

Abstract
The origins of the boat transition state preference in the Ireland-Claisen rearrangements studied experimentally by Kishi and co-workers have been explored computationally with Density Functional Theory and MO6-2X. Steric interactions in the chair transition states were identified as the principal reason for the boat transition state preference.
The Ireland-Claisen reaction1 has found wide use in synthesis. It belongs to the class of [3,3]-sigmatropic rearrangements, involving the rearrangement of a silyl ketene acetal. It is widely accepted that chair transition states are generally preferred over the boat transition states in [3,3]-sigmatropic shifts (Figure 1). This has been demonstrated by Doering and Roth2 and Hill and Gilman3 for the acyclic Cope rearrangement, for instance. Their findings suggested a 6 kcal/mol preference for the chair transition state, mirroring the free energy difference between the chair and boat conformers of cyclohexane.4 This has been confirmed by computational studies. The chair transition state in the Cope rearrangement of 1,5-hexadiene was calculated to be 11.4 kcal/mol lower than the boat transition state using (6,6)-CASPT2/6-31G(d),5a consistent with the corresponding experimental results (chair, ΔH‡= 33.5 ± 0.5 kcal/mol6; boat, ΔH‡= 44.7 ± 2.0 kcal/mol7). Similarly, the Claisen rearrangement of crotyl propenyl ether also prefers the chair transition state, but the preference for the chair is only 3 kcal/mol.8,9
Figure 1.

Possible transition states for Cope (X=CH2 ) and Claisen (X=O ) rearrangements.
In contrast to the acyclic systems described above, the preference for chair transition states in cyclic systems may be much less pronounced, and can even lead to the chair transition state being disfavored as a result of steric hindrance.10 This has been demonstrated by several groups,11 for instance in rearrangements of pyranoid and furanoid glucal systems.11 The groups of Neier, Houk and Aviyente investigated this in detail with both experimental and computational studies,12 extending the transition state proposals by Ireland13 (the latter are shown in Figure 2).
Figure 2.

Irelands’s proposed chair and boat transition-state models for an Ireland-Claisen rearrangement. Relative energies (kcal/mol) are given in the parenthesis.13
Some experimental studies support the existence of both chair and boat transition states; one of the most significant examples is the Ireland-Claisen rearrangement of cyclohexenyl silyl ketene acetals, which may follow either the boat or chair transition state, leading to moderate stereoselectivities.11
With an aim to improve the selectivity of the Ireland-Claisen rearrangement, Kishi and coworkers14 recently performed experiments to favor further the boat transition state.10,12 For this purpose, dihydropyran derivatives, such as 1 in Scheme 1, were synthesized and tested. The overall stereoselectivity of rearrangements was found to be largely consistent with the E/Z ratio of the produced enolate intermediate.14 Experiments under conditions involving mainly intermediate Z-2 led to 3 as the major product (8:1 ratio with 4), where experiments via E-2 gave predominantly 4 (5:1 ratio with 3) at 80 °C.14 This suggests that in both cases the major products arise from boat transition states (Scheme 1).
Scheme 1.

Pathways for the conversion of 1 into 3 and 4.14
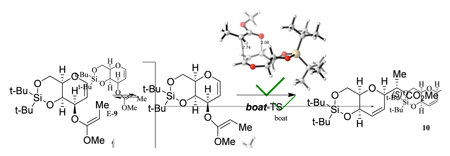
With the hypothesis that steric hindrance induced by the silyl moiety may improve selectivities, Kishi and co-workers prepared dihydropyran 5. The corresponding silyl ketene acetal 6 reacted to form a single diastereomer 7 (see Scheme 2). Furthermore, the E-stereoisomer of the silyl ketene acetal (E-6) was partially recovered from the reaction mixture. The authors thus suggested that the Claisen rearrangement proceeds through Z-6 but not through E-6, consistent with a mechanism that involves a boat transition state.14
Scheme 2.

Pathways for the conversion of 5 into 7 and 8.14
We have performed computational studies15,16 to examine the origins of modest stereoselectivity for the conversion of 1 to 3 and 4 (Scheme 1), and to understand the excellent stereoselectivity of the reaction of 5 to 7 (Scheme 2). For that purpose, model reactants 2' and 6' (models for 2 and 6, respectively) were investigated. OR and BnO were simplified in model 2', and a methoxy group was used instead of the OTBS group in 6', shown previously to be a valid approach.12
The supporting information describes additional computational models. Note, because of naming conventions, the Z-isomers of the experimentally investigated compounds correspond to the E-isomers of the computational models. For ease of comparison, we chose to use Z*-2', E*-2' (for the models of Z-2 and E-2) and Z*-6', E*-6' (for the models of Z- 6 and E-6).
The lowest energy conformations17 of the dihydropyran reactants, 2'and 6'are half chairs, in which the C1–C2 bond is in pseudo-axial and the C3–O4 bond is in pseudo-equatorial position (see Figure 3).18 Reactant conformers that have the reverse arrangement, i.e. C1–C2 in pseudo-equatorial and C3–C4 in pseudo-axial position are ca. 3 kcal/mol higher in energy (see SI).
Figure 3.

Model systems studied computationally. Relative enthalpy differences (ΔH in kcal/mol at 298 K) of E- and Z-isomers are given in parenthesis, calculated with B3LYP/6-31+G(d).17
The transition states for the rearrangements were subsequently calculated. The lowest-energy conformers of chair and boat transition states for model 2' are illustrated in Figure 4.18 Those of model 6' are shown in Figure 5. Table 1 summarizes the corresponding activation barriers at B3LYP/6-31+G(d).16 The activation barriers at MO6-2X//B3LYP/6-31+G(d)19 can be found in the supporting information.
Figure 4.

TSs for the rearrangements of 2'. ΔH‡ (in kcal/mol at 298 K) given in parenthesis, calculated with B3LYP/6-31+G(d).
Figure 5.

TSs for the rearrangements of 6'. ΔH‡ (kcal/mol) at 298 K given in parenthesis, calculated with B3LYP/6-31+G(d).
Table 1.
Activation parameters for models 2' and 6' in kcal/mol at 298K, calculated with B3LYP/6-31+G(d). ΔG‡ sol corresponds to the barriers in benzene at 353 K.
| TSs | ΔH‡ | ΔΔH‡ | ΔG‡ | ΔΔG‡ | ΔG‡sol |
|---|---|---|---|---|---|
| Z*-2'-boat-TS | 19.9 | 0.0 | 20.5 | 0.0 | 20.6 |
| Z*-2'-chair-TS | 21.6 | 1.7 | 22.6 | 2.1 | 22.2 |
| E*-2'-boat-TS | 21.6 | 1.7 | 22.6 | 2.1 | 22.4 |
| E*-2'-chair-TS | 23.7 | 3.8 | 24.9 | 4.4 | 24.0 |
| Z*-6'-boat-TS | 17.7 | 0.0 | 18.2 | 0.0 | 18.2 |
| Z*-6'-chair-TS | 25.4 | 6.9 | 26.8 | 8.6 | 25.8 |
| E*-6'-boat-TS | 25.3 | 6.8 | 25.7 | 7.5 | 25.2 |
| E*-6'-chair-TS | 32.6 | 14.1 | 33.3 | 15.1 | 31.2 |
Consistent with the experimental findings by Kishi and co-workers, our calculations predict the boat transition states to be preferred over the chair transition states for the rearrangements of E*-2' and Z*-2'. The principal reason for the boat-TS preference is the lower steric interaction between the OMe-substituents as indicated in Figure 4. To establish further that this repulsive interaction is crucial for the boat-TS preference, we calculated all TSs in the absence of methoxy groups. Those now resulted in a 0.8–1.3 kcal/mol preference of the chair TSs (see SI), supporting this steric hypothesis.
Turning to the sterically more encumbered model 6', it is apparent that the energy differences between boat and chair TSs increase substantially (Table 1). The preference for boat TSs is about 8 kcal/mol. The t-butyl silyl groups increase the repulsive interaction with the methoxy group, causing structural distortions in the molecules. With the introduction of the silyl substituents, the ring C–O bonds’ flexibilities are decreased (as compared to model 2') and forced into a more repulsive conformation. In the least favored E*-6'-chair-TS the C-O ring substituents now adopt pseudo-axial positions in a twisted chair TS, which was already shown to be disfavored for the reactant conformers. This contributes further to the larger boat-chair TS energy gap.
These rearrangements are highly exergonic (ΔGrxn = -34.6 kcal/mol for Z*-6'-chair, for example), showing that the rearrangement is irreversible.20 Thus, the principal reason for the high stereoselectivity in the reaction of 6 is the large steric interaction in the chair-TSs and E-boat TS. The Z-reactant isomer (Z*-6') will react via the most favorable boat-TS pathway. For the E-isomer, the low-energy rearrangement pathway is via the boat TS which is significantly disfavored compared to the Z*-6'-boat-TS. However, the E*-6'-boat-TS would lead to 8 - the stereoisomer opposite than observed experimentally (compare Scheme 2). This suggests that the E→Z isomerization, followed by reaction via Z*-6'-boat-TS, is favored over the high energy direct rearrangement to 8.
This raises the question of the mechanism and facility of the E/Z-isomerization. Kishi and co-workers heated a mixture of ‘crude’ silyl ketene acetal containing E- and Z-isomers for 3d at 80°C. Complete isomerization to the Z-isomer was indeed observed.14 Our calculation gives an unfavorable barrier of ΔG‡ = 51.5 kcal/mol (ΔH‡ = 51.0 kcal/mol) at B3LYP/6-31+G(d) for the E→Z isomerization of 6', suggesting that it is only feasible if catalyzed. Tanaka and Fuji21 have previously studied the isomerization of a silyl ketene acetal derived from methyl phenylacetate. They have shown that in the presence of LiCl (which is formed during the preparation of a silyl ketene acetal), the Z-isomer becomes the thermodynamically preferred species. Using 1H-NMR spectroscopy over a course of 40 h, they established that E/Z-isomerization is a facile process at 20°C in the presence of LiCl, but would not take place in the absence of LiCl.21 This supports the hypothesis that E→Z isomerization of 6 occurs prior to rearrangement via the boat TS under the ‘crude’ reaction conditions.
For system 2, Z-boat and E-boat TSs are both relatively low in energy, and the pathways via those TSs are equally accessible. Selectivity in that case (substrate 2) thus depends on the E/Z ratio of enol ethers in the reaction mixture.
Our computations thus support the hypotheses and conclusions drawn previously by Kishi and co-workers.14
Supplementary Material
ACKNOWLEDGMENT
We are grateful to the NIH-FIRCA project (TW007177) and the National Institute of General Medical Sciences, National Institutes of Health (GM 36700) for financial support. F.S. gratefully acknowledges the Alexander von Humboldt Foundation for a Feodor Lynen Fellowship. Computations were performed on the UCLA Hoffman2 cluster, and using the TUBITAK-ULAKBIM High Performance Computing Center and the National Center for High Performance Computing in Turkey (UYBHM), under the grant number 20532009. S.G. and V.A. thank the Boğaziçi Üniversitesi Bilimsel Araştırma Projeleri (BAP) for financial support.
Footnotes
SUPPORTING INFORMATION Full reference 15 and Cartesian coordinates and energies of all stationary points. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.(a) Ireland RE, Mueller RH. J. Am. Chem. Soc. 1972;94:5897–5898. [Google Scholar]; (b) Ireland RE, Mueller RH, Willard AK. J. Am. Chem. Soc. 1976;98:2868–2877. [Google Scholar]; (c) Claisen L. Chem. Ber. 1912;45:3157–3166. [Google Scholar]
- 2.Doering WvE, Roth WR. Tetrahedron. 1962;18:67–74. [Google Scholar]
- 3.Hill RK, Gilman NW. Chem. Commun. 1967:619–620. [Google Scholar]
- 4.(a) Allinger NL, Freiburg LA. J. Am. Chem. Soc. 1960;82:2393–2394. [Google Scholar]; (b) Johnson WS, Bauer VJ, Margrave JL, Frisch MA, Dreger LH, Hubbard WN. J. Am. Chem. Soc. 1961;83:606–611. [Google Scholar]
- 5.(a) Hrovat DA, Borden WT. J. Am. Chem. Soc. 2001;123:4069–4072. doi: 10.1021/ja004209o. [DOI] [PubMed] [Google Scholar]; See also: Ventura E, do Monte SA, Dallos M, Lischka H. J. Phys. Chem. A. 2003;107:1175–1180. ; (c) Navarro-Vàzquez A, Prall M, Schreiner PR. Org. Lett. 2004;6:2981–2984. doi: 10.1021/ol0488340. [DOI] [PubMed] [Google Scholar]; (d) Staroverov VN, Davidson ER. J. Am. Chem. Soc. 2000;122:7377–7385. [Google Scholar]
- 6.Doering WvE, Toscano VG, Beasley GH. Tetrahedron. 1971;27:5299–5306. [Google Scholar]
- 7.(a) Goldstein MJ, Benzon MS. J. Am. Chem. Soc. 1972;94:5119–5121. [Google Scholar]; (b) Goldstein MJ, Benzon MS. J. Am. Chem. Soc. 1972;94:7147–7149. [Google Scholar]
- 8.(a) Vittorelli P, Winkler T, Hansen HJ, Schmid H. Helv. Chim. Acta. 1968;51:1457–1461. [Google Scholar]; (b) Vittorelli P, Hansen HJ, Schmid H. Helv. Chim. Acta. 1975;58:1293–1309. [Google Scholar]; (c) Vance RL, Rondan NG, Houk KN, Jensen F, Borden WT, Komornicki A, Wimmer E. J. Am. Chem. Soc. 1988;110:2314–2315. [Google Scholar]
- 9.Recent related computational studies: Rehbein J, Leick S, Hiersemann M. J. Org. Chem. 2009;74:1531–1540. doi: 10.1021/jo802303m. ; Rehbein J, Hiersemann M. J. Org. Chem. 2009;74:4336–4342. doi: 10.1021/jo900635k. [DOI] [PubMed] [Google Scholar]
- 10.Velker J, Roblin JP, Neels A, Tesouro A, Evans HS, Klaerner FG, Gehrke JS, Neier R. Synlett. 1999;S1:925–929. [Google Scholar]
- 11.For reviews, see: Ziegler FE. Chem. Rev. 1988;88:1423–1452. ; (b) Enders D, Knopp M, Schiffers R. Tetrahedron: Asymmetry. 1996;7:1847–1882. [Google Scholar]; (c) Chai Y, Hong S-P, Lindsay HA, McFarland C, Mclntosh MC. Tetrahedron. 2002;58:2905–2928. [Google Scholar]; (d) Martin Castro AM. Chem. Rev. 2004;104:2939–3002. doi: 10.1021/cr020703u. [DOI] [PubMed] [Google Scholar]; (e) McFarland CM, McIntosh MC. Chapter 4. In: Hiersemann M, Nubbemeyer U, editors. The Claisen Rearrangement. Wiley-VCH; 2007. pp. 117–210. [Google Scholar]
- 12.Khaledy MM, Kalani MYS, Khuong KS, Houk KN, Aviyente V, Neier R, Soldermann N, Velker J. J. Org. Chem. 2003;68:572–577. doi: 10.1021/jo020595b. [DOI] [PubMed] [Google Scholar]
- 13.Ireland RE, Wipf P, Xiang JN. J. Org. Chem. 1991;56(11):3572–3582. [Google Scholar]
- 14.Chen C, Namba K, Kishi Y. Org. Lett. 2009;11:409–412. doi: 10.1021/ol8027225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frisch MJ, et al. Gaussian 03, Revision D.01. Wallingford CT: Gaussian, Inc; 2004. [Google Scholar]
- 16.Applying B3LYP/6-31+G(d) methodology, which was previously shown to be adequate, see: reference 12. ; (b) Houk KN, Gustafson SM, Black KA. J. Am. Chem. Soc. 1992;114:8565. [Google Scholar]; (c) Wiest O, Black KA, Houk KN. J. Am. Chem. Soc. 1994;116:10336–10337. [Google Scholar]; (d) Goumans TPM, Ehlers AW, Lammertsma K, Würthwein E-U, Grimme S. Chem. Eur. J. 2004;10:6468–6475. doi: 10.1002/chem.200400250. [DOI] [PubMed] [Google Scholar]
- 17.Conformational search was done at AM1 level of theory using SPARTAN (©Wavefunction, Inc). All obtained conformers within a 5 kcal/mol energy range were subsequently optimized with B3LYP/6-31+G(d) in Gaussian03.
- 18.Figures created with: CYLview, 1.0b. Legault, CY: Université de Sherbrooke; 2009. http://www.cylview.org. [Google Scholar]
- 19.Zhao Y, Truhlar DG. Theor. Chem. Acc. 2008;120:215–241. [Google Scholar]
- 20.We calculated the silyl analogue of E’-6-boat-TS with OTMS instead of OMe. The activation barrier of the rearrangement is 25.8 kcal/mol and the free energy of reaction is −32.9 kcal/mol, indicating that the OMe model is adequate.
- 21.Tanaka F, Fuji K. Tetrahedron Lett. 1992;33:7885–7888. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.