involving the backbone nitrogen atom of the γtDmp residue and the N-H of the NHMe blocking group. This structure is unfavored by 3.4 kcal/mol with respect to the global minimum, while the ΔEgp values of the other two cis conformers are 6.5 (c-εL[d]) to 8.0 kcal/mol (c-εL[u]).
Eight minimum energy conformations were characterized for Ac-γcDmp-NHMe. The lowest energy one corresponds to the t-γL[u] (Figure 4h), while the other structures with ω0 in trans, t-γL[d] and t-αL[u], are unfavored by 2.0 and 3.0 kcal/mol, respectively. These three conformations are stabilized by an intramolecular hydrogen bond. Thus, a seven-membered hydrogen bonded ring involving the NHMe and Ac blocking groups is shown by the two t-γL structures, whereas in the t-αL[u] conformation the nitrogen of the γcDmp residue and the N-H of the NHMe group forms a five-membered intramolecular hydrogen bonded ring. Comparison with the minima listed in Table 1 for Ac-γcAmp-NHMe indicates that the incorporation of the methyl groups into the amino substituent mainly affects to the puckering of the pyrrolidine ring, i.e. the relative stability between t-γL[u] and t-γL[d] is exchanged and the t-αL[u] minimum transform into the t-αL[d].
On the other hand, ω0 is arranged in cis in the remaining five minima of Ac-γcDmp-NHMe, the most stable cis structure being the c-αL[u] (Figure 4i). This conformation is 2.7 kcal/mol less stable than the global minimum, and is stabilized by an intramolecular hydrogen bond similar to that described for the t-αL[u] minimum. Interestingly, the Ac-γcDmp-NHMe dipeptide shows two minima with c-αL[d] conformation, which differ in the nitrogen atom that acts as acceptor in the stabilizing intramolecular hydrogen bond. In the most favored conformation that is 3.2 kcal/mol less stable than the global minimum, the nitrogen of the γcDmp residue participates in such interaction, whereas the interaction in the second conformation which is 0.4 kcal/mol less favored than the first one, involves the nitrogen of the dimethylamino side group. Finally, the ΔEgp values of the c-εL[u] and c-εL[d] structures are 5.5 and 8.9 kcal/mol, respectively.
The ΔGgp values listed in Table 5 show that the effects produced by the incorporation of the ZPVE and the thermal and entropic corrections are less dramatic for Dmp-containing dipeptides than for the Amp ones. Thus, the addition of these statistical terms represents relative variations typically smaller than 1 kcal/mol. Specifically, the largest change found in Ac-βtDmp-NHMe, Ac-βcDmp-NHMe, Ac-γtDmp-NHMe and Ac-γcDmp-NHMe, is -0.9 (c-εL[u]), -1.1 (c-αL[d]), -1.3 (c-εL[u]) and -1.4 kcal/mol (c-εL[u]), respectively. Inspection of the relative free energies in carbon tetrachloride, also displayed in Table 5, indicates that in general, solute-solvent interactions tend to stabilize the minimum energy conformations with ω0 in cis. In spite of this, the lowest energy conformation in carbon tetrachloride solution and in the gas-phase is the same for the four Dmp-containing dipeptides. This is an important difference with respect to the four Amp-containing dipeptides since, as we previously showed, this organic solvent is able to alter the conformational preferences of Ac-βcAmp-NHMe and Ac-γcAmp-NHMe (Table 2).
The stabilization of conformers with the peptide bond in cis is enhanced in chloroform solution (Table 5). Thus, the lowest energy minimum in this solvent for Ac-βtDmp-NHMe, Ac-γtDmp-NHMe and Ac-γcDmp-NHMe corresponds to the c-εL[u] (Figure 4c), c-εL[d] (Figure 5a) and c-εL[u] (Figure 5b), respectively. It is worth noting that the tendency to adopt the c-εL conformer in chloroform solution was also detected in Ac-βtAmp-NHMe and Ac-γtAmp-NHMe (Table 2), while Ac-γcAmp-NHMe preferred a c-αL helical structure. In contrast, the t-γL[d] (Figure 4d) is the lowest energy conformation of Ac-βcDmp-NHMe, which was also the global minimum in both the gas-phase and carbon tetrachloride solution. Amazingly this exceptional behavior is identical to that observed in the previous section for Ac-βcAmp-NHMe.
The most stable conformation in both methanol and aqueous solutions of Ac-βtDmp-NHMe, Ac-γtDmp-NHMe and Ac-γcDmp-NHMe also corresponds to the c-εL conformer (Figures 5c, 5a and 5b, respectively), even though the puckering of the pyrrolidine ring depends on the position of the substituent. Again, this result is fully consistent with that obtained for the Amp-containing dipeptides. Regarding the Ac-βcDmp-NHMe dipeptide, the lowest energy conformations in methanol and aqueous solutions are the c-εL[d] (Figure 5d) and t-αL[u] (Figure 5e), respectively. However, it should be noted that in the latter environment the former conformation is destabilized by only 0.4 kcal/mol. This indicates that in the presence of polar solvents the four Dmp-containing dipeptides follow a similar behavior.
The free energies relative to the lowest energy minimum of the most stable Dmp-containing isomer are listed in Table 6. The most stable isomer in gas-phase is Ac-γtDmp-NHMe, even though the Ac-γcDmp-NHMe is destabilized by only 1.0 kcal/mol. However, the latter is the most favored isomer in solution, independent of the polarity of the environment. Moreover, the relative order in terms of stability is: Ac-γcDmp-NHMe > Ac-γtDmp-NHMe > Ac-βcDmp-NHMe > Ac-βtDmp-NHMe. A detailed comparison of these results with those reported in the previous section (see Table 3) for the Amp-containing dipeptides reveals some important differences. Thus, the relative order found for the aminated dipeptides in aqueous and chloroform solution was Ac-γcAmp-NHMe > Ac-βtAmp-NHMe > Ac-γtAmp-NHMe > Ac-βcAmp-NHMe, while in methanol and carbon tetrachloride solutions the relative orders were Ac-γcAmp-NHMe ≅ Ac-βtAmp-NHMe > Ac-γtAmp-NHMe > Ac-βcAmp-NHMe and Ac-γtAmp-NHMe > Ac-βcAmp-NHMe > Ac-γcAmp-NHMe > Ac-βtAmp-NHMe, respectively.
Conclusions
Quantum mechanical calculations at the B3LYP/6-31+G(d,p) level have been used to explore the conformational preferences of Ac-βtAmp-NHMe, Ac-βcAmp-NHMe, Ac-γtAmp-NHMe, Ac-γcAmp-NHMe, Ac-βtDmp-NHMe, Ac-βcDDmp-NHMe, Ac-γtDmp-NHMe and Ac-γcDmp-NHMe. Comparison of the results with those obtained for Ac-l-Pro-NHMe at the same theoretical level allows us to draw the following conclusions:
Incorporation of the amino group into the β- or γ-position of the pyrrolidine ring reduces the intrinsically low conformational flexibility of conventional Pro. Specifically, the four Amp-containing dipeptides investigated in this work only show one energetically accessible minimum in the gas-phase, i.e. ΔEgp < 1.5 kcal/mol, when ω0 is arranged in trans. On the other hand, the stability of conformations with ω0 in cis is, in general, higher for Amp than for Pro. This is because some such conformations are stabilized by intramolecular hydrogen bonds in which the nitrogen of the amino substituent acts as acceptor.
In general the solvent enhances the stability of the conformers with ω0 in cis. Thus, the c-εL was the most stable conformation for Ac-βtAmp-NHMe, Ac-βcAmp-NHMe and Ac-γtAmp-NHMe in chloroform, methanol and aqueous solution, whereas for Ac-γtAmp-NHMe the most stable conformer in solution was the c-αL (chloroform) or the c-εL (methanol and water). However, PCM results must be analyzed with caution since this SCRF method overestimates the stability of the cis structures significantly, especially when protic solvents (as water or methanol) are considered.
Characterization of the minimum energy conformations of the four Dmp-containing peptides indicates that substitution of the amino by the dimethylamino reduces considerably the conformational restriction found in the N-acetyl-N′-methyl derivatives of Amp. This is indicated by both the increase in the number of minimum energy conformations and the decrease of relative conformational energies.
The stabilization of the structures with ω0 in cis in the Dmp-containing dipeptides is similar to that found for the Amp derivatives. Thus, this type of structures is the most favored in chloroform, methanol and aqueous solutions. However, the conformational preferences of the Dmp-containing dipeptides in the gas-phase and in carbon tetrachloride solution are identical.
A detailed energetic comparison of the dipeptides studied in this work indicates that the most favored Amp isomer in the gas-phase, chloroform, methanol and water solutions is the Ac-γcAmp-NHMe dipeptide. In contrast, Ac-γtDmp-NHMe is the most stable Dmp-containing dipeptide in the gas-phase, even although the Ac-γcDmp-NHMe is most favored isomer in solution, independently of the polarity of the environment.
Supplementary Material
Acknowledgments
Gratitude is expressed to the Centre de Supercomputació de Catalunya (CESCA) and to the Universitat de Lleida for computational facilities. Financial support from the Gobierno de Aragón (research group E40) and Ministerio de Educación y Ciencia– FEDER (project CTQ2004-5358) is gratefully acknowledged. This project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under contract number N01-CO-12400. The content of this publication does not necessarily reflect the view of the policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organization imply endorsement by the U.S. Government. This research was supported [in part] by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
Supporting Information: Structural and energy parameters obtained for the Pro-containing dipeptide at the B3LYP/6-31+G(d,p) level. Coordinates and energy of the minimum energy conformations characterized for all the Amp- and Dmp-containing dipeptides studied in this work. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Chakrabarti P, Pal D. Prog Biophys Mol Biol. 2001;76:1. doi: 10.1016/s0079-6107(01)00005-0. [DOI] [PubMed] [Google Scholar]; (b) Marraud M, Aubry A. Biopolymers. 1996;40:45. doi: 10.1002/(sici)1097-0282(1996)40:1<45::aid-bip3>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]; (c) MacArthur MW, Thornton JM. J Mol Biol. 1991;218:397. doi: 10.1016/0022-2836(91)90721-h. [DOI] [PubMed] [Google Scholar]; (d) Rose GD, Gierasch LM, Smith JA. Adv Protein Chem. 1985;37:1. doi: 10.1016/s0065-3233(08)60063-7. [DOI] [PubMed] [Google Scholar]
- 2.(a) Boussard G, Marraud M, Aubry A. Biopolymers. 1979;18:1297. [Google Scholar]; (b) Madison V, Kopple KD. J Am Chem Soc. 1980;102:4855. [Google Scholar]; (c) Liang GB, Rito CJ, Gellman SH. Biopolymers. 1992;32:293. doi: 10.1002/bip.360320309. [DOI] [PubMed] [Google Scholar]; (d) Beausoleil E, Lubell WD. J Am Chem Soc. 1996;118:12902. [Google Scholar]
- 3.(a) Kang YK. J Phys Chem B. 2006;110:21338. doi: 10.1021/jp0647481. [DOI] [PubMed] [Google Scholar]; (b) Kang YK, Byun BJ. J Phys Chem B. 2007;111:5377. doi: 10.1021/jp067826t. [DOI] [PubMed] [Google Scholar]; (c) Rivail JL, Bouchy A, Loos PF. J Arg Chem Soc. 2006;94:19. [Google Scholar]
- 4.(a) Dugave C, Demange L. Chem Rev. 2003;103:2475. doi: 10.1021/cr0104375. [DOI] [PubMed] [Google Scholar]; (b) Pal D, Chakrabarti P. J Mol Biol. 1999;294:271. doi: 10.1006/jmbi.1999.3217. [DOI] [PubMed] [Google Scholar]; (c) Stewart DE, Sarkar A, Wampler JE. J Mol Biol. 1990;214:253. doi: 10.1016/0022-2836(90)90159-J. [DOI] [PubMed] [Google Scholar]; (d) Grathwohl C, Wüthrich K. Biopolymers. 1981;20:2623. [Google Scholar]
- 5.Mac Arthur MW, Thornton JM. J Mol Biol. 1991;218:397. doi: 10.1016/0022-2836(91)90721-h. [DOI] [PubMed] [Google Scholar]
- 6.(a) Sapse AM, Mallah-Levy L, Daniels SD, Erickson BW. J Am Chem Soc. 1987;109:3526. [Google Scholar]; (b) Ramachandran GN. Int J Peptide Prot Res. 1988;31:1. [PubMed] [Google Scholar]
- 7.Andreotti AH. Biochemistry. 2003;42:9515. doi: 10.1021/bi0350710. [DOI] [PubMed] [Google Scholar]
- 8.(a) Wedemeyer WJ, Welker E, Scheraga HA. Biochemistry. 2002;41:14637. doi: 10.1021/bi020574b. [DOI] [PubMed] [Google Scholar]; (b) Dugave C, Demange L. Chem Rev. 2003;103:2475. doi: 10.1021/cr0104375. [DOI] [PubMed] [Google Scholar]
- 9.(a) Zimmerman SS, Pottle MS, Nemethy G, Scheraga HA. Macromolecules. 1977;10:1. doi: 10.1021/ma60055a001. [DOI] [PubMed] [Google Scholar]; (b) Fischer S, Dunbrack RL, Jr, Karplus M. J Am Chem Soc. 1994;116:11931. [Google Scholar]; (c) Kang YK. J Phys Chem. 1996;100:11589. [Google Scholar]; (d) Improta R, Benzi C, Barone V. J Am Chem Soc. 2001;123:12568. doi: 10.1021/ja010599i. [DOI] [PubMed] [Google Scholar]; (e) Benzi C, Improta R, Scalmani G, Barone V. J Comput Chem. 2002;23:341. doi: 10.1002/jcc.10015. [DOI] [PubMed] [Google Scholar]; (f) Kang YK. J Phys Chem B. 2002;106:2074. [Google Scholar]; (g) Hudáky I, Baldoni HA, Perczel A. J Mol Struct (THEOCHEM) 2002;582:233. [Google Scholar]; (h) Hudáky I, Perczel A. J Mol Struct (THEOCHEM) 2003;630:135. [Google Scholar]; (i) Allen WD, Czinki E, Császár AG. Chem Eur J. 2004;10:4512. doi: 10.1002/chem.200400112. [DOI] [PubMed] [Google Scholar]; (j) Kang YK, Park HS. J Mol Struct (Theochem) 2005;718:17. [Google Scholar]; (k) Czinki E, Csaszar AG. Chem Eur J. 2003;9:1008. doi: 10.1002/chem.200390103. [DOI] [PubMed] [Google Scholar]
- 10.Sahai MA, Kehoe AK, Koo JCP, Setiadi DH, Chass GA, Viskolcz B, Penke B, Pai EF, Csizmadia IG. J Phys Chem A. 2005;109:2660. doi: 10.1021/jp040594i. [DOI] [PubMed] [Google Scholar]
- 11.(a) Kang YK, Park HS.Biophys Chem 20051139315617814 [Google Scholar]; (b) Kang YK, Jhon JS, Park HS. J Phys Chem B. 2006;110:17645. doi: 10.1021/jp0629792. [DOI] [PubMed] [Google Scholar]; (c) Kang YK. J Mol Struct. 2004;675:37. [Google Scholar]; (e) Kang YK, Choi HY. Biophys Chem. 2004;111:135. doi: 10.1016/j.bpc.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 12.(a) Flores-Ortega A, Casanovas J, Zanuy D, Nussinov R, Alemán C. J Phys Chem B. 2007;111:5475. doi: 10.1021/jp0712001. [DOI] [PubMed] [Google Scholar]; (b) Flores-Ortega A, Jiménez AI, Cativiela C, Nussinov R, Alemán C, Casanovas J. J Org Chem. 2008;73:3418. doi: 10.1021/jo702710x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Benzi C, Improta R, Scalmani G, Barone V. J Comput Chem. 2002;23:341. doi: 10.1002/jcc.10015. [DOI] [PubMed] [Google Scholar]; (b) Lam JSW, Koo JCP, Hudáky I, Varro A, Papp JG, Penke B, Csizmadia IG. J Mol Struct (Theochem) 2003;666-667:285–289. [Google Scholar]
- 14.(a) Alemán C, Zanuy D, Jiménez AI, Cativiela C, Haspel N, Zheng J, Casanovas J, Wolfson H, Nussinov R. Phys Biol. 2006;3:S54. doi: 10.1088/1478-3975/3/1/S06. [DOI] [PubMed] [Google Scholar]; (b) Tsai CJ, Zheng J, Zanuy D, Haspel N, Wolfson H, Alemán C, Nussinov R. Proteins. 2007;68:1. doi: 10.1002/prot.21413. [DOI] [PubMed] [Google Scholar]
- 15.(a) Haspel N, Zanuy D, Alemán C, Wolfson H, Nussinov R. Structure. 2006;14:1137. doi: 10.1016/j.str.2006.05.016. [DOI] [PubMed] [Google Scholar]; (b) Zheng J, Zanuy D, Haspel N, Tsai CJ, Alemán C, Nussinov R. Biochemistry. 2007;46:1205. doi: 10.1021/bi061674a. [DOI] [PubMed] [Google Scholar]; (c) Zanuy D, Jiménez AI, Cativiela C, Nussinov R, Alemán C. J Phys Chem B. 2007;111:3236. doi: 10.1021/jp065025k. [DOI] [PubMed] [Google Scholar]
- 16.Zanuy D, Flores-Ortega A, Casanovas J, Curcó D, Nussinov R, Alemán C. J Phys Chem B. 2008;112:8692. doi: 10.1021/jp711477k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Porter EA, Wang X, Schmitt MA, Gellman SH. Org Lett. 2002;4:3317. doi: 10.1021/ol0266370. [DOI] [PubMed] [Google Scholar]; (b) Farrera-Sinfreu J, Zaccaro L, Vidal D, Salvatella X, Giralt E, Pons M, Albericio F, Royo M. J Am Chem Soc. 2004;126:6048. doi: 10.1021/ja0398621. [DOI] [PubMed] [Google Scholar]
- 18.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Vreven T, Jr, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, CStrain M, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03, Revision B.02. Gaussian, Inc.; Pittsburgh PA: 2003. [Google Scholar]
- 19.Becke AD. J Chem Phys. 1993;98:1372. [Google Scholar]
- 20.Lee C, Yang W, Parr RG. Phys Rev B. 1993;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 21.McLean AD, Chandler GS. J Chem Phys. 1980;72:5639. [Google Scholar]
- 22.(a) Alemán C, Jiménez AI, Cativiela C, Pérez JJ, Casanovas J. J Phys Chem B. 2002;106:11849. [Google Scholar]; (b) Casanovas J, Zanuy D, Nussinov R, Alemán C. Chem Phys Lett. 2006;429:528. doi: 10.1021/jp062804s. [DOI] [PubMed] [Google Scholar]; (c) Casanovas J, Jiménez AI, Cativiela C, Pérez JJ, Alemán C. J Phys Chem B. 2006;110:5762. doi: 10.1021/jp0542569. [DOI] [PubMed] [Google Scholar]
- 23.(a) Tomasi J, Mennucci B, Cammi R. Chem Rev. 2005;105:2999. doi: 10.1021/cr9904009. [DOI] [PubMed] [Google Scholar]; (b) Tomasi J, Persico M. Chem Rev. 1994;94:2027. [Google Scholar]; (c) Miertus S, Tomasi J. Chem Phys. 1982;65:239. [Google Scholar]; (d) Miertus M, Scrocco E, Tomasi J. Chem Phys. 1981;55:117. [Google Scholar]
- 24.Perczel A, Angyan JG, Kajtar M, Viviani W, Rivail JL, Marcoccia JF, Csizmadia IG. J Am Chem Soc. 1991;113:6256. [Google Scholar]
- 25.(a) Hudáky I, Perczel A. J Mol Struct (THEOCHEM) 2003;630:135. [Google Scholar]; (b) Hudáky I, Baldoni HA, Perczel A. J Mol Struct (THEOCHEM) 2002;582:233. [Google Scholar]
- 26.(a) Iribarren JI, Casanovas J, Zanuy D, Alemán C. Chem Phys. 2004;302:77. [Google Scholar]; (b) Jang YH, Goddard WA, III, Noyes KT, Sowers LC, Hwang S, Chung DS. J Phys Chem B. 2003;107:344. [Google Scholar]; (c) Hawkins GD, Cramer CJ, Truhlar DG. J Chem Phys B. 1998;102:3257. [Google Scholar]; (d) Orozco M, Luque FJ. J Am Chem Soc. 1995;117:1378. [Google Scholar]; (e) Alemán C, Navarro E, Puiggalí J. J Org Chem. 1995;60:6135. [Google Scholar]
- 27.(a) Assfeld X, Ruiz-Lopez MF, González J, Lopez R, Sordo JA, Tomas TL. J Comp Chem. 1994;15:479. [Google Scholar]; (b) Assfeld X, Ruiz-Lopez MF, García JI, Mayoral JA, Salvatella L. J Chem Soc Chem Comm. 1995;13:1371. [Google Scholar]
- 28.Delaney NG, Madison V. J Am Chem Soc. 1982;104:6635. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.