

Abstract
Transmissible Spongiform Encephalopathies are fatal and infectious neurodegenerative diseases characterized by extensive neuronal apoptosis and the accumulation of an abnormally folded form of the cellular prion protein (PrP), denoted PrPSC. Compelling evidence suggests the involvement of several signaling pathways in prion pathogenesis, including proteasome dysfunction, alterations in the protein maturation pathways and the unfolded protein response. Recent reports indicate that endoplasmic reticulum stress due to the PrP misfolding may be a critical factor mediating neuronal dysfunction in prion diseases. These findings have applications for developing novel strategies for treatment and early diagnosis of transmissible spongiform encephalopathies and other neurodegenerative diseases.
Keywords: Prion related disorders, apoptosis, PrPSC, proteasome, ER stress, glucose-regulated proteins, caspase-12, PrPSC-like, aggresomes
INTRODUCTION
Transmissible spongiform encephalopathies (TSEs), also known as prion disorders are a group of diseases that affect humans and animals and present a long incubation period. Once the first clinical signs appear, the disease progression is relatively fast and death occurs in a short time. Prion diseases are characterized by neurological dysfunction that may include dementia, ataxia and psychiatric disturbances. The central molecular event in the pathogenesis of prion diseases is the conversion of the normal cellular prion protein, termed PrPC, into the pathological form denoted PrPSC (for scrapie associated PrP) [1]. Etiologically, prion diseases can be classified as infectious (derived from the exposure to material contaminated with infectious prions), sporadic (spontaneous origin) or hereditary (inherited in an autosomal dominant manner). Human familial TSEs include some forms of Creutzfeldt-Jacob disease (CJD), Gertmann-Straussler-Sheinker (GSS) syndrome and fatal familial insomnia [2]. Inherited prion diseases in humans are genetically linked to different point mutations or increased number of octapeptide repeats within the PrP open reading frame. Around 90% of the CJD cases are sporadic. Infectious TSE diseases include kuru, which was propagated by cannibalism, and iatrogenic CJD, which is spread by tissue transplantation, contamination of surgical tools or inoculation with materials derived from CJD-infected tissues [1]. In animals, scrapie is the equivalent disease in sheep and goats, and in cattle the bovine spongiform encephalopathy (BSE) better known as “mad cow disease” [3]. The new variant form of CJD (vCJD) is the newest and most frightening member of the TSE group in humans. Its appearance has been undoubtedly linked to consumption of cattle infected with BSE.
In infectious forms of the disease, PrPSC formation from wild-type PrPC is initiated by the exposition to exogenous infectious agents, promoting a conformational transition from α-helical to β-sheet structure, resulting in the formation of PrPSC [4]. The biological function of PrPC is unclear [5], but its expression is essential for the development of prion diseases since PrPC-knockout mice are resistant to prion infection [6]. Under certain conditions, the conversion of PrPC into PrPSC can be achieved in a cell-free conversion assays [7–9] in a highly efficient manner [10]. Although the nature of the infectious agent has not been completely elucidated, PrPSC seems to be the main constituent. In fact, recent reports demonstrated that in vitro generated PrPSC in two different cell-free systems induce the disease when injected into the brain of healthy animals [11,12] (Fig. 1A).
Fig. 1.
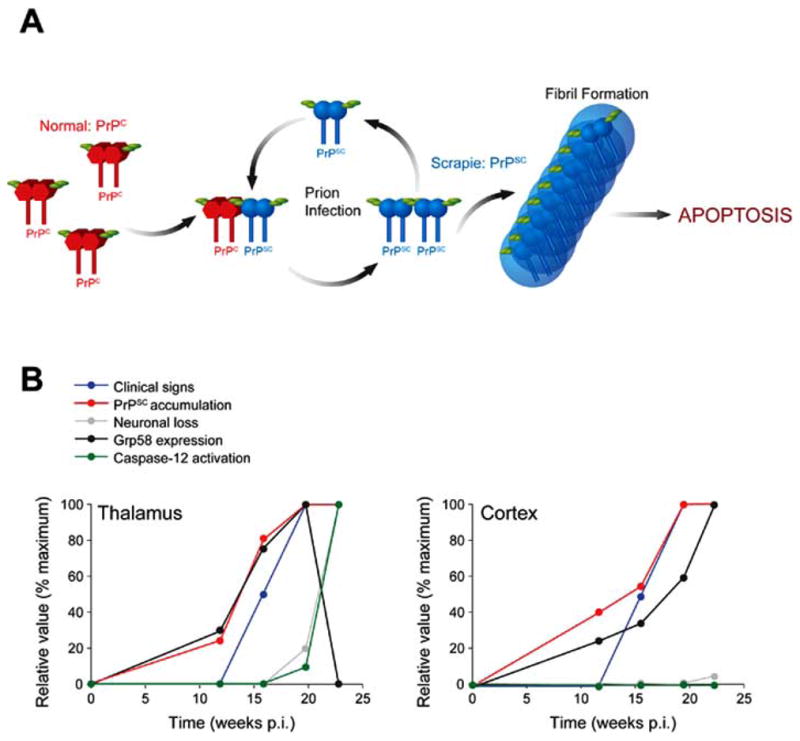
(A) A model for TSE progression. The hallmark event in the disease is the misfolding of PrPC to form the -sheet-rich PrPSC. Upon interaction and aggregation of endogenous PrPC with infectious PrPSC, PrPC is misfolded into the pathological form. This process proceeds in a cyclic manner during the course of the disease, generating increasing amounts of PrPSC. Deposition of PrPSC protein aggregates leads to the formation of amyloid prion rods. The final cause of clinical symptoms associated with TSEs is the extensive spongiform degeneration of the brain due to neuronal dysfunction. (B) Correlation between protein misfolding, ER stress and disease features in a murine-scrapie model. The kinetic of neuronal loss, PrPSC accumulation, caspase-12 activation and the expression levels of Grp58 in thalamus and cortex is presented over time. Also, the progression of clinical signs of the disease is shown. To facilitate the comparison, the data is shown as relative values normalized as a percentage of the maximum value observed for each parameter (data from [25]).
A number of studies indicate that neuronal death in humans and animals affected with TSEs occurs through apoptosis and is observed mostly in terminally ill individuals [13]. Neuronal dysfunction and synaptic alterations are likely earlier events responsible for the initial disease symptoms [14]. Different strategies have been developed to understand the relationship between PrP misfolding and neuronal dysfunction [13]. Hereditary prion diseases are linked to point or insertional mutations in the PrP gene and transgenic mouse models expressing the human-associated PrP mutant allele have proved the involvement of PrP in the disease process. For example, a transgenic mice model of familial prion diseases expressing the PrP homologue of a nine-octapeptide insertional mutation showed accumulation of protease-resistant PrPSC and apoptotic cell death of the cerebellar granule cells, in addition to progressive ataxia [15]. Another transgenic mice expressing PrP fragments die spontaneously by ataxia, showing an accumulation of protease resistant PrP within neuronal dendrites and cell bodies, apparently causing apoptosis [16,17].
Attempts to understand the molecular basis of neuronal dysfunction in prion diseases have led to the search for in vitro models to analyze the exact role of PrPSC in neurodegeneration. Preliminary characterization employed the PrP fragment spanning the sequence 106–126 (PrP106-126), which has been extensively used to induce cell death in neuronal cultures [18–20]. The use of PrP106–126 has been questioned as an appropriate model of prion toxicity because it has never been found in vivo. Besides, several days of incubation and high concentrations of this peptide are needed to observe cell death (between 50 to 100 μM). As an alternative and more relevant approach, the toxicity of purified PrPSC from scrapie-infected brains was explored by different groups. Brain derived-PrPSC purified from scrapie-infected animals is cytotoxic in vitro at nanomolar concentrations [21–25]. However, immortalized neuronal and neuroglial cell lines persistently infected by prions generally show no overt signs of cytotoxicity, although producing readily detectable amounts of PrPSC and infectivity [26,27]. A possible explanation for the lack of toxicity of endogenous PrPSC produced in N2a neuroblastoma cells is that since these cells divide rapidly, they do not accumulate enough PrPSC to lead to cell death. In agreement with this idea, scrapie-infected neuroblastoma cells are more susceptible to apoptosis triggered by brain-derived PrPSC [24]. Moreover, a recent article described that scrapie-infection of primary cultures trigger apoptosis, strongly arguing for a direct role of PrPSC in the neurodegeneration process [28]. This subject had gained more complexity since in vivo, the acute neuron-targeted depletion of PrP in the brain of mice with ongoing infection is able to prevent neuronal loss and progression to disease. Moreover, a reversion of early spongiform change was observed despite marked accumulation of PrPSC in non-neuronal cells [29]. This finding suggested that the prion replication process per se is an important component of the pathology but the mechanism explaining these observations is not yet available.
PROTEIN QUALITY CONTROL AND PrPSC-LIKE GENERATION
PrPC is a glycophosphatidyl inositol (GPI) anchored protein located in membrane subdomains, denominated detergent resistant-membranes or lipid rafts [30]. The subcellular localization of PrPSC in this plasma membrane subcompartment is relevant for the replication process, because alterations on the cholesterol or sphingolipid composition or replacement of the GPI anchor signal, rendering the protein soluble, impact the ability of prion-chronically infected cells to replicate PrPSC [31]. In addition a similar treatment modulates PrPSC toxicity in vitro [23].
During wild type PrP synthesis and maturation, a GPI anchor is added in the ER, one intramolecular disulfide bond is formed and the amino and carboxy-terminal regions are cleaved in the signal sequences (review is [30,32]). After this process, mature glycosylations are added in the Golgi compartment at two different positions, and the mature protein traffic to the cell surface, mostly into lipid rafts structures (Fig. 2). Recent work suggested that the protein quality control may play an active role in modulating prion protein aggregation and neuronal cell death (review in [33]). Approximately 10% of nascent PrP molecules are subjected to ER-associated degradation (ERAD) pathway through the proteasome [34,35]. ER-chaperones, like Grp78/Bip, have been implicated in the process of recognition of misfolded PrP for proteasomal degradation, playing a role in the maintenance of PrP “quality control” [36]. The first evidence for a role of the proteasome system in the pathogenesis of prion diseases came from studies using cell lines expressing a mutated form of PrP associated with GSS Syndrome [37]. The authors described that the mutation PrPY145 stop leads to an accumulation of PrP in intracellular compartments. Pharmacological inhibition of the proteasome induces an increase in the amount of intracellular mutant PrP, mainly in the ER, Golgi and nucleus [37]. Part of the accumulated PrP escape the ER quality control and can be detected as aggregates in post-Golgi compartments, while a proportion of this protein never exit the ER. Another PrP mutant, PrPQ217R, was shown to be accumulated and aggregated in the ER [38] and is also subjected to ERAD, ubiquitinated and degraded by the proteasome system [36].
Fig. 2.
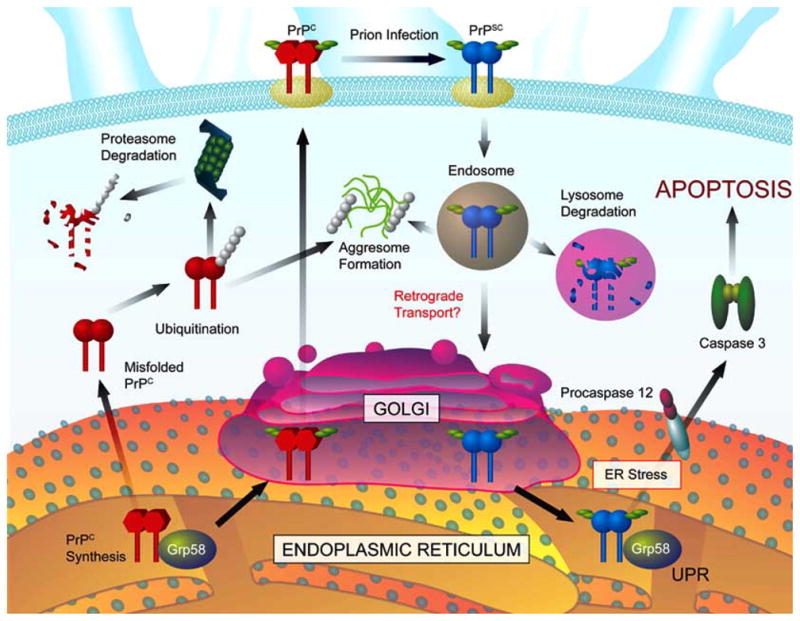
Cellular pathways involved in the synthesis, folding, secretion, trafficking, clearance and neurotoxicity of the prion protein. During synthesis, immature PrP is proteolytically processed in the amino- and carboxy-terminus signal peptide sequence in the ER, and GPI membrane anchor is added. After its transit through the Golgi compartment, mature glycosylations are attached to the protein, and PrP is exported to the cell surface. During the normal cycle, PrP can be internalized by endocytosis and re-targeted to the plasma membrane or delivered to degradation in lysosomes. A fraction of PrPC is usually misfolded during this process, undergoing degradation through the proteasome pathway. Scrapie infection triggers PrPSC generation at the plasma membrane and in early endocytic vesicles. PrPSC may trigger ER stress due to intracellular accumulation or proteasomal dysfunction. Extensive ER stress activates specific cell death pathways, which are negatively regulated by different components of the UPR including Grp58 and many other chaperones, folding enzymes and formation of aggresomes.
Susan Lindquist’s group proposed that proteasome inhibition leads to the accumulation of fully matured PrP in the cytoplasm that exhibits some of the biochemical properties of PrPSC, such as insolubility in non-ionic detergents and partial resistance to protease degradation (here termed PrPSC–like) [35,39,40]. Transgenic mouse expressing a cytosolic PrP mimicking the PrP form generated through the ERAD process (lacking the N-terminal signal peptide and C-terminal GPI addition sequence anchor), developed severe ataxia with cerebellar degeneration and gliosis [41]. However, another report argued that neither wild type or mutant forms of PrP are subjected to retrograde transport prior to proteasome degradation [42]. The authors described that only a small number of PrP molecules, both mutant (PrP14 and PrPD177N) and wild type PrP, are degraded by the proteasome. These PrP molecules represent aberrant chains that have been translated in the cytoplasm and have not been translocated into the ER lumen. Despite these, mutant PrP forms are significantly delayed in their transit along the early part of the secretory pathway, being possible that the differential alteration of PrP maturation upon mutations may be the triggering step in the toxicity of abnormal PrP molecules. Leblanc and co-workers suggested that cytosolic PrP is not toxic to primary neuronal cultures and do not show any PrPSC-like properties as those observed in neuroblastoma cell lines [43]. Conversely, they showed that PrPC has an anti-apoptotic activity (review in [5]), even when it is expressed in the cytosol [43]. The analysis of proteasome involvement should be performed in more relevant diseases-associated models or in individuals affected with naturally occurring forms of prion diseases to solve this controversy in the field.
A recent report suggested that misfolded PrP might be targeted to aggresomes contributing to prion-mediated neuronal dysfunction [44]. The authors described that mild proteasome inhibition leads to apoptosis only in cells chronically infected with scrapie prions and not in control cells. More interestingly, this toxic effect was associated with the formation of intracellular protein aggregates in aggresomes containing PrPSC, chaperones, ubiquitin and proteasome subunits [44]. Aggresomes are subcellular structures in which misfolded protein aggregates accumulate inside the cell, which are then ubiquitinated and delivered to the proteasome for degradation [45,46]. Ubiquitination of PrP was described in the brain of scrapie-infected mice at the terminal stage of the disease [47]. An alternative explanation for PrP toxicity might be that the accumulation of misfolded prion protein may leads to proteasome dysfunction due to an inability of the cells to degrade this highly protease-resistant protein (Fig. 2). Proteasome dysfunction may in turn lead to ER stress through a general accumulation of misfolded proteins normally generated by the ERAD pathway, poisoning the cells with cytotoxic protein aggregates. Indeed, proteasome inhibition triggers ER stress [48].
ER STRESS AND NEURONALDYSFUNCTION IN TSEs
The viability of a cell strictly depends on the functional and structural integration of intracellular organelles. Alteration of ER function leads to the accumulation of misfolded proteins in this organelle, triggering an stress response termed the unfolded protein response (UPR) [49,50]. The UPR is an adaptive process that aims the restoration of homeostasis and affects the expression of proteins involved in nearly every aspect of the secretory pathway. The UPR has basically two components [49]: A pro-survival component mediated by the up-regulation of multiple chaperones, folding enzymes, in addition to an increase in the efficiency of the ERAD pathway and a general decrease in protein translation into the ER. Depending on the cellular system, different subset of chaperones and folding enzymes have been shown to be upregulated during an UPR, including chaperones of the glucose-regulated proteins (GRPs), disulfide isomerases, heat-shock proteins (HSPs) and others [51]. These proteins may recover the homeostasis of the ER by attenuating the toxicity of misfolded proteins, by correcting the misfolding or by increasing their degradation [51].
When the stress level overcomes the ability of the cell to maintain the overload of misfolded proteins in the ER, specific pro-apoptotic components trigger cellular death. This pathway includes the activation of the ER-resident caspase-12 [52] (in rodents and its putative human homolog caspase-4 [53]), the induction of the transcriptional factor CHOP/GADD153 [54,55], the down regulation of particular ER-chaperones and the activation of several pro-apoptotic kinases such as c-Jun-N terminal Kinase [56,57]. Pro-caspase-12 can be activated by the calcium dependent protease, m-calpain. Active caspase-12 leads to the processing and activation of downstream caspases triggering cell death by apoptosis [52,58]. However, the role of caspase-12 and caspase-4 in ER stress-mediated apoptosis is still controversial [59] and requires more in vivo validation. CHOP/GADD153 is known to induce the down regulation of the anti-apoptotic protein Bcl-2 [60] and also induces cell death by promoting protein synthesis and oxidation in the stressed ER [54]. Many other components are known to be involved in ER stress-mediated apoptosis (for reviews see [61]).
The up-regulation of UPR responsive chaperones, such as Grp78/BiP, Grp94 and Grp58, was observed in the cortex of patients affected with variant CJD and sporadic CJD validating the idea that prion replication triggers ER stress [24]. Another study, using proteomic analysis, also described that Grp58 is highly expressed in the cerebellum of humans patients affected with sporadic CJD [62]. Highly purified PrPSC from the brain of scrapie-infected mice induces ER stress and caspase-12-dependent apoptosis in vitro [24]. This was associated with a release of calcium from the ER and upregulation of GRPs. Specific targeting of the anti-apoptotic protein Bcl-2 to the ER membrane, or expression of a dominant negative form of caspase-12 decrease the sensitivity of neuroblastoma cells to PrPSC cytotoxicity. In addition, scrapie-infected neuroblastoma cells were shown to be more susceptible to ER stress-induced apoptosis than non-infected cells, supporting the idea that PrPSC replication alters the homeostasis of the ER [24].
Mice infected with 139A-scrapie prions develop the first signs of ER stress during the pre-symptomatic phase of the disease. The first alterations observed soon after PrPSC formation include the induction of the disulfide isomerase Grp58 [25] (Fig. 1B). By contrast, a transient induction of Grp78 and Grp94 was observed in those mice at the beginning of the symptomatic phase. Unexpectedly, no indication of CHOP/GADD153, calnexin or Hsp70 induction was observed in 139A-scrapie infected mice brains, suggesting that PrP misfolding triggers a non-classical ER stress response [25]. In that model, only at the late stage of the disease neuronal loss is observed and correlates with pro-caspase-12 processing and inversely correlates with Grp58 expression [24] (Fig. 1B). In mice infected with the ME7 strain of scrapie, Brown and co-workers reported an expression profile analysis of pre-clinical scrapie-infected hippocampus showing that most of the genes affected by prion infection are related with proteins implicated in ER stress and oxidative stress [63].
In vitro studies using N2a neuroblastoma cells demonstrated that components of the UPR have a drastic effect in maintaining cellular viability upon accumulation of misfolded PrP. For example, the expression of the disulfide isomerase Grp58 protect cells against PrPSC toxicity [25]. An interaction between Grp58 and PrP was observed, that was increased in cells infected with scrapie prions or in cells treated with proteasome inhibitors, suggesting that this chaperone has a higher affinity for misfolded PrP. However, the ability of those cells to be infected with scrapie is not affected by the modulation of Grp58 expression levels (Hetz and Soto, unpublished results). Thus, PrP misfolding per se may be recognized as an ER stress factor triggering an irreversible response that culminates in neuronal loss.
It remains to be determined how the accumulation of misfolded PrP or PrPSC replication triggers ER stress. The possibility that abnormal prions target directly the ER remains to be explored. We have described that scrapie infection triggers an abnormal subcellular distribution of PrP at the terminal stage of the disease, where PrPSC is no longer located in lipid rafts structures [14]. Scrapie infection leads to a decrease in the glycosylation of PrP early in the disease progression, suggesting that the homeostasis of the secretory pathway may be altered [64,65]. On the other hand, the stimulation of retrograde transport toward ER increases the accumulation of PrPSC in prion-infected neuroblastoma cells [66] and several PrP mutants associated with inherited prion diseases are retained in this organelle [36,38]. In addition, expression of mutant PrP induces aggregation of wild type PrP in the ER [67]. Taking together, it is possible to suggest that alterations in prion protein intracellular metabolism are directly linked with the pathologenesis of TSEs.
CONCLUDING REMARKS
In this review, we have described a general overview of the biochemical and cellular events involved in the pathogenesis of TSEs. PrP maturation is a highly regulated process and the cellular signaling pathways responsible for the elimination of misfolded prion protein appear to play a key role in controlling the cellular fate. Compelling evidence indicate that the accumulation of misfolded PrP (mutant PrP or infectious PrPSC) irreversibly damage the homeostasis of the cell. Particularly relevant seems to be the induction of chronic ER stress that leads to cellular damage. Irreversible stress to the ER has been implicated in the pathology of several neurodegenerative disorders associated with the accumulation of misfolded protein aggregates [50]. For example, the expression of mutant polyglutamine containing proteins (ataxin-3, androgen receptor and huntingtin) induce ER stress-dependent apoptosis in different experimental systems in vitro [68–71]. Transgenic mice expressing a mutant SOD1 allele involved in amyotrophic lateral sclerosis triggers ER stress in the spinal cord linked with caspase-12 activation [72–74]. In-toxin based models of Parkinson activation of the UPR is observed in vitro and in vivo in dopaminergic neurones [75–78]. Aggregated amyloid beta peptide (implicated in Alzheimer’s disease) triggers ER stress in vivo when injected into the brain of healthy animals [79,80]. In addition, in many other conditions of neuronal dysfunction, such as brain trauma and ischemia, the involvement of the UPR/ER stress pathway has been implicated [81]. Finally, mice homozygous for the woozy mutation develop adult-onset ataxia with cerebellar Purkinje cell loss [82]. This mutation affect a co-factor of the chaperone Grp78/BiP and leads to intracellular protein accumulations reminiscent of protein inclusions in both the ER and the nucleus [82]. In addition, upregulation of the UPR is observed in this mouse model, demonstrating that alteration of ER homeostasis can lead to neurodegenerative conditions.
It remains to be established at the molecular level how disease-related protein misfolding Shows ER stress and which is the exact contribution to neuronal dysfunction. A proper understanding of the molecular cross-talk between the different pathways that control protein folding, misfolding, trafficking and clearance is imperative to determine which cellular processes mediate neuronal dysfunction. This knowledge is crucial for the development of rationally-designed therapeutic strategies for TSEs and other neurodegenerative disorders. Assuming that protein misfolding is the triggering event in the development of these diseases, therapeutic approaches that prevent or reverse this process might have beneficial consequences for disease treatment.
Acknowledgments
We thank Eric Smith (Dana Farber Cancer Institute, Boston, USA) for graphics design. CH is supported by a Post Doctoral Fellowship from Damon Runyon Cancer Research Foundation. This work was supported in part by a NIH grant 1R01NS050349-01 to CS.
References
- 1.Prusiner SB. Proc Natl Acad Sci USA. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prusiner SB, Scott MR. Annu Rev Genet. 1997;286:593–606. doi: 10.1146/annurev.genet.31.1.139. [DOI] [PubMed] [Google Scholar]
- 3.Collinge J. Annu Rev Neurosci. 2001;24:519–550. doi: 10.1146/annurev.neuro.24.1.519. [DOI] [PubMed] [Google Scholar]
- 4.Pan KM, Baldwin M, Njuyen J, Gassett M, Serban A, Groth D, Mehlhorn I, Prusiner SB. Proc Natl Acad Sci USA. 1993;90:10962–10966. doi: 10.1073/pnas.90.23.10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hetz C, Maundrell K, Soto C. Trends Mol Med. 2003;9:237–243. doi: 10.1016/s1471-4914(03)00069-8. [DOI] [PubMed] [Google Scholar]
- 6.Bueler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, Weissmann C. Cell. 1993;73:1339–1347. doi: 10.1016/0092-8674(93)90360-3. [DOI] [PubMed] [Google Scholar]
- 7.Kocisko DA, Come JH, Priola SA, Chesebro B, Raymond GJ, Lansbury PT, Caughey B. Nature. 1994;370:471–474. doi: 10.1038/370471a0. [DOI] [PubMed] [Google Scholar]
- 8.Deleault NR, Lucassen RW, Supattapone S. Nature. 2003;425:717–720. doi: 10.1038/nature01979. [DOI] [PubMed] [Google Scholar]
- 9.Saborio GP, Permanne B, Soto C. Nature. 2001;411:810–813. doi: 10.1038/35081095. [DOI] [PubMed] [Google Scholar]
- 10.Castilla J, Saa P, Soto C. Nat Med. 2005;11:982–985. doi: 10.1038/nm1286. [DOI] [PubMed] [Google Scholar]
- 11.Castilla J, Saa P, Hetz C, Soto C. Cell. 2005;121:195–206. doi: 10.1016/j.cell.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 12.Legname G, Baskakov IV, Nguyen HO, Riesner D, Cohen FE, DeArmond SJ, Prusiner SB. Science. 2004;305:673–676. doi: 10.1126/science.1100195. [DOI] [PubMed] [Google Scholar]
- 13.Hetz C, Soto C. Cell Mol Life Sci. 2003;60:133–143. doi: 10.1007/s000180300009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Russelakis-Carneiro M, Hetz C, Maundrell K, Soto C. Am J Pathol. 2004;165:1839–1848. doi: 10.1016/S0002-9440(10)63439-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chiesa R, Drisaldi B, Quaglio E, Migheli A, Piccardo P, Ghetti B, Harris DA. Proc Natl Acad Sci USA. 2000;97:5574–5579. doi: 10.1073/pnas.97.10.5574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Supattapone S, Bouzamondo E, Ball HL, Wille H, Nguyen HO, Cohen FE, DeArmond SJ, Prusiner SB, Scott M. Mol Cell Biol. 2001;21:2608–2616. doi: 10.1128/MCB.21.7.2608-2616.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Supattapone S, Bosque P, Muramoto T, Wille H, Aagaard C, Peretz D, Nguyen HO, Heinrich C, Torchia M, Safar J, Cohen FE, DeArmond SJ, Prusiner SB, Scott M. Cell. 1999;96(6):869–78. doi: 10.1016/s0092-8674(00)80596-6. [DOI] [PubMed] [Google Scholar]
- 18.Forloni G, Angeretti N, Chiesa R, Monzani E, Salmona M, Bugiani O, Tagliavini F. Nature. 1993;362:543–546. doi: 10.1038/362543a0. [DOI] [PubMed] [Google Scholar]
- 19.Thellung S, Florio T, Corsaro A, Arena S, Merlino M, Salmona M, Tagliavini F, Bugiani O, Forloni G, Schettini G. Int J Dev Neurosci. 2000;18:481–492. doi: 10.1016/s0736-5748(00)00005-8. [DOI] [PubMed] [Google Scholar]
- 20.O’Donovan CN, Tobin D, Cotter TG. J Biol Chem. 2001;276:43516–43523. doi: 10.1074/jbc.M103894200. [DOI] [PubMed] [Google Scholar]
- 21.Bate C, Reid S, Williams A. Neuroreport. 2001;12:2589–2594. doi: 10.1097/00001756-200108080-00059. [DOI] [PubMed] [Google Scholar]
- 22.Post K, Brown DR, Groschup M, Kretzschmar HA, Riesner D. Arch Virol Suppl. 2000:265–273. doi: 10.1007/978-3-7091-6308-5_25. [DOI] [PubMed] [Google Scholar]
- 23.Bate C, Salmona M, Diomede L, Williams A. J Biol Chem. 2004;279:14983–14990. doi: 10.1074/jbc.M313061200. [DOI] [PubMed] [Google Scholar]
- 24.Hetz C, Russelakis-Carneiro M, Maundrell K, Castilla J, Soto C. EMBO J. 2003;22:5435–5445. doi: 10.1093/emboj/cdg537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hetz C, Russelakis-Carneiro M, Walchli S, Carboni S, Vial-Knecht E, Maundrell K, Castilla J, Soto C. J Neurosci. 2005;25:2793–2802. doi: 10.1523/JNEUROSCI.4090-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rubenstein R, Carp RI, Callahan SM. J Gen Virol. 1984;65:2191–2198. doi: 10.1099/0022-1317-65-12-2191. [DOI] [PubMed] [Google Scholar]
- 27.Butler DA, Scott MRD, Bockman M, Borchelt DR, Taraboulos A, Hsiao KK, Kingsbury DT, Prusiner SB. J Virol. 1988;62:1558–1564. doi: 10.1128/jvi.62.5.1558-1564.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cronier S, Laude H, Peyrin JM. Proc Natl Acad Sci U S A. 2004;101:12271–12276. doi: 10.1073/pnas.0402725101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mallucci G, Dickinson A, Linehan J, Klohn PC, Brandner S, Collinge J. Science. 2003;302:871–874. doi: 10.1126/science.1090187. [DOI] [PubMed] [Google Scholar]
- 30.Hegde RS, Rane NS. Trends Neurosci. 2003;26:337–339. doi: 10.1016/S0166-2236(03)00143-7. [DOI] [PubMed] [Google Scholar]
- 31.Naslavsky N, Shmeeda H, Friedlander G, Yanai A, Futerman AH, Barenholz Y, Taraboulos A. J Biol Chem. 1999;274:20763–20771. doi: 10.1074/jbc.274.30.20763. [DOI] [PubMed] [Google Scholar]
- 32.Prusiner SB. Brain Pathol. 1998;8:499–513. doi: 10.1111/j.1750-3639.1998.tb00171.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dimcheff DE, Portis JL, Caughey B. Trends Cell Biol. 2003;13:337–340. doi: 10.1016/s0962-8924(03)00125-9. [DOI] [PubMed] [Google Scholar]
- 34.Yedidia Y, Horonchik L, Tzaban S, Yanai A, Taraboulos A. EMBO J. 2001;20:5383–5391. doi: 10.1093/emboj/20.19.5383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ma J, Lindquist S. Proc Natl Acad Sci USA. 2001;98:14955–14960. doi: 10.1073/pnas.011578098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jin T, Gu Y, Zanusso G, Sy M, Kumar A, Cohen M, Gambetti P, Singh N. J Biol Chem. 2000;275:38699–38704. doi: 10.1074/jbc.M005543200. [DOI] [PubMed] [Google Scholar]
- 37.Zanusso G, Petersen RB, Jin T, Jing Y, Kanoush R, Ferrari S, Gambetti P, Singh N. J Biol Chem. 1999;274:23396–23404. doi: 10.1074/jbc.274.33.23396. [DOI] [PubMed] [Google Scholar]
- 38.Singh N, Zanusso G, Chen SG, Fujioka H, Richardson S, Gambetti P, Petersen RB. J Biol Chem. 1997;272:28461–28470. doi: 10.1074/jbc.272.45.28461. [DOI] [PubMed] [Google Scholar]
- 39.Ma J, Lindquist S. Nat Cell Biol. 1999;1:358–361. doi: 10.1038/14053. [DOI] [PubMed] [Google Scholar]
- 40.Ma J, Lindquist S. Science. 2002;298:1784–1788. [Google Scholar]
- 41.Ma J, Wollmann R, Lindquist S. Science. 2002;298:1781–1785. doi: 10.1126/science.1073725. [DOI] [PubMed] [Google Scholar]
- 42.Drisaldi B, Stewart RS, Adles C, Stewart LR, Quaglio E, Biasini E, Fioriti L, Chiesa R, Harris DA. J Biol Chem. 2003;278:21732–21743. doi: 10.1074/jbc.M213247200. [DOI] [PubMed] [Google Scholar]
- 43.Roucou X, Guo Q, Zhang Y, Goodyer CG, LeBlanc AC. J Biol Chem. 2003;278:40877–40881. doi: 10.1074/jbc.M306177200. [DOI] [PubMed] [Google Scholar]
- 44.Kristiansen M, Messenger MJ, Klohn PC, Brandner S, adsworth JD, Collinge J, Tabrizi SJ. J Biol Chem. 2005;280(46):38851–61. doi: 10.1074/jbc.M506600200. [DOI] [PubMed] [Google Scholar]
- 45.Kopito RR, Sitia R. EMBO Rep. 2000;1:225–231. doi: 10.1093/embo-reports/kvd052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kopito RR. Trends Cell Biol. 2000;10:524–530. doi: 10.1016/s0962-8924(00)01852-3. [DOI] [PubMed] [Google Scholar]
- 47.Kang SC, Brown DR, Whiteman M, Li R, Pan T, Perry G, Wisniewski T, Sy MS, Wong BS. J Pathol. 2004;203:603–608. doi: 10.1002/path.1555. [DOI] [PubMed] [Google Scholar]
- 48.Jiang HY, Wek RC. J Biol Chem. 2005;280:14189–14202. doi: 10.1074/jbc.M413660200. [DOI] [PubMed] [Google Scholar]
- 49.Rutkowski DT, Kaufman RJ. Trends Cell Biol. 2004;14:20–28. doi: 10.1016/j.tcb.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 50.Rao RV, Bredesen DE. Curr Opin Cell Biol. 2004;16:653–662. doi: 10.1016/j.ceb.2004.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schroder M, Kaufman RJ. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 52.Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- 53.Hitomi J, Katayama T, Eguchi Y, Kudo T, Taniguchi M, Koyama Y, Manabe T, Yamagishi S, Bando Y, Imaizumi K, Tsujimoto Y, Tohyama M. J Cell Biol. 2004;165(3):347–56. doi: 10.1083/jcb.200310015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, Nagata K, Harding HP, Ron D. Genes Dev. 2004;18:3066–3077. doi: 10.1101/gad.1250704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL, Ron D. Genes Dev. 1998;12:982–995. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 57.Reinhard C, Shamoon B, Shyamala V, Williams LT. EMBO J. 1997;16:1080–1092. doi: 10.1093/emboj/16.5.1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rao RV, Castro-Obregon S, Frankowski H, Schuler M, Stoka V, del Rio G, Bredesen DE, Ellerby HM. J Biol Chem. 2002;277:21836–21842. doi: 10.1074/jbc.M202726200. [DOI] [PubMed] [Google Scholar]
- 59.Obeng EA, Boise LH. J Biol Chem. 2005;280:29578–29587. doi: 10.1074/jbc.M502685200. [DOI] [PubMed] [Google Scholar]
- 60.Ikeyama S, Wang XT, Li J, Podlutsky A, Martindale JL, Kokkonen G, van HR, Gorospe M, Holbrook NJ. J Biol Chem. 2003;278:16726–16731. doi: 10.1074/jbc.M300677200. [DOI] [PubMed] [Google Scholar]
- 61.Breckenridge DG, Germain M, Mathai JP, Nguyen M, Shore GC. Oncogene. 2003;22:8608–8618. doi: 10.1038/sj.onc.1207108. [DOI] [PubMed] [Google Scholar]
- 62.Yoo BC, Krapfenbauer K, Cairns N, Belay G, Bajo M, Lubec G. Neurosci Lett. 2002;334:196–200. doi: 10.1016/s0304-3940(02)01071-6. [DOI] [PubMed] [Google Scholar]
- 63.Brown AR, Rebus S, McKimmie CS, Robertson K, Williams A, Fazakerley JK. Biochem Biophys Res Commun. 2005;334:86–95. doi: 10.1016/j.bbrc.2005.06.060. [DOI] [PubMed] [Google Scholar]
- 64.Russelakis-Carneiro M, Saborio GP, Anderes L, Soto C. J Biol Chem. 2002;277(39):36872–7. doi: 10.1074/jbc.M202229200. [DOI] [PubMed] [Google Scholar]
- 65.Rudd PM, Endo T, Colominas C, Groth D, Wheeler SF, Harvey DJ, Wormald MR, Serban H, Prusiner SB, Kobata A, Dwek RA. Proc Natl Acad Sci USA. 1999;96 (23):13044–9. doi: 10.1073/pnas.96.23.13044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Beranger F, Mange A, Goud B, Lehmann S. J Biol Chem. 2002;277:38972–38977. doi: 10.1074/jbc.M205110200. [DOI] [PubMed] [Google Scholar]
- 67.Gu Y, Verghese S, Mishra RS, Xu X, Shi Y, Singh N. J Neurochem. 2003;84:10–22. doi: 10.1046/j.1471-4159.2003.01255.x. [DOI] [PubMed] [Google Scholar]
- 68.Kouroku Y, Fujita E, Jimbo A, Kikuchi T, Yamagata T, Momoi MY, Kominami E, Kuida K, Sakamaki K, Yonehara S, Momoi T. Hum Mol Genet. 2002;11(13):1505–15. doi: 10.1093/hmg/11.13.1505. [DOI] [PubMed] [Google Scholar]
- 69.Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K, Inoue K, Hori S, Kakizuka A, Ichijo H. Genes Dev. 2002;16:1345–1355. doi: 10.1101/gad.992302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tsai YC, Fishman PS, Thakor NV, Oyler GA. J Biol Chem. 2003;278:22044–22055. doi: 10.1074/jbc.M212235200. [DOI] [PubMed] [Google Scholar]
- 71.Thomas M, Yu Z, Dadgar N, Varambally S, Yu J, Chinnaiyan AM, Lieberman AP. J Biol Chem. 2005;280:21264–21271. doi: 10.1074/jbc.M500144200. [DOI] [PubMed] [Google Scholar]
- 72.Tobisawa S, Hozumi Y, Arawaka S, Koyama S, Wada M, Nagai M, Aoki M, Itoyama Y, Goto K, Kato T. Biochem Biophys Res Commun. 2003;303:496–503. doi: 10.1016/s0006-291x(03)00353-x. [DOI] [PubMed] [Google Scholar]
- 73.Wootz H, Hansson I, Korhonen L, Napankangas U, Lindholm D. Biochem Biophys Res Commun. 2004;322:281–286. doi: 10.1016/j.bbrc.2004.07.118. [DOI] [PubMed] [Google Scholar]
- 74.Turner BJ, Atkin JD, Farg MA, Zang dW, Rembach A, Lopes EC, Patch JD, Hill AF, Cheema SS. J Neurosci. 2005;25:108–117. doi: 10.1523/JNEUROSCI.4253-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ryu EJ, Harding HP, Angelastro JM, Vitolo OV, Ron D, Greene LA. J Neurosci. 2002;22:10690–10698. doi: 10.1523/JNEUROSCI.22-24-10690.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Holtz WA, O’Malley KL. J Biol Chem. 2003;278:19367–19377. doi: 10.1074/jbc.M211821200. [DOI] [PubMed] [Google Scholar]
- 77.Silva RM, Ries V, Oo TF, Yarygina O, Jackson-Lewis V, Ryu EJ, Lu PD, Marciniak SM, Ron D, Przedborski S, Kholodilov N, Green LA, Burke RE. J Neurochem. 2005;95 (4):974–986. doi: 10.1111/j.1471-4159.2005.03428.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Conn KJ, Gao W, McKee A, Lan MS, Ullman MD, Eisenhauer PB, Fine RE, Wells JM. Brain Res. 2004;1022:164–172. doi: 10.1016/j.brainres.2004.07.026. [DOI] [PubMed] [Google Scholar]
- 79.Ghribi O, Herman MM, DeWitt DA, Forbes MS, Savory J. Brain Res Mol Brain Res. 2001;96:30–38. doi: 10.1016/s0169-328x(01)00256-x. [DOI] [PubMed] [Google Scholar]
- 80.Ghribi O, Herman MM, Savory J. J Neurosci Res. 2003;71:853–862. doi: 10.1002/jnr.10511. [DOI] [PubMed] [Google Scholar]
- 81.Aufenberg C, Wenkel S, Mautes A, Paschen W. J Neurotrauma. 2005;22:1018–1024. doi: 10.1089/neu.2005.22.1018. [DOI] [PubMed] [Google Scholar]
- 82.Zhao L, Longo-Guess C, Harris BS, Lee JW, Ackerman SL. Nat Genet. 2005;37:974–979. doi: 10.1038/ng1620. [DOI] [PubMed] [Google Scholar]