

Abstract
The main purpose of this study was to investigate whether consumption of diets enriched in antioxidants attenuates the level of oxidative stress in the senescence-accelerated mouse (SAM). In separate and independent studies, two different dietary mixtures, one enriched with vitamin E, vitamin C, L-carnitine, and lipoic acid (Diet I) and another diet including vitamins E and C and 13 additional ingredients containing micronutrients with bioflavonoids, polyphenols, and carotenoids (Diet II), were fed for 8 and 10 months, respectively. The amounts of glutathione (GSH) and glutathione disulfides (GSSG) and GSH:GSSG ratios were determined in plasma, tissue homogenates, and mitochondria isolated from five different tissues of SAM (P8) mice. Both diets had a reductive effect in plasma; however Diet I had relatively little effect on the glutathione redox status in tissue homogenates or mitochondria. Remarkably, Diet II caused a large increase in the amount of glutathione and a marked reductive shift in glutathione redox state in mitochondria. Overall, the effects of Diet II were tissue and gender specific. Results indicated that the glutathione redox state in mitochondria and tissues can be altered by supplemental intake of a relatively complex mixture of dietary antioxidants that contains substances known to induce phase 2 enzymes, glutathione, and antioxidant defenses. Whether corresponding attenuations occur in age-associated deleterious changes in physiological functions or life span remains unknown.
Keywords: Antioxidants, Senescence-accelerated mouse (SAM), Glutathione, Mitochondria, Aging, Oxidative stress, Redox status
Introduction
The concept that reactive oxygen species (ROS) are generated as products of oxygen metabolism in aerobic organisms is now firmly established. It is also well recognized that despite the existence of an array of enzymatic and nonenzymatic antioxidants, steady-state amounts of the products of ROS interaction with macromolecules such as proteins, lipids, and nucleic acid are detectable even in young, healthy animals. The presence of such oxidatively modified macromolecules is generally regarded to suggest that the intracellular antioxidant defenses are outmatched by the ROS fluxes, whereby cells normally exist under a certain level of oxidative stress.
Accumulation of oxidatively damaged macromolecules with age and in association with some pathological conditions such as neurodegeneration and atherosclerosis, among others, has been well documented in several reports [1–4]. Other studies have shown that: (i) there is an increase in rates of mitochondrial ROS generation with age [4,5]; (ii) the maximum life span of the species is inversely correlated with rates of ROS generation and steady-state amounts of oxidative damage [6,7]; and (iii) experimental regimens that extend life span of animals such as restriction of caloric intake also cause a decrease in the rates of mitochondrial ROS generation and steady-state amounts of macromolecular oxidative damage [5,7,8]. Prompted by such knowledge, many efforts have been made to experimentally lower the level of oxidative stress, ostensibly to postpone or retard the onset of age-related deleterious alterations and/or some pathologic conditions [4,9–11]. A frequently employed strategy is the supplementation of food with low molecular antioxidants. Although some studies have shown positive effects on life span or severity of certain disease conditions [12–17], in other cases, beneficial effects were absent [4,9–11,18,19]. Such results have often been cited by critics to suggest that oxidative stress may be a consequence rather than a cause of aging or the pathological impairments.
It would seem that the issue of the efficacy of antioxidant supplementation cannot be properly resolved without addressing the antecedent question of whether or not intake of low molecular weight antioxidants indeed causes a lowering of the level of oxidative stress. Accordingly, the main purpose of this study was to determine whether long-term dietary supplementation with antioxidants affects indices of oxidative stress.
Although there is currrently no broad consensus about the availability of a reliable biochemical indicator of the physiological level of oxidative stress, there is increasing recognition that redox states, expressed by the amounts of glutathione (GSH) and glutathione disulfide (GSSG) and the ratio of GSH:GSSG, are sensitive and reliable measures of the overall level of oxidative stress [20–22]. Vina and colleagues were among the first to highlight relationship between mitochondria, aging and dietary intervention [23,24]. These early studies demonstrated that glutathione levels decreased and the glutathione redox state was more oxidized in mitochondria during aging. Gingko biloba extract EGb 761, which contains ∼24% bioflavonoids, reversed these age-related changes in mitochondrial glutathione [23,24]. Furthermore, it has been demonstrated that the glutathione redox state in humans, rats, mice, and fruit flies becomes more pro-oxidizing during aging, especially in mitochondria [25–29].
In this context, in the present study the effects of long-term dietary supplementation with two different mixtures of antioxidants or substances known to induce antioxidant activity on glutathione redox state were determined in homogenates and mitochondria of liver, heart, brain, hind limb skeletal muscle, and kidney of the senescence-accelerated mouse (SAM). The rationale for using mixtures is that the mechanisms and loci of action of different antioxidants vary and they interact in a redox network and thus a mixture is more likely to have a stronger effect on antioxidant defenses [30–32]. The SAM mouse has been shown to exhibit a relatively higher level oxidative stress and has an approximately 30% shorter life than the longer lived strain of mice such as C57BL/6 [33–36].
Materials and methods
Reagents
GSH, GSSG, and cysteine were obtained from Sigma Chemical Co. (St Louis, MO) and used as calibration standards. Acetonitrile, meta-phosphoric acid, and 1-octane sulfonic acid were from EM Science (Gibbstown, NJ). Deionized water was prepared using a Millipore Milli-Q System. All other chemicals were HPLC grade or of the highest purity available.
Animals
The colony of SAM (P8) mice was originally obtained from the Council for SAM Research, Kyoto, Japan and maintained at University of California, Berkley, and subsequent to 2000 was maintained at the University of Southern California. Mice were fed ad libitum on the specified diet and kept under standard vivarium conditions with appropriate supervision of resident veterinarians. Food was removed on the night before experiments. All procedures were approved by the animal use and care committee at the University of Southern California. Two separate experiments were conducted each with an independent control group. In one, the mice were fed a diet supplemented with vitamins E and C, carnitine, and lipoic acid (Table 1) for 8 month, i.e., from 5 to 13 months of age. In the other, mice were fed a more complex diet supplemented with several purified and natural substances (Table 1) for 10 month starting at 7 months of age. It should be made explicit that due to variations in age of mice and duration of dietary intake, the present experimental design does not permit a direct comparison of the effects of these two dietary mixtures.
Table 1.
Composition of control, Diet I, and Diet IIa
| Component | Control | Diet I | Diet II |
|---|---|---|---|
| Protein | 170 | 170 | 190 |
| Fat | 100 | 100 | 100 |
| Vitamin E | 200 ppmb | 500 ppmb | 500 ppmb |
| Vitamin C | <32 ppm | 80 ppmb | 80 ppmb |
| L-Carnitine | 10 ppmb | 300 ppmb | – |
| Lipoic acid | –e | 125 ppm | – |
| ±Broccolic | – | – | 15 |
| ±Rice branc | – | – | 10 |
| Marine oil | – | – | 8.8 |
| Glutamine dipeptided | – | – | 5 |
| Methionined | – | – | 1.7 |
| ±Selenium-yeasta | – | – | 0.3 |
| ±Algaea | – | – | 0.25 |
| L-Threonined | – | – | 0.25 |
| Lutein (5%) | – | – | 0.15 |
| Lycopene (5%) | – | – | 0.15 |
| Astaxanthin (8%) | – | – | 0.094 |
| β-Carotene (10%) | – | – | 0.075 |
| Curcurmin | – | – | 0.05 |
Components are expressed as g/kg diet.
Approximately.
General source of micronutrients and phytochemical antioxidants (as bioflavonoids and other polyphenols, carotenoids, vitamins C and E, lipoic acid, etc.).
Precursors for endogenous antioxidants.
None added.
Preparation of tissue homogenates and isolation of mitochondria
Liver, kidney, brain, heart, and skeletal muscle were quickly removed and placed in ice-cold buffer containing 50 mmol/L potassium phosphate buffer, pH 7.4, 2 mmol/L EDTA, and 0.1 mmol/L butylated hydroxytoluene). EDTA-containing blood (50 μl of 100 mmol/L EDTA solution for each ml of blood) was centrifuged at 2000g for 3 min to separate serum. Tissue homogenization procedures and buffers used for isolation of mitochondria were identical to those described previously [28,36]. Mitochondria from all tissues were isolated within 1–2 h after tissue dissection, except those from brain, which required a longer isolation time (up to 3 h) due to additional steps involving Percoll gradient centrifugation [37].
Sample preparation for glutathione analysis
Immediately after homogenization of the tissues, 200-μl aliquots of the crude homogenates were mixed with 200 μl of ice-cold 10% (w/v) meta-phosphoric acid, incubated for 30 min on ice, and centrifuged for 20 min at 14,000g at 4°C. Supernatants were transferred to autosample vials and either injected immediately or stored at −80°C until analysis. Storage of deproteinized tissue samples in 5% (w/v) MPA at −80°C for up to 6 months was found to have no effect on the concentrations of GSH and GSSG.
For measurements of mitochondrial glutathione content, aliquots (150–200 μl) of fresh mitochondrial preparations were washed once with a 100-fold excess of buffer and pelleted by centrifugation in order to remove any extra-mitochondrial GSH and GSSG. Mitochondria were then resuspended in 200 μl of 5 % (w/v) meta-phosphoric acid. After 20 min of incubation on ice, samples were centrifuged for 20 min at 18,000g at 4°C. Supernatants were transferred into autosample vials and immediately used for injections or stored at −80°C until analysis.
The protein content of homogenates and mitochondria was determined by the BCA protein assay, according to the manufacturer's instructions (Pierce, Rockford, IL).
HPLC-coulometric EC detection of aminothiols
The procedures for the measurement of GSH and GSSG and other aminothiols are described in detail in previous reports from this laboratory [28,29,36]. It was pointed out that technical problems associated with sample origin, preparation, and handling, together with analytical methodology employed, can potentially cause errors in the quantification of GSH/GSSG, and is also well recognized in the literature. A particular procedural hazard is the artificial oxidation of GSH to GSSG. The methodology used by us previously and in the present study resulted in minimal GSH oxidation and the GSH:GSSG ratios obtained are among the highest published values [22,28,39].
Briefly, aminothiol compounds (GSH, GSSG, cysteine, and methionine) were separated by HPLC, equipped with a Shimadzu Class VP solvent delivery system, using a reverse-phase C18 Luna (II) column (3 μm; 4.6 × 150 mm), obtained from Phenomenex (Torrance, CA). The mobile phase for isocratic elution consisted of 25 mmol/L monobasic sodium phosphate, 0.5 mmol/L of the ion-pairing agent 1-octane sulfonic acid, 2% (v/v) acetonitrile, pH 2.7, adjusted with 85% phosphoric acid. The flow rate was 0.7 ml/min. Under these conditions, the separation of aminothiols was completed in 35 min; cysteine was the first and GSSG was the last eluting peak, with retention times of 5 and 30 min, respectively. Deproteinated samples were injected directly onto the column using a Shimadzu autosampler. Calibration standards were prepared by dilution of 2 mmol/L stock solutions of cysteine, GSH, or GSSG in 5% (w/v) meta-phosphoric acid, and were injected at regular intervals to ensure uniform standardization. Each sample was injected twice, and the average of the peak areas was used for calculations of the aminothiol concentrations. Following HPLC separation, aminothiols were detected with a Model 5600 CoulArray electrochemical detector (ESA, Inc., Chelmsford, MA), equipped with a four-channel analytical cell. Increasing potentials of +400, +600, +750, and +875 mV were applied on channels 1–4, respectively. Cysteine and GSH were detected on channel 3 (+750 mV), whereas GSSG was detected on channel 4 (+875 mV). Low potentials on channels 1 (+400 mV) and 2 (+600 mV) were used as an oxidative screen to eliminate interfering compounds that oxidize at a lower voltage than the GSH and GSSG. Redox potentials of the glutathione redox couple (2GSH → GSSG + 2e− + 2H+) in mitochondria were calculated by using experimentally determined concentrations of GSH and GSSG in the Nernst equation [28].
Statistics
Statistical significance was determined by Student's t tests. P < 0.05 was considered to be significant.
Results
Life span
As recently reported [36] the average life span of male SAM(P8) mice was 17.8 ± 4.3 months and the age at which 90% mortality had occurred was 23 months.
Effects of Diet I on glutathione redox state
Diet I (Table 1) was administered to mice starting at 5 months of age until 13 months of age, when the amounts of GSH and GSSG were measured in plasma and in homogenates and mitochondria from five different tissues, liver, kidney, heart, brain, and skeletal muscle of male mice. In addition, the concentrations of cysteine and methionine were determined in blood serum.
Serum
Administration of Diet I had no effect on GSH content; however, GSSG concentration was decreased and consequently the GSH:GSSG ratio was elevated. The supplemented mice showed a 2.6-fold increase in cysteine and a 20% decrease in methionine content as compared to the controls (Fig. 1).
Fig. 1.
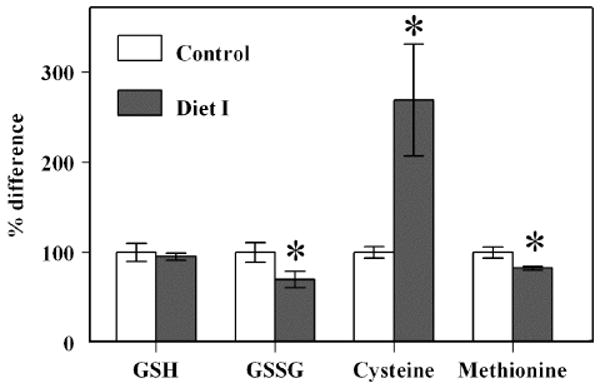
Content of aminothiols in blood serum of SAM mice fed with Diet I. Percentage differences in concentrations of GSH, GSSG, cysteine, and blood serum of control (n = 5) and experimental (n = 4) SAM male mice fed Diet I. The absolute concentrations of GSH, GSSG, cysteine, and methionine in blood serum of the control SAM mouse were 1.25 ± 0.13, 0.102 ± 0.011, 1.99 ± 0.12, and 42.85 ± 2.61 μmol/L, respectively. *A significant difference (P < 0.05) based on unpaired t test.
Tissue homogenates
In control mice, there were approximately 10- and 4-fold variations in GSH and GSSG concentrations, respectively, among different tissues (Table 2). The rank order of GSH content was liver > brain > heart > skeletal muscle > kidney, and GSSG amounts were heart > liver = kidney > brain > skeletal muscle. Neither the GSSG amount nor the GSH:GSSG ratio were correlated with the GSH concentration in the corresponding tissue. Such interorgan variations in GSH and GSSG concentrations suggest the existence of highly varied glutathione redox status of different tissues.
Table 2.
Concentrations of GSH and GSSG in homogenates of different tissues of SAM mice fed Diet I
| Tissues | GSH (nmol/mg protein) | GSSG (nmol/mg protein) |
|---|---|---|
| Liver | ||
| Control | 25.23 ± 2.12 | 0.179 ± 0.058 |
| Diet I | 26.51 ± 2.12 | 0.149 ± 0.036 |
| Kidney | ||
| Control | 1.44 ± 0.26 | 0.179 ± 0.023 |
| Diet I | 1.60 ± 0.26 | 0.161 ± 0.008 |
| Heart | ||
| Control | 6.26 ± 0.39 | 0.28 ± 0.09 |
| Diet I | 8.59 ± 0.47a | 0.17 ± 0.03 |
| Brain | ||
| Control | 12.58 ± 2.22 | 0.098 ± 0.02 |
| Diet I | 14.27 ± 1.16 | 0.108 ± 0.03 |
| Skeletal muscle | ||
| Control | 2.62 ± 0.17 | 0.077 ± 0.007 |
| Diet I | 2.57 ± 0.09 | 0.074 ± 0.002 |
Significant difference (P < 0.05) between control (n = 5) and experimental (n = 4) mice.
Diet I administration enhanced GSH content only in the heart homogenate, where it elevated the GSH:GSSG ratio from 26 ± 10 to 53 ± 9 (P < 0.005) (Fig. 2). There was no effect on GSSG content or GSH:GSSG ratio in other tissues examined. Thus, the effects of the diet on GSH and GSSG content observed in the serum were not apparent in the homogenates of most of the organs.
Fig. 2.
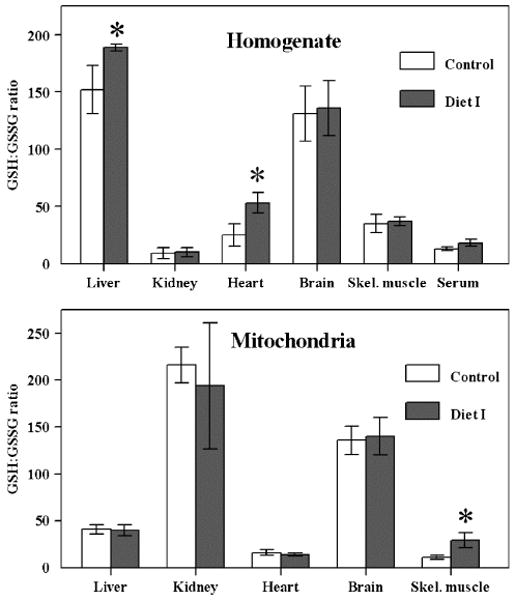
GSH:GSSG ratio in tissue homogenates and mitochondria from SAM mice fed with Diet I. GSH:GSSG ratio in homogenates (top panel) and mitochondria (bottom panel) of liver, kidney, heart, brain, skeletal muscle, and blood serum of control (n = 5) and experimental (n = 4) male SAM mice fed Diet I. *A significant difference (P < 0.05) based on unpaired t test.
Mitochondria
Diet I intake had no effect on mitochondrial GSH content in any of the tissues (Table 3). The GSSG concentration decreased only in the skeletal muscle, which consequently elevated the GSH:GSSG ratio.
Table 3.
Concentrations of GSH, GSSG, and total glutathione (GSH + GSSG) in mitochondria of SAM mice fed with Diet I
| Tissue | GSH (nmol/mg protein) |
GSSG (nmol/mg protein) |
GSH + GSSG (nmol/mg protein) |
Total glutathione (% change) |
|---|---|---|---|---|
| Liver | ||||
| Control | 7.17 ± 0.48 | 0.176 ± 0.025 | 7.25 ± 0.50 | |
| Diet I | 7.21 ± 0.24 | 0.182 ± 0.027 | 7.40 ± 0.27 | NSa |
| Kidney | ||||
| Control | 1.79 ± 0.15 | 0.008 ± 0.002 | 1.80 ± 0.15 | |
| Diet I | 1.88 ± 0.27 | 0.010 ± 0.002 | 1.89 ± 0.27 | NS |
| Heart | ||||
| Control | 4.54 ± 0.33 | 0.298 ± 0.039 | 4.84 ± 0.37 | |
| Diet I | 4.03 ± 0.45 | 0.281 ± 0.052 | 4.31 ± 0.52 | NS |
| Brain | ||||
| Control | 4.22 ± 0.25 | 0.031 ± 0.005 | 4.25 ± 0.25 | |
| Diet I | 4.29 ± 0.23 | 0.031 ± 0.004 | 4.32 ± 0.23 | NS |
| Skeletal muscle | ||||
| Control | 1.12 ± 0.09 | 0.102 ± 0.012 | 1.22 ± 0.10 | |
| Diet I | 1.12 ± 0.10 | 0.040 ± 0.007b | 1.16S ± 0.11 | NS |
NS, not significant.
Significant difference (P < 0.05) between control (n = 5) and experimental (n = 4) mice.
Effect of Diet II on glutathione redox state
Mice were fed Diet II starting at 7 months of age until 17 months of age, when concentrations of GSH and GSSG together with serum cysteine and methionine levels were measured in liver, kidney, heart, brain, and skeletal muscle of male and female control and experimental mice. In general, there were no significant differences between the male and the emale controls in the amount of GSH, GSSG, serum cysteine, or methionine in any of the tissues or their mitochondria; however, there were some notable sex-related differences in the effects of Diet II administration on GSH and GSSG content.
Serum
In both sexes Diet II intake resulted in a significant increase in plasma GSH content and a decrease in GSSG amount, thereby enhancing the GSH:GSSG ratio (Fig. 3). Compared to controls the methionine content was higher in experimental male mice while cysteine amount was not affected.
Fig. 3.
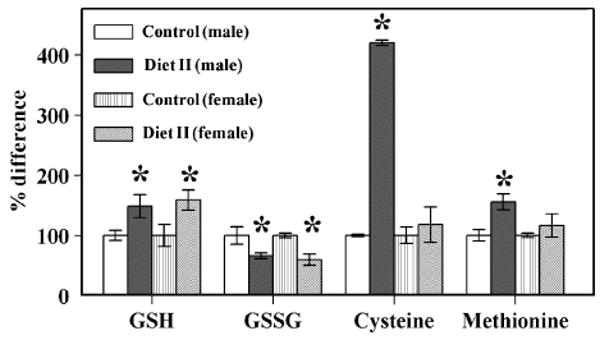
Content of aminothiols in blood serum of SAM mice fed with Diet II. Percentage differences in concentrations of GSH, GSSG, cysteine, and methionine status in blood serum of control male, control female, experimental male, and experimental female SAM mice fed Diet II (n = 4 for each group). The absolute concentrations of GSH, GSSG, cysteine, and methionine in blood serum of the control male SAM mouse were 0.45 ± 0.04, 0.145 ± 0.021, 3.39 ± 0.07, and 48.1 ± 4.6, respectively. The absolute concentrations of GSH, GSSG, cysteine, and methionine in blood serum of the control female SAM mouse were 0.49 ± 0.09, 0.125 ± 0.005, 4.42 ± 0.61, and 40.8 ± 1.4, respectively. *A significant difference (P < 0.05) based on paired t test.
Tissue homogenates
Diet II intake had only a limited effect on GSH and GSSG content (Table 4). For instance, in liver, there was no effect on GSH or GSSG content in males or females; in kidney, there was an elevation in GSH and a decrease in GSSG amount in males only; in heart, there was no effect; in brain, the GSH content was elevated and GSSG was decreased in females only; and in skeletal muscle, there was elevation in GSH and a decrease in GSSG amount in males only.
Table 4.
Concentrations of GSH and GSSG in tissue homogenates of and female SAM mice fed Diet II
| Tissues | GSH (nmol/mg protein) | GSSG (nmol/mg protein) |
|---|---|---|
| Liver | ||
| Control (male) | 19.66 ± 2.75 | 0.214 ± 0.021 |
| Diet II (male) | 20.93 ± 2.71 | 0.199 ± 0.015 |
| Control (female) | 19.74 ± 2.58 | 0.170 ± 0.017 |
| Diet II (female) | 19.53 ± 0.53 | 0.148 ± 0.008 |
| Kidney | ||
| Control (male) | 1.96 ± 0.10 | 0.176 ± 0.023 |
| Diet II (male) | 2.32 ± 0.17a | 0.113 ± 0.012a |
| Control (female) | 2.05 ± 0.15 | 0.192 ± 0.024 |
| Diet II (female) | 2.53 ± 0.30 | 0.194 ± 0.018 |
| Heart | ||
| Control (male) | 7.63 ± 0.39 | 0.368 ± 0.039 |
| Diet II (male) | 8.70 ± 0.90 | 0.439 ± 0.045 |
| Control (female) | 9.16 ± 0.55 | 0.294 ± 0.016 |
| Diet II (female) | 8.25 ± 0.94 | 0.310 ± 0.024 |
| Brain | ||
| Control (male) | 12.97 ± 0.64 | 0.141 ± 0.020 |
| Diet II (male) | 13.10 ± 0.48 | 0.165 ± 0.018 |
| Control (female) | 13.06 ± 0.62 | 0.207 ± 0.015 |
| Diet II (female) | 14.46 ± 0.39a | 0.188 ± 0.011 |
| Skeletal muscle | ||
| Control (male) | 1.64 ± 0.11 | 0.084 ± 0.007 |
| Diet II (male) | 1.88 ± 0.26 | 0.043 ± 0.008a |
| Control (female) | 1.36 ± 0.05 | 0.055 ± 0.007 |
| Diet II (female) | 1.30 ± 0.12 | 0.058 ± 0.006 |
Significant difference (P < 0.05, n = 4) between control and experimental groups of SAM mice.
Mitochondria
The amounts of mitochondrial GSH and GSSG were relatively more widely affected in response to Diet II intake than in the tissue homogenates (Table 5). In both sexes, in comparison to the controls, mitochondrial GSH levels were enhanced in all the tissues of the experimental animals except the liver. Compared to controls, the amounts of mitochondrial GSSG were lower in all the tissues of the female experimental mice whereas in male experimental mice this effect was observed only in heart and brain. Thus, in comparison to controls, the mitochondrial GSH:GSSG ratio was elevated in all the tissues of the females, and only in heart and brain of males (Fig. 4).
Table 5.
Concentrations of GSH, GSSG, and total glutathione (GSH + GSSG) in mitochondria of SAM mice fed Diet II
| Gender/tissues | GSH (nmol/mg protein) |
GSSG (nmol/mg protein) |
GSH + GSSG (nmol/mg protein) |
Total glutathione (% of change) |
|---|---|---|---|---|
| Male | ||||
| Liver | ||||
| Control | 6.10 ± 0.07 | 0.175 ± 0.019 | 6.27 ± 0.09 | |
| Diet II | 5.65 ± 0.36 | 0.162 ± 0.019 | 5.81 ± 0.38 | NSa |
| Kidney | ||||
| Control | 2.23 ± 0.27 | 0.021 ± 0.004 | 2.25 ± 0.27 | |
| Diet II | 2.86 ± 0.27b | 0.018 ± 0.002 | 2.88 ± 0.27b | 28% |
| Heart | ||||
| Control | 4.25 ± 0.45 | 0.322 ± 0.009 | 4.57 ± 0.46 | |
| Diet II | 7.09 ± 0.44b | 0.290 ± 0.020b | 7.38 ± 0.46b | 61% |
| Brain | ||||
| Control | 4.86 ± 0.15 | 0.059 ± 0.001 | 4.92 ± 0.15 | |
| Diet II | 6.02 ± 0.32b | 0.030 ± 0.001b | 6.05 ± 0.32b | 23% |
| Skeletal muscle | ||||
| Control | 1.76 ± 0.10 | 0.113 ± 0.021 | 1.87 ± 0.12 | |
| Diet II | 2.49 ± 0.40b | 0.125 ± 0.027 | 2.61 ± 0.43b | 40% |
| Female | ||||
| Liver | ||||
| Control | 6.20 ± 0.53 | 0.428 ± 0.063 | 6.63 ± 0.59 | |
| Diet II | 6.74 ± 0.35 | 0.175 ± 0.024b | 6.91 ± 0.37 | NS |
| Kidney | ||||
| Control | 2.42 ± 0.48 | 0.021 ± 0.001 | 2.44 ± 0.48 | |
| Diet II | 3.72 ± 0.20b | 0.012 ± 0.001b | 3.73 ± 0.20b | 53% |
| Heart | ||||
| Control | 4.08 ± 0.28 | 0.349 ± 0.011 | 4.43 ± 30.29 | |
| Diet II | 4.55 ± 0.44 | 0.319 ± 0.002b | 4.87 ± 0.44b | 10% |
| Brain | ||||
| Control | 5.29 ± 0.26 | 0.034 ± 0.002 | 5.32 ± 0.26 | |
| Diet II | 6.23 ± 0.20b | 0.027 ± 0.001b | 6.50 ± 0.20b | 22% |
| Skeletal muscle | ||||
| Control | 1.80 ± 0.06 | 0.147 ± 0.016 | 1.95 ± 0.08 | |
| Diet | 2.43 ± 0.39b | 0.098 ± 0.010b | 2.53 ± 0.40b | 30% |
NS, not significant.
Significant difference (P < 0.05, n = 4) between control and experimental groups of SAM mice.
Fig. 4.
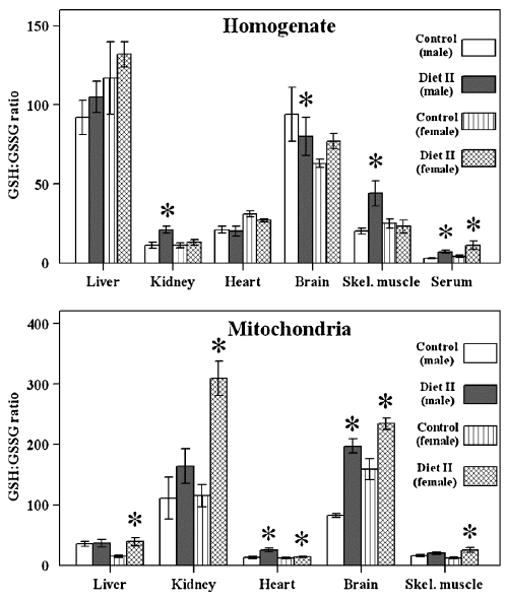
GSH:GSSG ratio in tissue homogenates and mitochondria from SAM mice fed with Diet II. GSH:GSSG ratio in homogenates (top panel) and mitochondria (bottom panel) of liver, kidney, heart, brain, skeletal muscle, and blood serum of control male, control female, experimental male, and experimental female SAM mice fed Diet II (n = 4 for each group). *A significant difference (P < 0.05) based on paired t test.
Effect of Diets I and II on mitochondrial glutathione redox potential
Diet I administration has no affect on the mitochondrial glutathione redox potential in most of the tissues examined, except heart, where it shifted from −208.4 ± 10.7 to −225.9 ± 5.8 (P < 0.05) (Fig. 5A). Compared to controls, the mitochondrial glutathione redox potential in Diet II administered mice was significantly (P < 0.005) more pro-reducing in the liver, kidney, brain, and skeletal muscle of female mice and in heart, brain, and skeletal muscle of male mice (Fig. 5B).
Fig. 5.
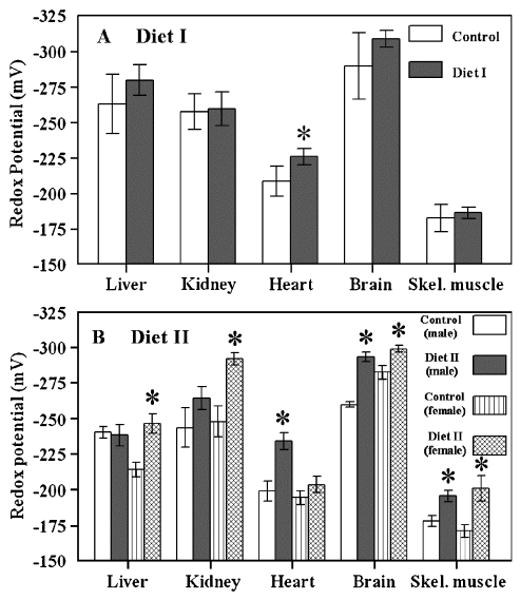
Glutathione redox potentials in mitochondria isolated from liver, kidney, heart, brain, and skeletal muscle of Sam mice fed Diet I (A) or Diet II (B). Redox values were calculated using the Nernst equation and the experimentally determined concentrations of mitochondrial GSH and GSSG, as described in Ref. [23]. All values represent the mean ± standard deviation of n = 4. *A significant difference (P < 0.05) based on paired t test.
Discussion
Glutathione redox state
Based on the finding that the GSH:GSSG ratio varies in different tissues and even within different intracellular compartments, it can be inferred that characteristic redox states exist in different tissues. Intertissue variations in GSH and GSSG amounts observed in this study can be reasonably assumed to stem from corresponding differences in some of the factors that modulate their relative amounts. For instance, GSH levels in cells are primarily dependent upon the rates of biosynthesis and utilization in oxidation/reduction reactions. Amounts of the precursor amino acids such as cysteine and the activity of the enzyme glutamate-cysteine ligase are the key factors affecting GSH synthesis. Whereas depletion of GSH occurs due to its involvement in a variety of reactions requiring reducing equivalents, including direct interaction with various radical species, enzymatic reduction of hydroperoxides, metabolism of xenobiotics, regeneration of reduced forms of redox pairs such as cysteine/cystine, NADPH/NADP+, and thioredoxinred/thioredoxinox, and protection/regulation of proteins occur by forming thiol disulfide bonds with cysteinyl residues [20–22,38–40]. Significant interorgan differences in amounts of glutamate-cysteine ligase activity, rates of mitochondrial generation, and activities of various enzymatic and nonenzymatic antioxidative defenses are indeed well documented [8,41,42].
Dietary modification of glutathione redox state
Results demonstrating significantly enhanced GSH:GSSG ratios were observed in tissue homogenates and mitochondria of SAM mice fed with Diet II. This diet is enriched with vitamins C and E, botanical and yeast extracts containing micronutrients, sulphorophane (an isothiocyntate derived from hydrolysis of glucaphanin), polyphenols, and bioflavonoids, plus four carotenoids and curcumin. These phytochemicals are known to induce phase 2 antioxidant enzymes and thus to increase the biosynthesis of antioxidant and detoxification enzymes and major cellular antioxidants especially glutathione [43]. Isothiocyanates as sulphorophane from broccoli and curcumin from tumeric are among the best documented phase 2 enzyme inducers with anticancer properties whose molecular mechanisms have been extensively investigated [44–46].
Diet II caused significant alterations in glutathione redox state and interestingly the effects were tissue and sex specific. For instance, Diet II caused an increase in serum cysteine concentration in females but not in males, and GSH elevation occurred in homogenates of only kidney and skeletal muscle of males and the brain of females. The decrease in mitochondrial GSSG content and increase in redox potential in response to Diet II intake occurred in more tissues of females than in males. Burger and Promislow [47] have recently listed a number of instances where males and females responded differently to experimental interventions that affect longevity. Although the genetic basis of this phenomenon is not well understood, differences in nutrient needs, hormonal distribution, and expression of X-linked and Y-linked genes could be some of the underlying factors. Notwithstanding, the present results emphasize that effects of consumption of an antioxidant-enriched diet observed in one gender cannot a priori be extrapolated to the other gender.
Vitamins E and C and lipoic acid, and carnitine-enriched Diet I, have been demonstrated in a variety of studies to counteract experimental oxidative stress [48,49]. However, an effect on the glutathione redox state, a sensitive indicator of oxidative stress, was not observed in most of the tissues under normal physiological conditions. One explanation of the lack of effects is the low concentrations of these ingredients included in the diet. The reason why Diet II is particularly effective in altering the glutathione redox state may be that it contains a variety of the antioxidants, which would permit theoretically quenching of a wide array of radical species, with the overall effect of lowering the oxidant load and inducing antioxidant defense mechanisms. In addition, curcumin and sulphorophanes in Diet II can regulate genes of phase 2 enzymes as well as have various effects on signaling. The effectiveness of Diet II can be also due to a variety of antioxidants and micronutrients in botanical extracts. It is also noteworthy that while both Diet I and Diet II had a significant effect on GSH:GSSG ratio in the plasma, their effects differed in different tissues. For instance, Diet I had no effect on most of the tissue homogenates and mitochondria. Changes in plasma redox state do not necessarily reflect those occurring in the tissues. It therefore seems essential that effects of antioxidant interventions should be verified by sampling the redox state in multiple tissues.
The redox state of cells is known to exert profound influence on cellular functions. For instance, the obvious effects of depletion of GSH would be an increase in the production of ROS and a decrease in the glutathione S-transferase-mediated elimination of electrophilic xenobiotics and some of the end-products of lipid peroxidation. Alterations in glutathione redox state can initiate activation/inactivation of many redox-sensitive proteins belonging to different functional categories such as signal transduction, enzyme catalysis, transcription factors, growth factors, among others (reviewed in [25,50–52]).
Glutathione redox state has been demonstrated to become exponentially more pro-oxidizing during aging in diverse species such as Drosophila, mouse, and man, suggesting that aging may be associated with a progressive shift in the redox set point [26–29]. Whether the reductive shift induced by interventions, such as Diet II, did indeed modify the aging process cannot be determined on the basis of the present study. Notwithstanding, improvements in cognitive and/or motor performance have been reported in experimental animals in response to intake of antioxidant mixtures [12–17]. Diets enriched with blueberry and spinach extracts, among others, have been demonstrated to enhance the performance of rats in tests of cognition [53]. Comprehensive studies in dogs have also shown that a diet enriched in both antioxidants and mitochondrial cofactors, such as α-lipoic acid and L-carnitine, can retard age-associated decline in spatial memory [18,19]. However, such ameliorative effects are usually observed in older and not in young healthy animals, suggesting that redox effects of exogenous antioxidants are more effective under conditions of elevated oxidative stress, thereby reflecting an enhanced requirement for antioxidants by the aged animals.
Conclusion
The main findings of this study are that (a) there is a several-fold variation in GSH and GSSG content and glutathione redox potential among different tissues of the mouse; (b) neither mitochondrial GSH and GSSG content nor GSH:GSSG ratio were correlated with those in the corresponding tissue homogenate; (c) intake of Diet I had a relatively little overall effect on GSH:GSSG ratio and redox potential, whereas Diet II caused a widespread reductive shift, notably in mitochondria; (d) effects of Diet II intake on glutathione were tissue and gender specific; (e) changes in GSH and GSSG content in response to antioxidant intake in the plasma do not correspond to those in the tissues.
Acknowledgments
This research was supported by the National Institute on Aging-National Institutes of Health (Grant RO1AG 13563) and the Hill's Pet Nutrition, Inc., Topeka, Kansas.
Abbreviations
- GSH
reduced glutathione
- GSSG
oxidized glutatione or glutathione disulfide
- ROS
reactive oxygen species
- SAM
senescence-accelerated mouse
References
- 1.Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 2.Beckman KB, Ames BN. The free radical theory of aging matures. Physiol Rev. 1998;78:547–581. doi: 10.1152/physrev.1998.78.2.547. [DOI] [PubMed] [Google Scholar]
- 3.Levine RL, Stadtman ER. Oxidative modification of proteins during aging. Exp Gerontol. 2001;36:1495–1502. doi: 10.1016/s0531-5565(01)00135-8. [DOI] [PubMed] [Google Scholar]
- 4.Sohal RS, Mockett RJ, Orr WC. Mechanisms of aging: an appraisal of the oxidative stress hypothesis. Free Radic Biol Med. 2002;33:575–586. doi: 10.1016/s0891-5849(02)00886-9. [DOI] [PubMed] [Google Scholar]
- 5.Sohal RS, Ku HH, Agarwal S, Forster MJ, Lal H. Oxidative damage, mitochondrial oxidant generation, and antioxidant defenses during aging and in response to food restriction in the mouse. Mech Ageing Dev. 1994;74:121–137. doi: 10.1016/0047-6374(94)90104-x. [DOI] [PubMed] [Google Scholar]
- 6.Ku HH, Brunk UT, Sohal TS. Relationship between mitochondrial superoxide and hydrogen peroxide production and longevity of mammalian species. Free Radic Biol Med. 1994;15:621–627. doi: 10.1016/0891-5849(93)90165-q. [DOI] [PubMed] [Google Scholar]
- 7.Barja G. Rate of generation of oxidative stress-related damage on animal longevity. Free Radic Biol Med. 2002;33:1167–1172. doi: 10.1016/s0891-5849(02)00910-3. [DOI] [PubMed] [Google Scholar]
- 8.Sohal RS, Weindruch R. Oxidative stress, caloric restriction, and aging. Science. 1996;273:59–63. doi: 10.1126/science.273.5271.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kohn RR. Effects of antioxidants on the life span of C57BL mice. J Gerontol. 1971;26:378–380. doi: 10.1093/geronj/26.3.378. [DOI] [PubMed] [Google Scholar]
- 10.Bayne ACV, Sohal RS. Effects of superoxide dismutase/catalase mimetics on life span and oxidative stress resistance in the housefly, musca domestica. Free Radic Biol Med. 2002;32:1229–1234. doi: 10.1016/s0891-5849(02)00849-3. [DOI] [PubMed] [Google Scholar]
- 11.Keaney M, Matthijssens F, Sharpe M, Vabfleteren J, Gems D. Superoxide dismutase mimetics elevate superoxide dismutase activity in vivo but do not retard aging in the nematode Caenorhabditis elegans. Free Radic Biol Med. 2004;37:239–250. doi: 10.1016/j.freeradbiomed.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 12.Joseph JA, Shukitt-Hale B, Denisova NA, Prior RL, Cao G, Martin A, Taglialatela G, Bickford PC. Long-term dietary strawberry, spinach, or vitamin E supplementation retards the onset of age-related neuronal signal-transduction and cognitive behavioral deficits. J Neurosci. 1998;18:8047–8055. doi: 10.1523/JNEUROSCI.18-19-08047.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shukitt-Hale B, Smith DE, Meydani M, Joseph JA. The effects of dietary antioxidants on psychomotor performance in aged mice. Exp Gerontol. 1999;34:797–808. doi: 10.1016/s0531-5565(99)00039-x. [DOI] [PubMed] [Google Scholar]
- 14.Milgram NW, Zicker SC, Head E, Muggenburg BA, Murphey H, Ikeda-Douglas CJ, Cotman CW. Dietary enrichment counteracts age-associated cognitive dysfunction in canines. Neurobiol Aging. 2002;32:737–745. doi: 10.1016/s0197-4580(02)00020-9. [DOI] [PubMed] [Google Scholar]
- 15.Bickford PC, Gould T, Briederick L. Antioxidant-rich diets improve cerebellar physiology and motor learning in aged rats. Brain Res. 2000;866:211–217. doi: 10.1016/s0006-8993(00)02280-0. [DOI] [PubMed] [Google Scholar]
- 16.Socci DJC, Crandall BM, Arendash GW. Chronic antioxidant treatment improves the cognitive performance of aged rats. Brain Res. 1995;693:88–94. doi: 10.1016/0006-8993(95)00707-w. [DOI] [PubMed] [Google Scholar]
- 17.Cotman CW, Head E, Muggenburg BA, Zicker S, Milgram NW. Brain aging in the canine: a diet enriched in antioxidants reduces cognitive dysfunction. Neurobiol Aging. 2002;25:809–818. doi: 10.1016/s0197-4580(02)00073-8. [DOI] [PubMed] [Google Scholar]
- 18.Sumien N, Heinrich KR, Sohal RS, Forster MJ. Short-term vitamin E intake fails to improve cognitive or psychomotor performance of aged mice. Free Radic Biol Med. 2004;36:1424–1433. doi: 10.1016/j.freeradbiomed.2004.02.081. [DOI] [PubMed] [Google Scholar]
- 19.Meydani M. Nutrition interventions in aging and age-associated disease. Ann N Y Acad Sci. 2001;928:226–235. doi: 10.1111/j.1749-6632.2001.tb05652.x. [DOI] [PubMed] [Google Scholar]
- 20.Kirlin WG, Cai J, Thompson SA, Diaz D, Kavanagh TJ, Jones DP. Glutathione redox potential in response to differentiation and enzyme inducers. Free Radic Biol Med. 1999;27:1208–1218. doi: 10.1016/s0891-5849(99)00145-8. [DOI] [PubMed] [Google Scholar]
- 21.Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 22.Jones DP. Redox potential of GSH/GSSG couple: assay and biological significance. Methods Enzymol. 2002;348:93–112. doi: 10.1016/s0076-6879(02)48630-2. [DOI] [PubMed] [Google Scholar]
- 23.Sastre J, Millan A, Garcia de la Asuncion J, Pla R, Juan G, Pallardo FV, O'Connor E, Martin JA, Droy-Lefaix MT, Vina J. A Ginkgo biloba extract (EGb 761) prevents mitochondrial aging by protecting against oxidative stress. Free Radic Biol Med. 1998;24:298–304. doi: 10.1016/s0891-5849(97)00228-1. [DOI] [PubMed] [Google Scholar]
- 24.Sastre J, Pallardo FV, Garcia de la Asuncion J, Vina J. Mitochondria, oxidative stress and aging. Free Radic Res. 2000;32:189–198. doi: 10.1080/10715760000300201. [DOI] [PubMed] [Google Scholar]
- 25.Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 26.Jones DP, Mody VC, Carlson JL, Lynn MJ, Sternberg P. Redox analysis of human plasma allows separation of pro-oxidant events of aging from decline in antioxidant defenses. Free Radic Biol Med. 2002;33:1290–1300. doi: 10.1016/s0891-5849(02)01040-7. [DOI] [PubMed] [Google Scholar]
- 27.de la Asuncion GJ, Millan A, Pla R, Bruseghini L, Esteras A, Pallardo FV, Sastre J, Vina J. Mitochondrial glutathione oxidation correlates with age-associated oxidative damage to mitochondrial DNA. FASEB J. 1996;10:333–338. doi: 10.1096/fasebj.10.2.8641567. [DOI] [PubMed] [Google Scholar]
- 28.Rebrin I, Kamzalov S, Sohal RS. Effects of age and caloric restriction on glutathione redox state in mice. Free Radic Biol Med. 2003;35:626–635. doi: 10.1016/s0891-5849(03)00388-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rebrin I, Bayne ACV, Mockett RJ, Sohal RS. Free aminothiols, glutathione redox state and protein mixed disulfides in aging drosophila melanogaster. Biochem J. 2004;382:131–136. doi: 10.1042/BJ20040506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Packer L. Oxidants, antioxidant nutrients and the athlete. J Sports Sci. 1997;15:353–363. doi: 10.1080/026404197367362. [DOI] [PubMed] [Google Scholar]
- 31.Kagan VE, Shvedova A, Serbinova E, Khan S, Swanson C, Powell R, Packer L. Dihydrolipoic acid—A universal antioxidant both in the membrane and in the aqueous phase. Reduction of peroxyl, ascorbyl and chromanoxyl radicals. Biochem Pharmacol. 1992;44:1637–1649. doi: 10.1016/0006-2952(92)90482-x. [DOI] [PubMed] [Google Scholar]
- 32.Kagan VE, Witt E, Goldman R, Scita G, Packer L. Ultraviolet light-induced generation of vitamin E radicals and their recycling. A possible photosensitizing effect of vitamin E in skin. Free Radic Res Commun. 1992;16:51–64. doi: 10.3109/10715769209049159. [DOI] [PubMed] [Google Scholar]
- 33.Edamatsu R, Mori A, Packer L. The spin-trap N-tert-alpha-phenyl-butylnitrone prolongs the life span of the senescence accelerated mouse. Biochem Biophys Res Commun. 1995;211:847–849. doi: 10.1006/bbrc.1995.1889. [DOI] [PubMed] [Google Scholar]
- 34.Nakahara H, Kanno T, Inai Y, Utsuni K, Hiramatsu M, Mori A, Paker L. Mitochondrial dysfunction in the senescence accelerated mouse (SAM) Free Radic Biol Med. 1998;24:85–92. doi: 10.1016/s0891-5849(97)00164-0. [DOI] [PubMed] [Google Scholar]
- 35.Hosokawa M. A higher oxidative status accelerates senescence and aggravates age- dependent disorders in SAMP strains of mice. Mech Ageing Dev. 2002;123:1553–1561. doi: 10.1016/s0047-6374(02)00091-x. [DOI] [PubMed] [Google Scholar]
- 36.Rebrin I, Sohal RS. Comparison of thiol redox state of mitochondria and homogenates of various tissues between two strains of mice with different longevities. Exp Gerontol. 2004;39:1513–1519. doi: 10.1016/j.exger.2004.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sims NR. Mitochondrial isolation from brain: strategy, techniques, and criteria of purity. In: Lash LH, Jones DP, editors. Mitochondrial dysfunction. San Diego: Academic Press; 1993. pp. 29–41. [Google Scholar]
- 38.Reed DJ. Regulation of reductive processes by glutathione. Biochem Pharmacol. 1986;35:7–13. doi: 10.1016/0006-2952(86)90545-9. [DOI] [PubMed] [Google Scholar]
- 39.Ziegler DM. Role of reversible oxidation-reduction of enzyme thiols-disulfides in metabolic regulation. Annu Rev Biochem. 1985;52:711–760. doi: 10.1146/annurev.bi.54.070185.001513. [DOI] [PubMed] [Google Scholar]
- 40.Jones DP, Go YM, Anderson CL, Ziegler TR, Kinkade JM, Kirlin WG. Cysteine/cystine couple is a newly recognized node in the circuitry for biologic redox signaling and control. FASEB J. 2004;18:1246–1248. doi: 10.1096/fj.03-0971fje. [DOI] [PubMed] [Google Scholar]
- 41.Sohal RS, Agarwal S, Sohal BH. Oxidative stress and aging in the Mongolian gerbil (Meriones unguiculatus) Mech Ageing Dev. 1995;81:15–25. doi: 10.1016/0047-6374(94)01578-a. [DOI] [PubMed] [Google Scholar]
- 42.Forman HJ, Lui RM, Shi MM. Glutathione synthesis in oxidative stress. In: Paker L, Cadenas E, editors. Biothiols in Health and Disease. New York: Dekker; 1995. pp. 189–212. [Google Scholar]
- 43.Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y, Yamamoto M, Talalay P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci USA. 2002;99:11908–11913. doi: 10.1073/pnas.172398899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Talalay P, Fahey JW. Phytochemicals from cruciferous plants protect against cancer by modulating carcinogen metabolism. J Nutr. 2001;131(Suppl):3027S–3033S. doi: 10.1093/jn/131.11.3027S. [DOI] [PubMed] [Google Scholar]
- 45.Dinkova-Kostova AT, Massiah MA, Bozak RE, Hicks RJ, Talalay P. Potency of Michael reaction acceptors as inducers of enzymes that protect against carcinogenesis depends on their reactivity with sulfhydryl groups. Proc Natl Acad Sci USA. 2001;98:3404–3409. doi: 10.1073/pnas.051632198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Biswas SK, McClure D, Jimenez LA, Megson IL, Rahman I. Curcumin induces glutathione biosynthesis and inhibits NF-kappaB activation and interleukin-8 release in alveolar epithelial cells: mechanism of free radical scavenging activity. Antioxid Redox Signal. 2005;7:32–41. doi: 10.1089/ars.2005.7.32. [DOI] [PubMed] [Google Scholar]
- 47.Burger JMS, Promislow DEL. Sex-specific effects of interventions that extend fly life span. Sci Aging Know Environ. 2004;28:30–38. doi: 10.1126/sageke.2004.28.pe30. [DOI] [PubMed] [Google Scholar]
- 48.Packer L. Antioxidant defenses in biological systems: an overview. In: Packer L, Traber M, Xin W, editors. Proceedings of the International Symposium on Natural Antioxidants: Molecular Mechanisms and Health Effects; Champaigh: AOCS Press; 1996. pp. 9–23. [Google Scholar]
- 49.Hagen TM, Liu J, Wehr CM, Ingersoll RT, Vinarsky V, Bartholomew JC, Ames BN. Feeding acetyl-L-carnitine and lipoic acid to old rats significantly improves metabolic function while decreasing oxidative stress. Proc Natl Acad Sci USA. 2002;99:1870–1875. doi: 10.1073/pnas.261708898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cotgreave IA, Gerdes RG. Recent trends in glutathione biochemistry. Glutathione-protein interactions: a molecular link between oxidative stress and cell proliferation? Biochem Biophys Res Commun. 1998;242:1–9. doi: 10.1006/bbrc.1997.7812. [DOI] [PubMed] [Google Scholar]
- 51.Klatt P, Lamas S. Regulation of protein function by S-glutathiolation in response to oxidative and nitrosative stress. Eur J Biochem. 2000;267:4928–4944. doi: 10.1046/j.1432-1327.2000.01601.x. [DOI] [PubMed] [Google Scholar]
- 52.Finkel T. Oxidant signals and oxidative stress. Curr Opin Cell Biol. 2003;15:247–254. doi: 10.1016/s0955-0674(03)00002-4. [DOI] [PubMed] [Google Scholar]
- 53.Joseph JA, Shukitt-Halle B, Denisova NA, Bielinski D, Martin A, McEwen JJ, Bickford PC. Reversals of age-related declines in neuronal signal transduction, cognitive and motor behavioral deficits with blueberry, spinach, or strawberry dietary supplementation. J Neurosci. 1999;19:8114–8121. doi: 10.1523/JNEUROSCI.19-18-08114.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]